Absolute Configurations and Chitinase Inhibitions of Quinazoline-Containing Diketopiperazines from the Marine-Derived Fungus Penicillium polonicum


Abstract
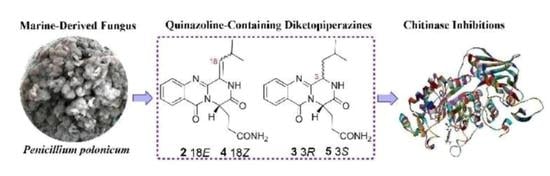
1. Introduction
2. Results
3. Discussion
4. Experimental Section
4.1. General Experimental Procedures
4.2. Isolation of the Fungal Material
4.3. General Computational Procedure
4.4. Preparation and Analysis of Marfey’s Derivatives
4.5. Enzymes Inhibitory Activity Assay
4.6. Molecular Docking
4.7. Cytotoxic Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Xu, W.; Dhingra, S.; Chooi, Y.H.; Calvo, A.M.; Lin, H.C.; Tang, Y. The fumagillin biosynthetic gene cluster in Aspergillus fumigatus encodes a cryptic terpene cyclase involved in the formation of β-trans-bergamotene. J. Am. Chem. Soc. 2013, 135, 4616–4619. [Google Scholar]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2019, 36, 122–173. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.M.; Liang, T.M.; Guo, Z.Y.; Wang, C.Y.; Shao, C.L. Discovery, absolute assignments, and total synthesis of asperversiamides A–C and their potent activity against Mycobacterium marinum. Chem. Commun. 2019, 55, 1104–1107. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Yang, M.Y.; Zhang, Y.H.; Shao, C.L.; Wang, C.Y.; Hu, L.D.; Cao, F.; Zhu, H.J. Absolute configurations of 14,15-hydroxylated prenylxanthones from a marine-derived Aspergillus sp. fungus by chiroptical methods. Sci. Rep. 2018, 8, 10621–10630. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Shao, C.L.; Liu, Y.F.; Zhu, H.J.; Wang, C.Y. Cytotoxic serrulatane-type diterpenoids from the gorgonian euplexaura sp. and their absolute configurations by vibrational circular dichroism. Sci. Rep. 2017, 7, 12548–12555. [Google Scholar] [CrossRef] [PubMed]
- Xin, Z.H.; Fang, Y.C.; Du, L.; Zhu, T.J.; Duan, L.; Chen, J.; Gu, Q.Q.; Zhu, W.M. Aurantiomides A–C, quinazoline alkaloids from the sponge-derived fungus Penicillium aurantiogriseum SP0-19. J. Nat. Prod. 2007, 70, 853–855. [Google Scholar] [CrossRef] [PubMed]
- Boyes-Korkis, J.M.; Gurney, K.A.; Penn, J.; Mantle, P.G.; Bilton, J.N.; Sheppard, R.N. Anacine, a new benzodiazepine metabolite of Penicillium aurantiogriseum produced with other alkaloids in submerged fermentation. J. Nat. Prod. 1993, 56, 1707–1717. [Google Scholar] [CrossRef]
- Wang, H.; Sim, M.M.J. Total solid phase syntheses of the quinazoline alkaloids: verrucines A and B and anacine. Nat. Prod. 2001, 64, 1497–1501. [Google Scholar] [CrossRef] [PubMed]
- Grimblat, N.; Zanardi, M.; Sarotti, A.J. Beyond DP4: An improved probability for the stereochemical assignment of isomeric compounds using quantum chemical calculations of NMR shifts. Org. Chem. 2015, 80, 12526–12534. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Yi, X.X.; Zhou, Y.; Su, X.; Peng, Y.; Gao, C.H. An update on 2,5-diketopiperazines from marine organisms. Mar. Drugs 2014, 12, 6213–6235. