Viriditoxin Stabilizes Microtubule Polymers in SK-OV-3 Cells and Exhibits Antimitotic and Antimetastatic Potential


Abstract
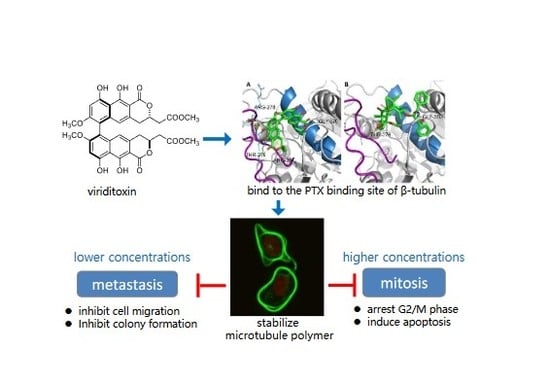
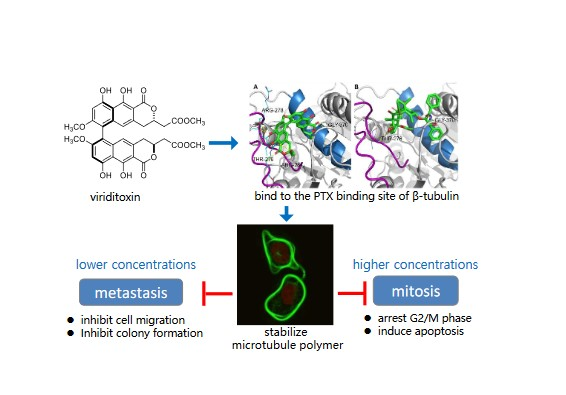
:
1. Introduction
2. Results and Discussion
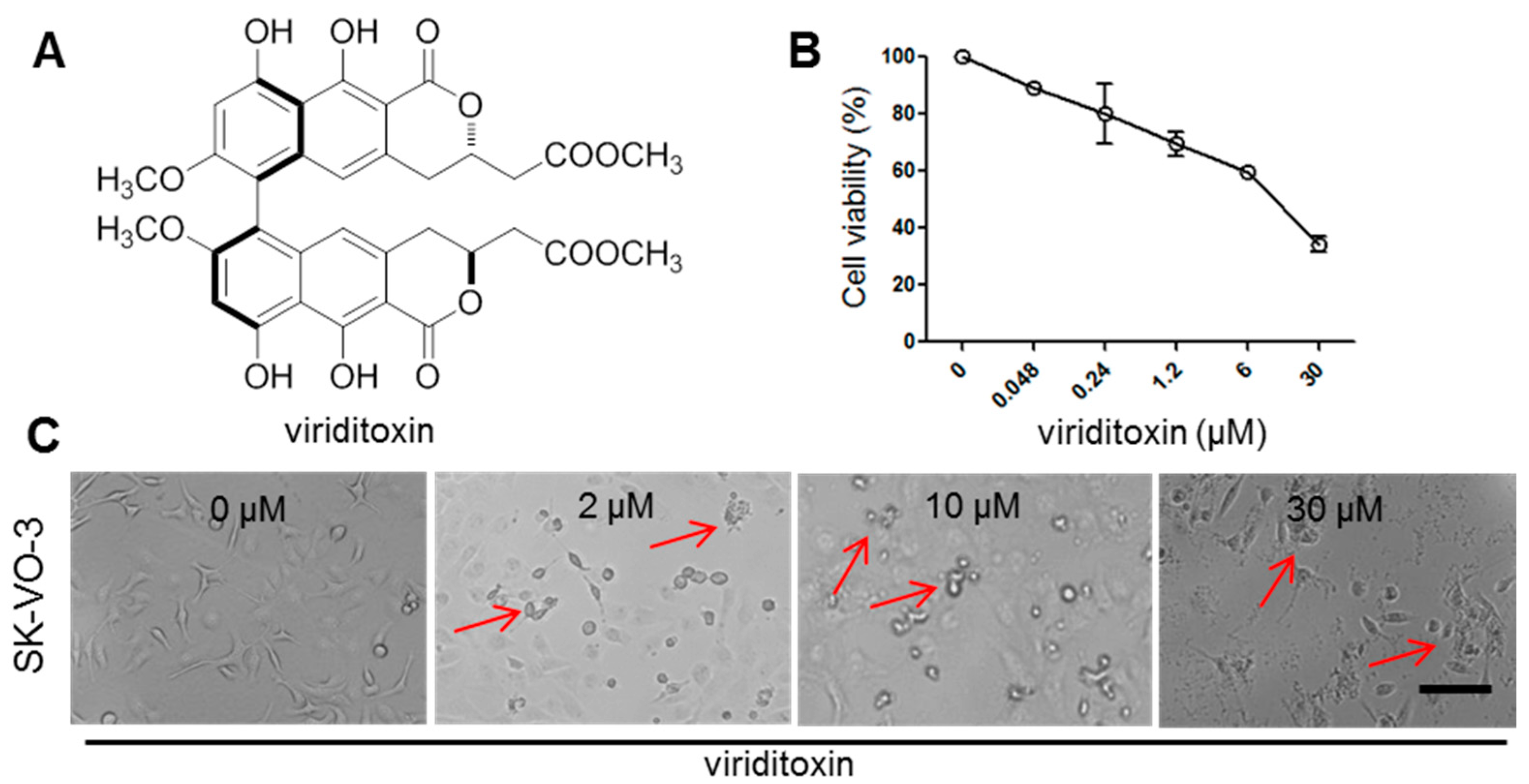
2.1. Viriditoxin Inhibited Proliferation of SK-OV-3 Cells
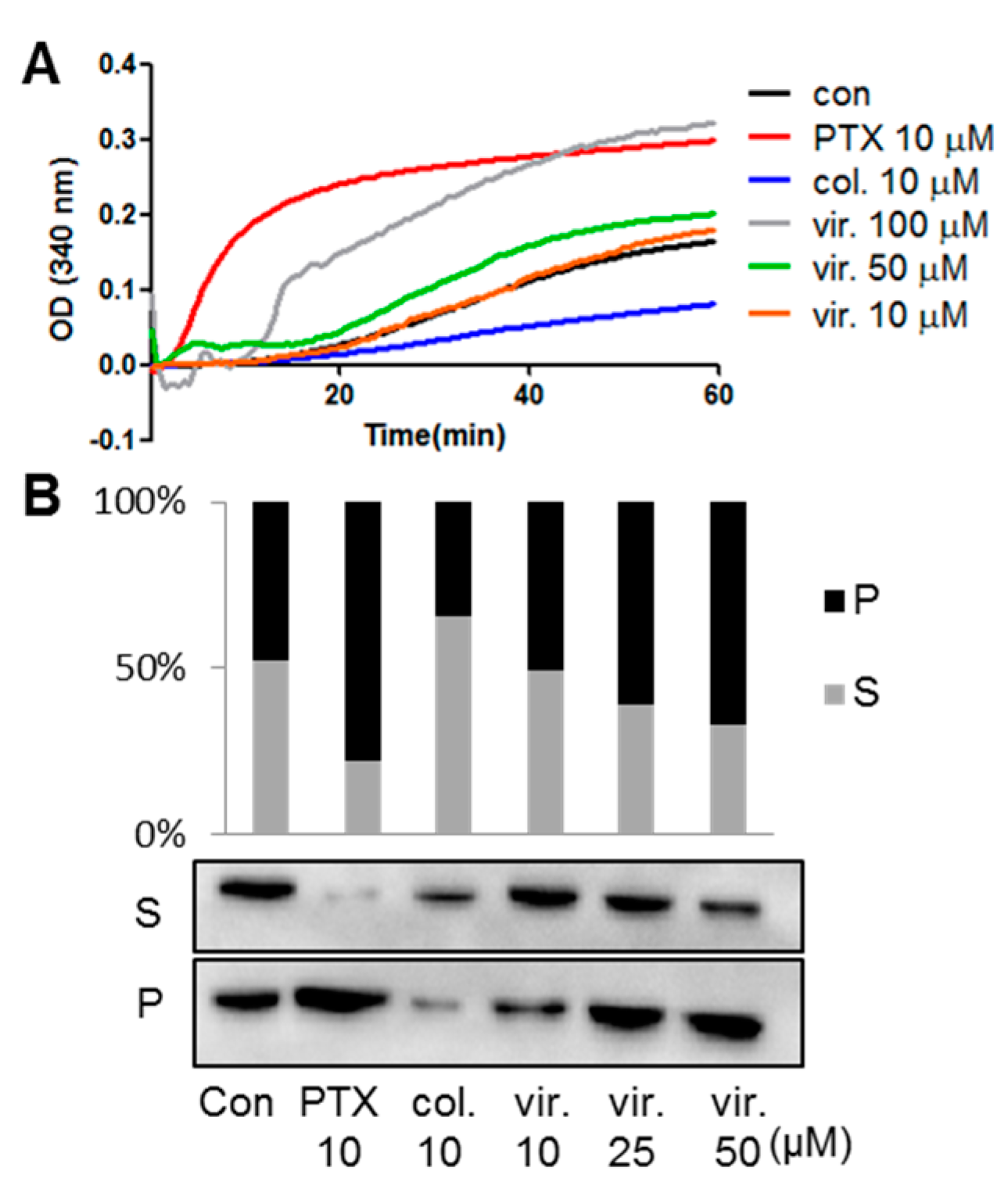
2.2. Viriditoxin Enhanced Tubulin Assembly
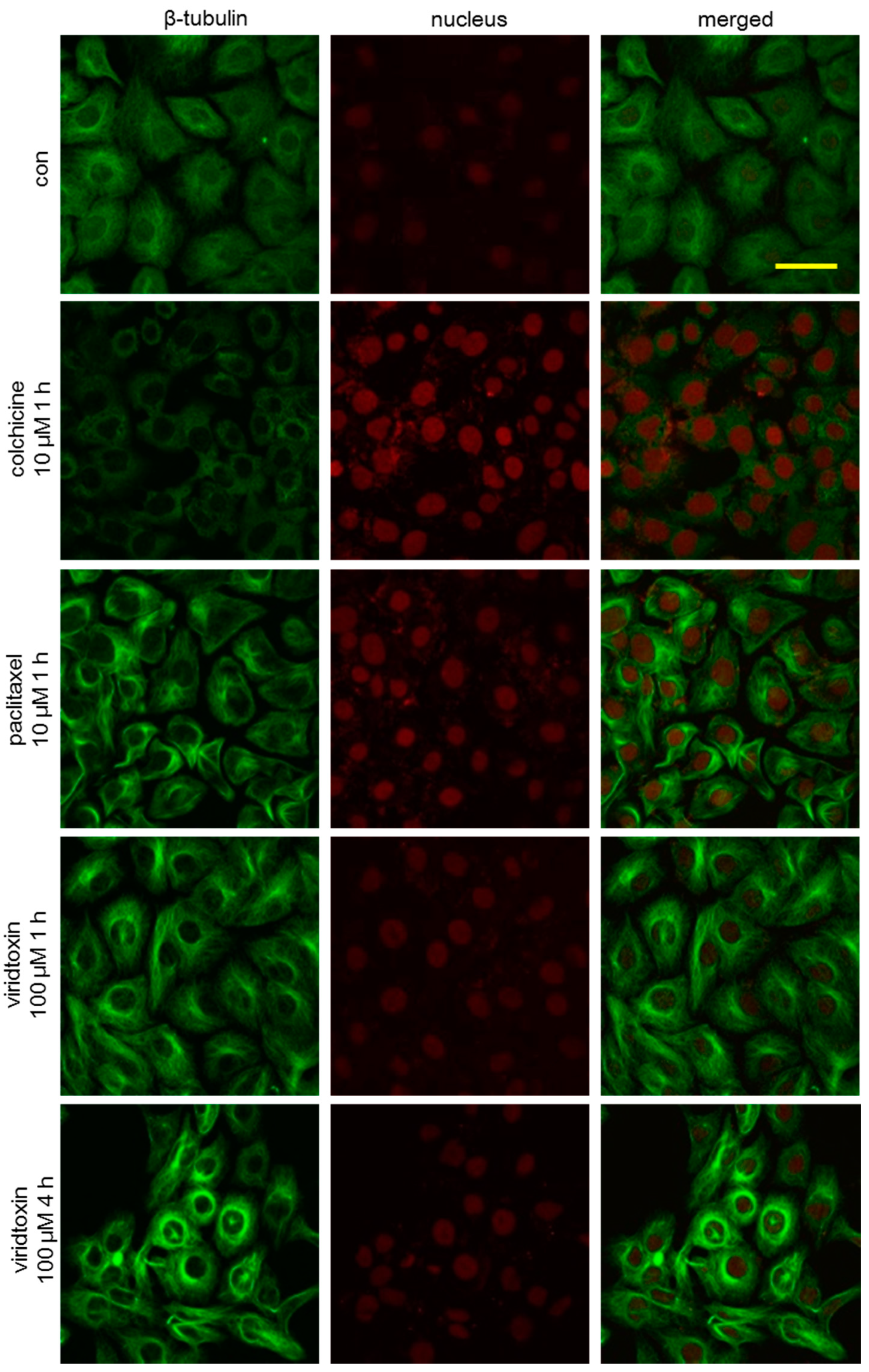
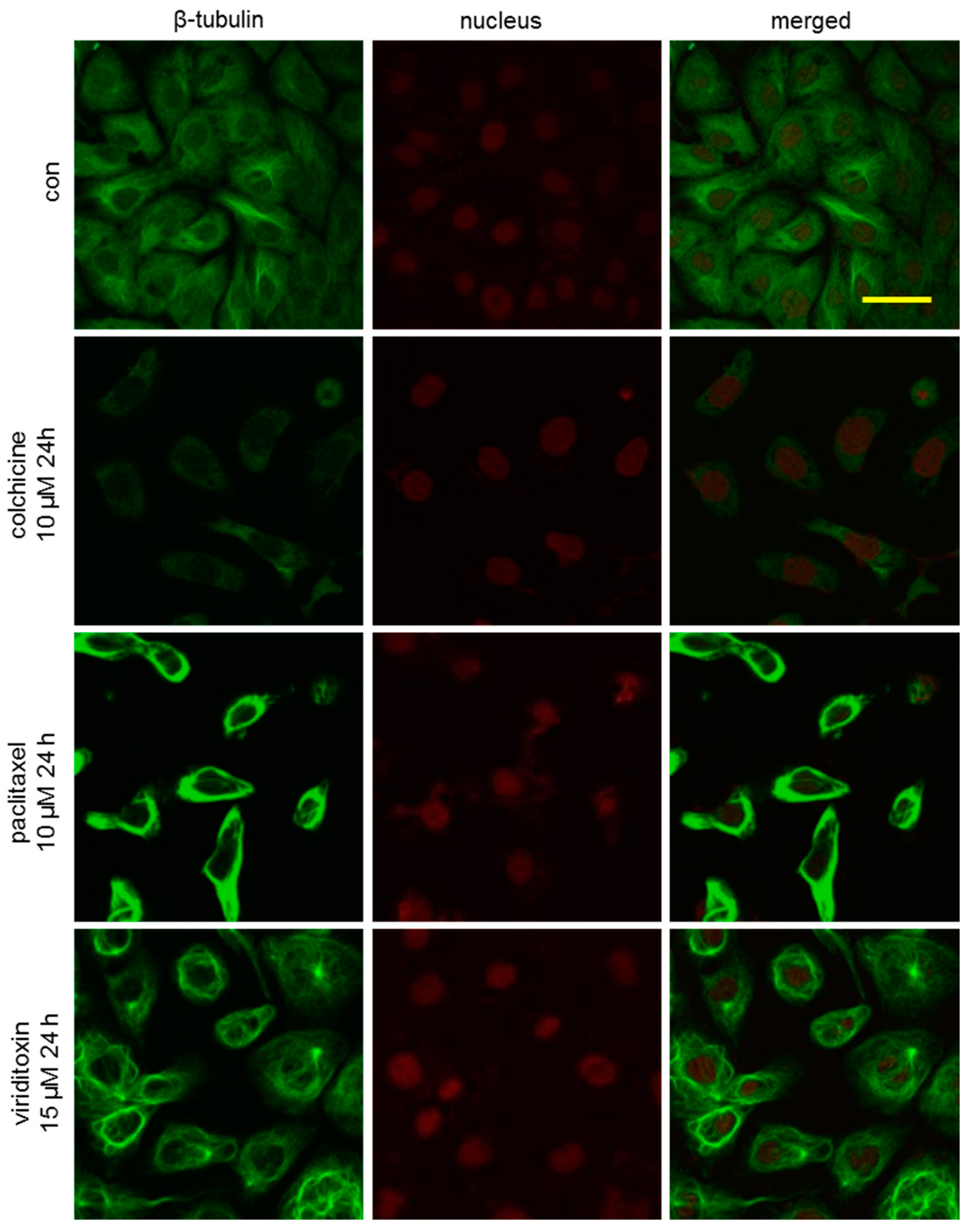
2.3. Viriditoxin Stabilized Microtubule Polymers in SK-OV-3 Cells
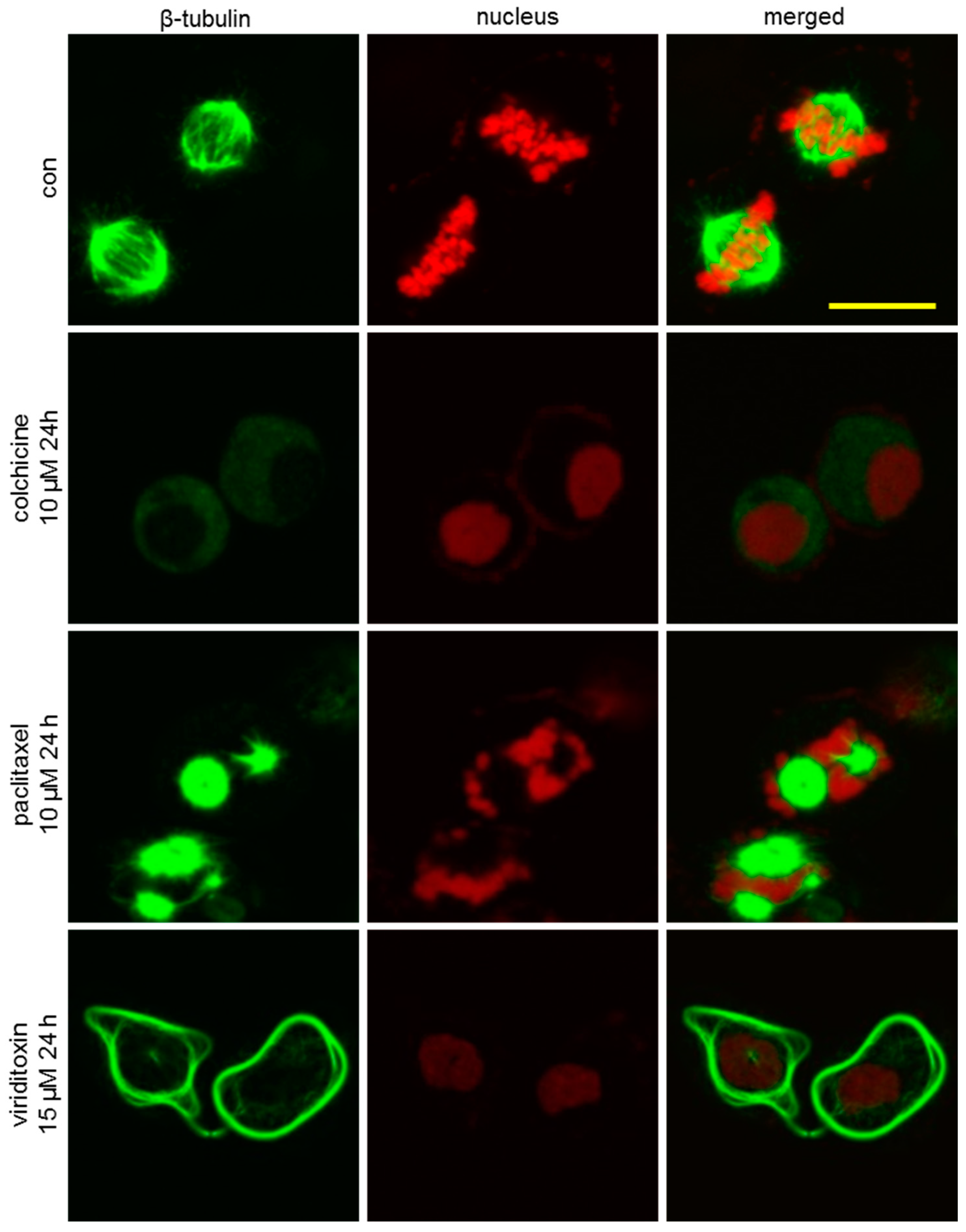
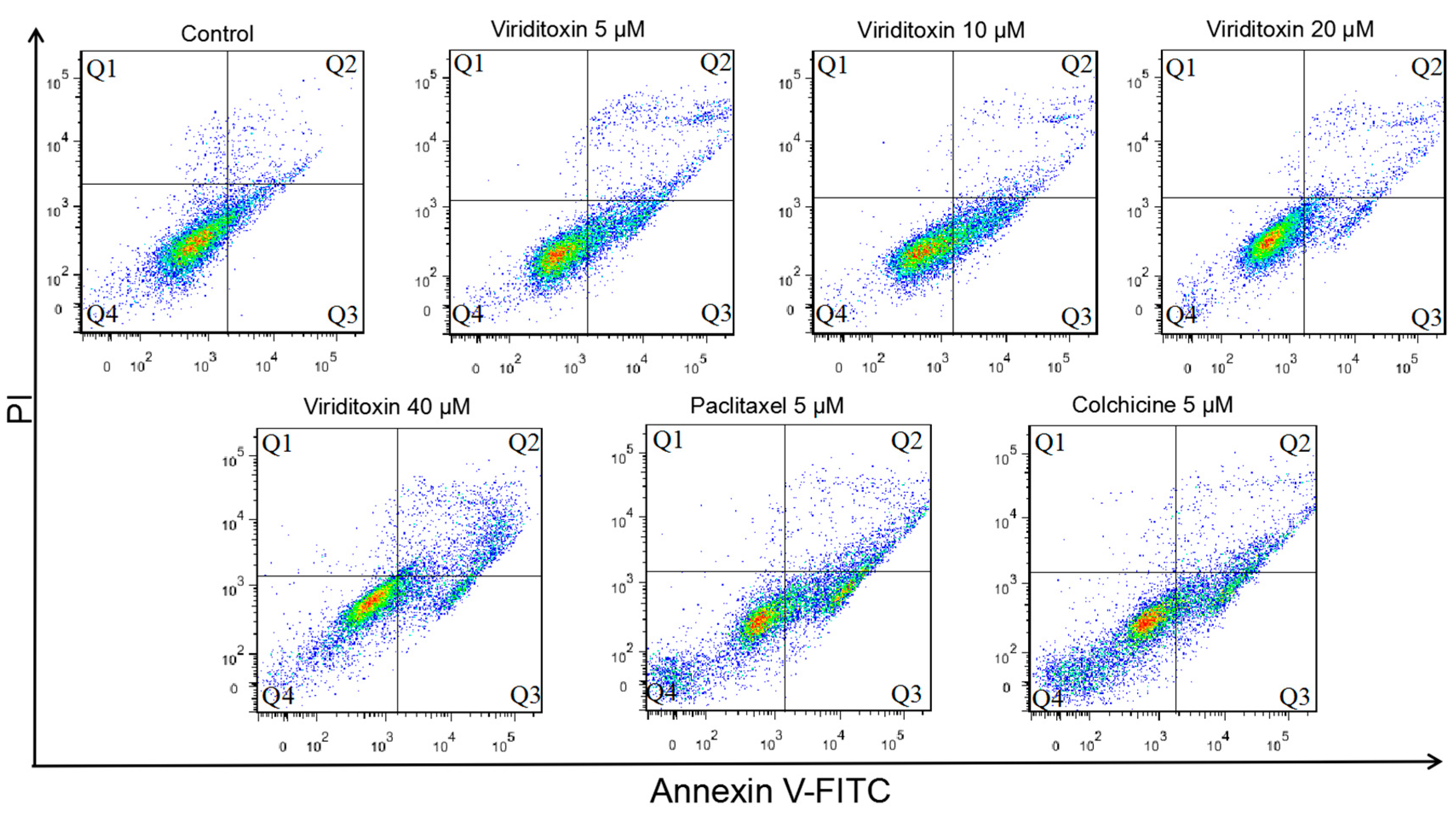
2.4. Viriditoxin Induced G2/M Phase Arrest and Apoptosis in SK-OV-3 Cells
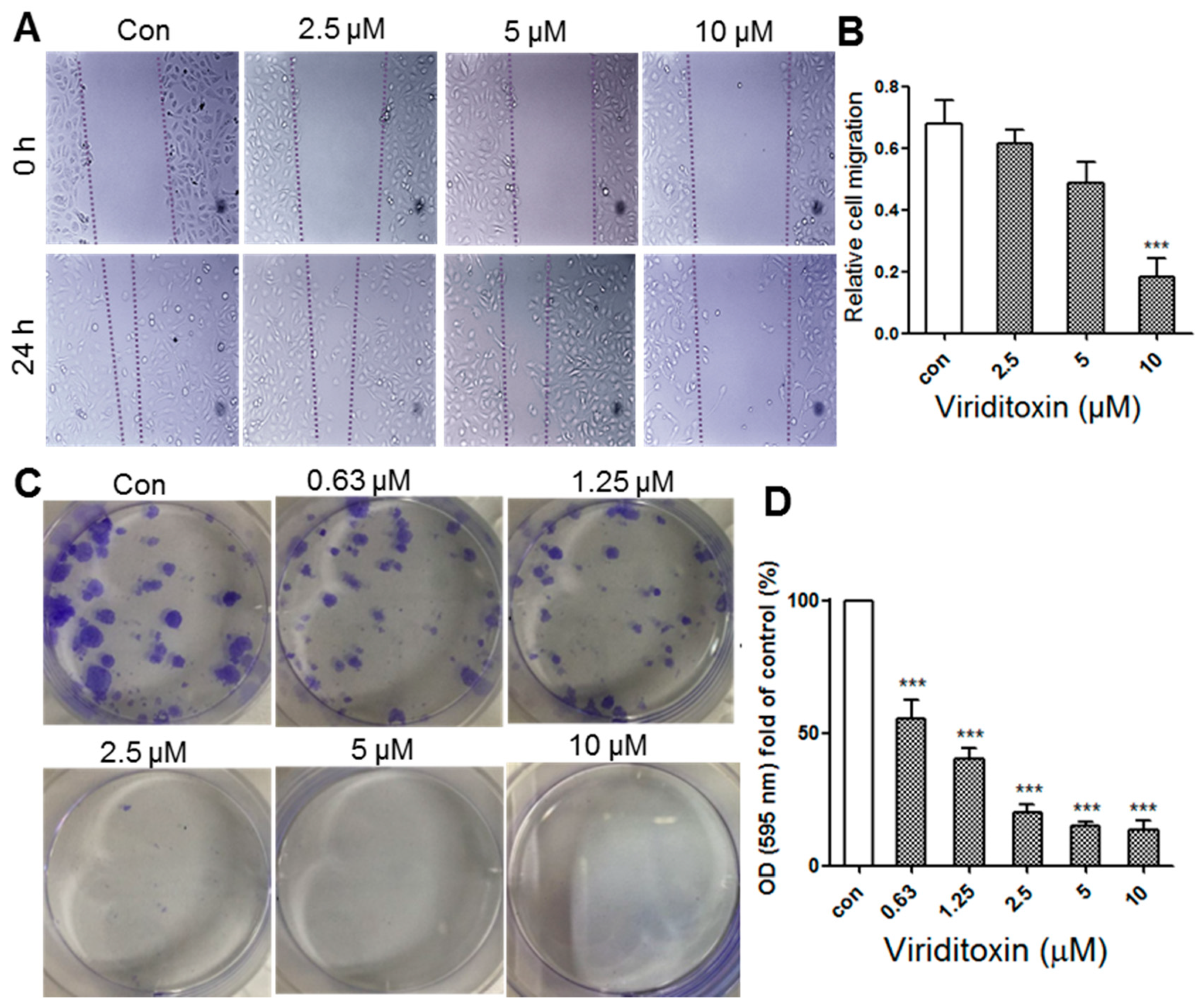
2.5. Viriditoxin Inhibited Migration and Proliferation of SK-OV-3 Cells
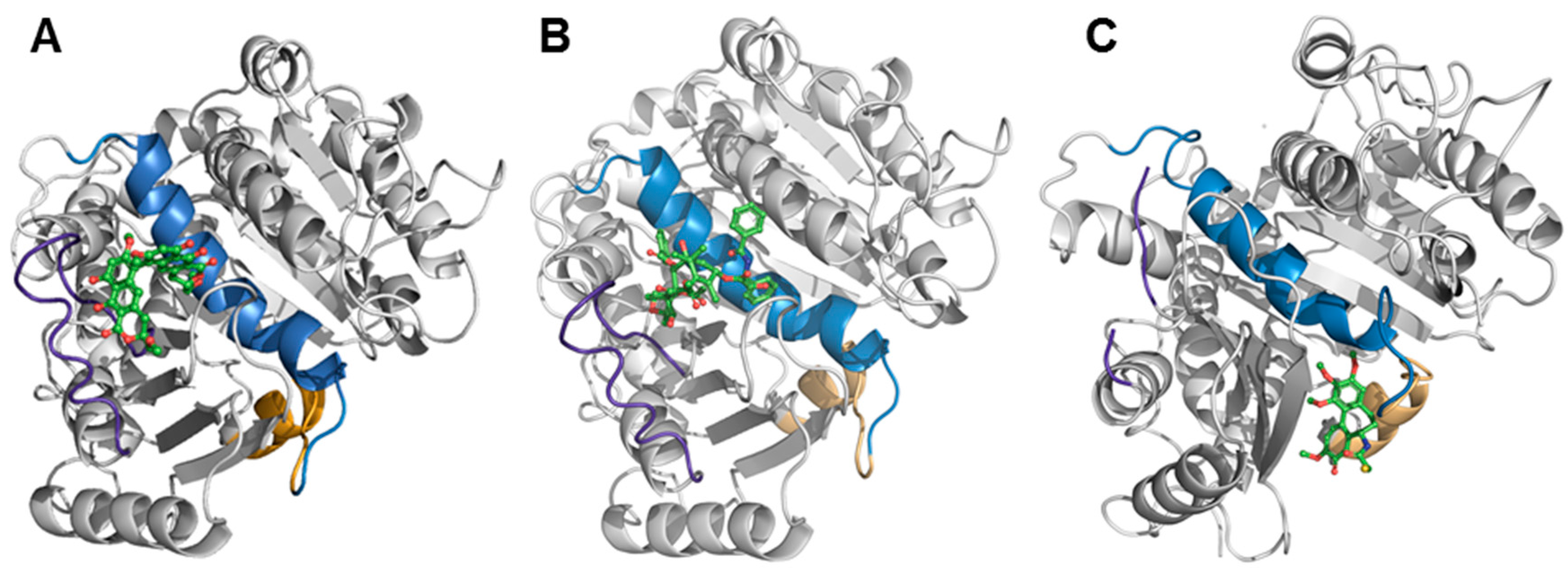
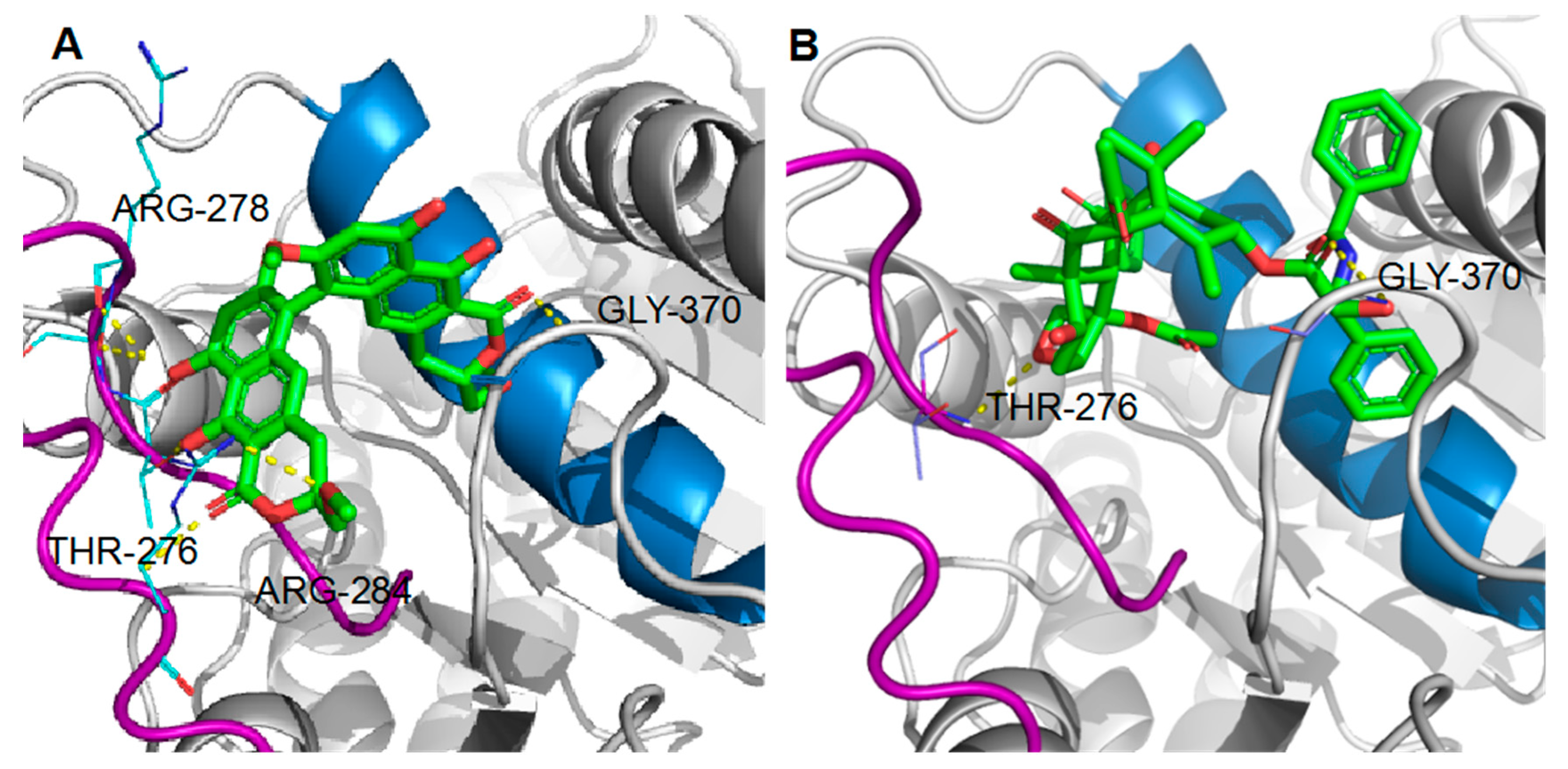
2.6. Binding of Viriditoxin to β-Tubulin
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Cell Lines
3.3. Cell Viability Assay
3.4. In Vitro Tubulin Polymerization Assay
3.5. Western Blot Analysis
3.6. Immunofluorescence
3.7. Cell Cycle Analysis
3.8. Annexin V-FITC Binding Assay
3.9. Cell Migration and Clonogenic Assay
3.10. Microtubule Binding Assay
3.11. Molecular Docking Study