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.F.; Mao, N.; Xue, X.J.; Qi, Y.X.; Wei, M.Y.; Wang, C.Y.; Shao, C.L. Structures and absolute configurations of diketopiperazine alkaloids chrysopiperazines A–C from the gorgonian-derived Penicillium chrysogenum Fungus. Mar. Drugs 2019, 17, 250. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Meng, Z.H.; Mu, X.; Yue, Y.F.; Zhu, H.J. Absolute configuration of bioactive azaphilones from the marine-derived fungus Pleosporales sp. CF09-1. J. Nat. Prod. 2019, 82, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.J. Organic Stereochemistry-Experimental and Computational Methods; KGaA; Wiley-VCH Verlag GmbH and Co.: Weinheim, Germany, 2015. [Google Scholar]
- Cao, F.; Meng, Z.H.; Wang, P.; Luo, D.Q.; Zhu, H.J. Dipleosporalones A and B, Dimeric azaphilones from a marine-derived Pleosporales sp. fungus. J. Nat. Prod. 2020, 83, 1283–1287. [Google Scholar] [CrossRef] [PubMed]
- Bruhn, T.; Schaumlöffel, A.; Hemberger, Y.; Bringmann, G. SpecDis: Quantifying the comparison of calculated and experimental electronic circular dichroism spectra. Chirality 2013, 25, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Liao, L.J.; You, M.J.; Chung, B.K.; Oh, D.C.; Oh, K.B.; Shin, J.J. Alkaloidal metabolites from a marine-derived Aspergillus sp. fungus. Nat. Prod. 2015, 78, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Dong, L.; Shen, S.; Chen, W.; Lu, H.; Xu, D.; Jin, S.; Yang, Q.; Zhang, J. Glycosyl triazoles as novel insect β-N-acetylhexosaminidase OfHex1 inhibitors: Design, synthesis, molecular docking and MD simulations. Bioorg. Med. Chem. 2019, 27, 2315–2322. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, H.; Liu, F.; Chen, L.; Shen, X.; Yang, Q. Active-pocket size differentiating insectile from bacterial chitinolytic β-N-acetyl-d-hexosaminidases. Biochem. J. 2011, 438, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Chen, L.; Zhou, Y.; Jiang, X.; Duan, Y.; Yang, Q.J. Structure, catalysis, and inhibition of OfChi-h, the lepidoptera-exclusive insect chitinase. Biol. Chem. 2017, 292, 2080–2088. [Google Scholar] [CrossRef] [PubMed]
- Koska, J.; Spassov, V.Z.; Maynard, A.J.; Yan, L.; Austin, N.; Flook, P.K.; Venkatachalam, C.M. Fully automated molecular mechanics based induced fit protein-ligand docking method. J. Chem. Inf. Model. 2008, 48, 1965–1973. [Google Scholar] [CrossRef] [PubMed]
- Scudiere, D.A.; Shoemaker, R.H.; Paull, K.D.; Monks, A.; Tierney, S.; Nofziger, T.H.; Currens, M.J.; Seniff, D.; Boyd, M.R. Evaluation of a soluble tetrazolium/formazan assay for cell growth and drug sensitivity in culture using human and other tumor cell lines. Cancer Res. 1988, 48, 4827–4833. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | 1 | 2 | 3 | 5 |
|---|---|---|---|---|
| 2 | 10.49, brs | 10.49, brs | 8.53, brs | 8.91, d (4.2) |
| 3 | - | - | 4.74, dd (7.8, 3.6) | 4,40–4.43, m |
| 7 | 7.69, d (7.8) | 7.65, d (8.4) | 7.68, d (8.4) | 7.66, d (7.8) |