3.12. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Jordan, A.; Hadfield, J.A.; Lawrence, N.J.; McGown, A.T. Tubulin as a target for anticancer drugs: Agents which interact with the mitotic spindle. Med. Res. Rev. 1998, 18, 259–296. [Google Scholar] [CrossRef]
- Luduena, R.F.; Banerjee, A. The tubulin superfamily. In The Role of Microtubules in Cell Biology, Neurobiology, and Oncology. Cancer Drug Discovery and Development; Fojo, T., Ed.; Humana Press: Totowa, NJ, USA, 2008. [Google Scholar]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Sorger, P.K.; Dobles, M.; Tournebize, R.; Hyman, A.A. Coupling cell division and cell death to microtubule dynamics. Curr. Opin. Cell Biol. 1997, 9, 807–814. [Google Scholar] [CrossRef]
- Chen, L.; Zhao, Y.-Y.; Lan, R.-F.; Du, L.; Wang, B.-S.; Zhou, T.; Li, Y.-P.; Zhang, Q.-Q.; Ying, M.-G.; Zheng, Q.-H. Dicitrinone D, an antimitotic polyketide isolated from the marine-derived fungus Penicillium citrinum. Tetrahedron 2017, 73, 5900–5911. [Google Scholar] [CrossRef]
- Mooberry, S.L.; Tien, G.; Hernandez, A.H.; Plubrukarn, A.; Davidson, B.S. Laulimalide and isolaulimalide, new paclitaxel-like microtubule-stabilizing agents. Cancer Res. 1999, 59, 653–660. [Google Scholar] [PubMed]
- Prota, A.E.; Bargsten, K.; Northcote, P.T.; Marsh, M.; Altmann, K.H.; Miller, J.H.; Díaz, J.F.; Steinmetz, M.O. Structural basis of microtubule stabilization by laulimalide and peloruside A. Angew. Chem. Int. Edit. 2014, 53, 1621–1625. [Google Scholar] [CrossRef]
- Liu, J.; Li, F.M.; La Kim, E.; Hong, J.K.; Jung, J.H. Viriditoxin, from a Jellyfish-derived Fungus, is Antibiotic to Fish Pathogens. Nat. Prod. Sci. 2013, 19, 61–65. [Google Scholar]
- Kundu, S.; Kim, T.H.; Yoon, J.H.; Shin, H.-S.; Lee, J.; Jung, J.H.; Kim, H.S. Viriditoxin regulates apoptosis and autophagy via mitotic catastrophe and microtubule formation in human prostate cancer cells. Int. J. Oncol. 2014, 45, 2331–2340. [Google Scholar] [CrossRef]
- Park, J.H.; Noh, T.H.; Wang, H.; Kim, N.D.; Jung, J.H. Viriditoxin induces G2/M cell cycle arrest and apoptosis in A549 human lung cancer cells. Nat. Prod. Sci. 2015, 21, 282–288. [Google Scholar] [CrossRef]
- Wang, J.; Galgoci, A.; Kodali, S.; Herath, K.B.; Jayasuriya, H.; Dorso, K.; Vicente, F.; González, A.; Cully, D.; Bramhill, D. Discovery of a small molecule that inhibits cell division by blocking FtsZ, a novel therapeutic target of antibiotics. J. Biol. Chem. 2003, 278, 44424–44428. [Google Scholar] [CrossRef] [Green Version]
- Erickson, H.P. FtsZ, a tubulin homologue in prokaryote cell division. Trends Cell Biol. 1997, 7, 362–367. [Google Scholar] [CrossRef]
- Erickson, H.P. Atomic structures of tubulin and FtsZ. Trends Cell Biol. 1998, 8, 133–137. [Google Scholar] [CrossRef]
- Anderson, D.E.; Kim, M.B.; Moore, J.T.; O’Brien, T.E.; Sorto, N.A.; Grove, C.I.; Lackner, L.L.; Ames, J.B.; Shaw, J.T. Comparison of small molecule inhibitors of the bacterial cell division protein FtsZ and identification of a reliable cross-species inhibitor. ACS Chem. Biol. 2012, 7, 1918–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bramhill, D. Bacterial cell division. Annu. Rev. Cell Dev. Biol. 1997, 13, 395–424. [Google Scholar] [CrossRef] [PubMed]
- Rothfield, L.; Justice, S.; Garcia-Lara, J. Bacterial cell division. Annu. Rev. Genet. 1999, 33, 423–448. [Google Scholar] [CrossRef]
- Bramhill, D.; Thompson, C.M. GTP-dependent polymerization of Escherichia coli FtsZ protein to form tubules. Proc. Natl. Acad. Sci. USA 1994, 91, 5813–5817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamch, Ł.; Bazylińska, U.; Kulbacka, J.; Pietkiewicz, J.; Bieżuńska-Kusiak, K.; Wilk, K.A. Polymeric micelles for enhanced Photofrin II® delivery, cytotoxicity and pro-apoptotic activity in human breast and ovarian cancer cells. Photodiagnosis Photodyn. Ther. 2014, 11, 570–585. [Google Scholar] [CrossRef]
- Tiberio, P.; Cavadini, E.; Cleris, L.; Dallavalle, S.; Musso, L.; Daidone, M.G.; Appierto, V. Sodium 4-Carboxymethoxyimino-(4-HPR) a Novel Water-Soluble Derivative of 4-Oxo-4-HPR Endowed with In Vivo Anticancer Activity on Solid Tumors. Front. Pharmacol. 2017, 8, 226. [Google Scholar] [CrossRef]
- Pai, H.C.; Kumar, S.; Shen, C.-C.; Liou, J.P.; Pan, S.L.; Teng, C.M. MT-4 suppresses resistant ovarian cancer growth through targeting tubulin and HSP27. PLoS ONE 2015, 10, e0123819. [Google Scholar] [CrossRef]
- Adams, G.; Zhou, J.; Wang, W.; Wu, H.; Quan, J.; Liu, Y.; Xia, P.; Wang, Z.; Zhou, S.; Jiang, J. The microtubule plus end tracking protein TIP150 interacts with cortactin to steer directional cell migration. J. Biol. Chem. 2016, 291, 20692–20706. [Google Scholar] [CrossRef] [Green Version]
- Mukhtar, E.; Adhami, V.M.; Sechi, M.; Mukhtar, H. Dietary flavonoid fisetin binds to β-tubulin and disrupts microtubule dynamics in prostate cancer cells. Cancer Lett. 2015, 367, 173–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinmetz, M.O.; Prota, A.E. Microtubule-targeting agents: Strategies to hijack the cytoskeleton. Trends Cell Biol. 2018, 28, 776–792. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, Y.; Li, G.-B.; Li, S.-A.; Wu, C.; Gigant, B.; Qin, W.; Chen, H.; Wu, Y.; Chen, Q. Mechanism of microtubule stabilization by taccalonolide AJ. Nat. Commun. 2017, 8, 15787. [Google Scholar] [CrossRef]
- Löwe, J.; Li, H.; Downing, K.; Nogales, E. Refined structure of αβ-tubulin at 3.5 Å resolution. J. Mol. Biol. 2001, 313, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Simmerling, C.; Ojima, I. Recent advances in the study of the bioactive conformation of taxol. ChemMedChem 2009, 4, 719–731. [Google Scholar] [CrossRef] [Green Version]
- Prota, A.E.; Bargsten, K.; Zurwerra, D.; Field, J.J.; Díaz, J.F.; Altmann, K.-H.; Steinmetz, M.O. Molecular mechanism of action of microtubule-stabilizing anticancer agents. Science 2013, 339, 587–590. [Google Scholar] [CrossRef] [Green Version]
- Ravelli, R.B.; Gigant, B.; Curmi, P.A.; Jourdain, I.; Lachkar, S.; Sobel, A.; Knossow, M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [Google Scholar] [CrossRef]
- Su, M.; Cao, J.; Huang, J.; Liu, S.; Im, D.; Yoo, J.-W.; Jung, J. The in vitro and in vivo anti-inflammatory effects of a phthalimide PPAR-γ agonist. Mar. Drugs 2017, 15, 7. [Google Scholar] [CrossRef] [Green Version]
- Noh, T.H.; Sen, L.; Hong, J.; Lee, J.-H.; Moon, H.R.; Jung, J.H. Antibacterial activities of viriditoxin congeners and synthetic analogues against fish pathogens. Bioorg. Med. Chem. Lett. 2017, 27, 4970–4974. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) | ||
|---|---|---|
| Cell | Viriditoxin | Paclitaxel |
| SK-OV-3 | 14.3 | 0.24 |
| A549 [10] | 5.1 | 1.9 |
| KB [10] | 2.3 | |
| HCT116 [10] | 18.0 | |
| SH-SY5Y [10] | 12.0 | |
| LNCaP [9] | 14.8 | |
| DU145 [9] | 18.4 | |
| PC3 [9] | 18.7 | |
| G0/G1 | S | G2/M | ||
|---|---|---|---|---|
| Control | 63.7 ± 5.0 | 10.6 ± 2.0 | 19.3 ± 0.7 | |
| Viriditoxin | 5 μM | 70.7 ± 1.1 | 7.6 ± 0.9 | 17.6 ± 0.6 |
| 10 μM | 65.7 ± 2.8 | 8.7 ± 0.7 | 19.8 ± 0.7 | |
| 20 μM | 59.7 ± 7.7 | 11.1 ± 1.4 | 22.7 ± 4.0 | |
| 40 μM | 52.0 ± 7.7 | 12.0 ± 1.6 | 27.2 ± 3.5 | |
| paclitaxel (5 μM) | 5.7 ± 1.4 | 7.4 ± 3.1 | 72.7 ± 3.7 | |
| colchicine (5 μM) | 3.1 ± 0.8 | 7.7 ± 4.8 | 74.3 ± 8.8 | |
| Live | Early Apoptosis | Late Apoptosis | Necrosis | ||
|---|---|---|---|---|---|
| Control | 90.1 ± 2.6 | 6.6 ± 3.3 | 1.6 ± 0.7 | 1.7 ± 1.5 | |
| Viriditoxin | 5 μM | 82.9 ± 6.2 | 14.6 ± 6.8 | 4.2 ± 1.7 | 0.8 ± 0.4 |
| 10 μM | 80.2 ± 7.0 | 14.8 ± 7.1 | 3.7 ± 0.7 | 1.3 ± 1.1 | |
| 20 μM | 84.3 ± 2.9 | 8.8 ± 1.6 | 5.0 ± 1.0 | 1.9 ± 1.3 | |
| 40 μM | 76.0 ± 3.9 | 10.9 ± 0.9 | 11.0 ± 4.1 | 2.1 ± 1.2 | |
| paclitaxel (5 μM) | 73.8 ± 6.6 | 16.9 ± 4.5 | 8.0 ± 2.5 | 1.3 ± 0.5 | |
| colchicine (5 μM) | 77.4 ± 5.6 | 15.1 ± 3.6 | 6.4 ± 2.4 | 1.0 ± 0.4 | |
| Distribution (%) | Incubated with a Single Ligand | Incubated with Two Ligands | ||
|---|---|---|---|---|
| Viriditoxin a | Paclitaxel b | Viriditoxin a | Paclitaxel b | |
| microtubule pellet | 99.95 ± 0.07 | 61.44 ± 13.98 | 91.97 ± 13.83 | 58.99 ± 12.31 |
| supernatant | 0 ± 0.07 | 38.56 ± 13.98 | 8.03 ± 13.83 | 41.00 ± 12.31 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Su, M.; Zhao, C.; Li, D.; Cao, J.; Ju, Z.; Kim, E.L.; Jung, Y.-S.; Jung, J.H. Viriditoxin Stabilizes Microtubule Polymers in SK-OV-3 Cells and Exhibits Antimitotic and Antimetastatic Potential. Mar. Drugs 2020, 18, 445. https://doi.org/10.3390/md18090445
Su M, Zhao C, Li D, Cao J, Ju Z, Kim EL, Jung Y-S, Jung JH. Viriditoxin Stabilizes Microtubule Polymers in SK-OV-3 Cells and Exhibits Antimitotic and Antimetastatic Potential. Marine Drugs. 2020; 18(9):445. https://doi.org/10.3390/md18090445
Chicago/Turabian StyleSu, Mingzhi, Changhao Zhao, Dandan Li, Jiafu Cao, Zhiran Ju, Eun La Kim, Young-Suk Jung, and Jee H. Jung. 2020. "Viriditoxin Stabilizes Microtubule Polymers in SK-OV-3 Cells and Exhibits Antimitotic and Antimetastatic Potential" Marine Drugs 18, no. 9: 445. https://doi.org/10.3390/md18090445
APA StyleSu, M., Zhao, C., Li, D., Cao, J., Ju, Z., Kim, E. L., Jung, Y.-S., & Jung, J. H. (2020). Viriditoxin Stabilizes Microtubule Polymers in SK-OV-3 Cells and Exhibits Antimitotic and Antimetastatic Potential. Marine Drugs, 18(9), 445. https://doi.org/10.3390/md18090445