| 8 | 7.84, dd (7.8, 7.2) | 7.86, dd (8.4, 7.2) | 7.85, dd (8.4, 7.2) | 7.84, dd (7.8, 7.2) |
| 9 | 7.52, dd (7.8, 7.2) | 7.57, dd (7.8, 7.2) | 7.56, dd (7.8, 7.2) | 7.54, dd (7.8, 7.2) |
| 10 | 8.13, d (7.8) | 8.16, d (7.8) | 8.15, d (7.8) | 8.15, d (7.8) |
| 14 | 5.19, dd (6.6, 6.0) | 5.12, dd (6.6, 6.0) | 5.09, dd (7.8, 6.6) | 4.86, dd (9.0, 6.0) |
| 15 | 2.12–2.17, m | 2.02–2.06, m | 2.17–2.20, m | 2.09–2.14, m |
| - | 2.02–2.07, m | - | 2.12–2.14, m | 2.01–2.06, m |
| 16 | 2.38–2.43, m | 2.11–2.14, m | 2.21–2.26, m | 2.30–2.35, m |
| - | 2.32–2.37, m | - | - | 2.38–2.44, m |
| 18 | 6.22, d (10.8) | 5.54, d (9.6) | 2.28–2.31, m | 1.74–1.81, m |
| - | - | - | 1.64–1.68, m | - |
| 19 | 2.94–3.00, m | 3.74–3.79, m | 2.07–2.11, m | 1.87–1.93, m |
| 20 | 1.05, d (6.6) | 1.02, d (6.6) | 0.97, d (6.6) | 0.97, d (6.6) |
| 21 | 1.08, d (6.6) | 1.20, d (6.6) | 0.98, d (6.6) | 0.99, d (6.6) |
| 17-OCH3 | 3.42, s, 3H | - | - | - |
| 17-NH2 | - | 7.26, s | 7.29, s | 7.36, s |
| - | - | 6.71, s | 6.75, s | 6.78, s |
| No. | 1 | 2 | 3 | 5 |
|---|---|---|---|---|
| 1 | 165.2, C | 165.5, C | 167.9, C | 166.6, C |
| 3 | 126.6, C | 124.8, C | 55.7, CH | 54.9, CH |
| 4 | 145.5, C | 144.8, C | 152.2, C | 152.0, C |
| 6 | 147.0, C | 146.7, C | 146.6, C | 147.0, C |
| 7 | 126.3, CH | 127.5, CH | 127.3, CH | 126.7, CH |
| 8 | 134.7, CH | 134.7, CH | 134.7, CH | 134.7, CH |
| 9 | 126.7, CH | 127.2, CH | 127.0, CH | 126.7, CH |
| 10 | 125.3, CH | 126.3, CH | 126.3, CH | 126.2, CH |
| 11 | 119.7, C | 119.7, C | 119.8, C | 119.7, C |
| 12 | 159.9, C | 159.7, C | 160.1, C | 160.1, C |
| 14 | 54.3, CH | 54.7, CH | 50.7, CH | 53.8, CH |
| 15 | 27.2, CH2 | 28.0, CH2 | 25.6, CH2 | 29.4, CH2 |
| 16 | 29.2, CH2 | 30.9, CH2 | 31.3, CH2 | 32.2, CH2 |
| 17 | 171.8, C | 172.4, C | 172.7, C | 172.8, C |
| 18 | 127.1, CH | 131.0, CH | 39.0 a, CH2 | 47.2, CH2 |
| 19 | 25.0, CH | 26.5, CH | 23.8, CH | 24.0, CH |
| 20 | 22.0, CH3 | 23.1, CH3 | 23.3, CH3 | 23.0, CH3 |
| 21 | 22.3, CH3 | 22.4, CH3 | 21.7, CH3 | 21.4, CH3 |
| 17-OCH3 | 51.3, CH3 | - | - | - |
| Compounds | Inhibition Rate (%) | |||
|---|---|---|---|---|
| OfHex1 | OfChi-h | |||
| 10.0 μM | 10.0 μM | 1.0 μM | 0.2 μM | |
| 1 | 0.7 | 91.9 | 75.1 | 28.1 |
| 2 | 3.8 | 79.1 | 74.3 | 4.0 |
| 3 | 1.4 | 86.1 | 73.1 | 15.8 |
| 4 | 10.3 | 95.4 | 85.5 | 23.2 |
| 5 | 6.8 | 92.3 | 75.9 | 20.1 |
| 6 | 7.4 | 90.5 | 83.9 | 3.2 |
| 7 | 5.9 | 85.7 | 77.6 | 21.7 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, X.-C.; Zhang, Y.-H.; Gao, W.-B.; Pan, L.; Zhu, H.-J.; Cao, F. Absolute Configurations and Chitinase Inhibitions of Quinazoline-Containing Diketopiperazines from the Marine-Derived Fungus Penicillium polonicum. Mar. Drugs 2020, 18, 479. https://doi.org/10.3390/md18090479
Guo X-C, Zhang Y-H, Gao W-B, Pan L, Zhu H-J, Cao F. Absolute Configurations and Chitinase Inhibitions of Quinazoline-Containing Diketopiperazines from the Marine-Derived Fungus Penicillium polonicum. Marine Drugs. 2020; 18(9):479. https://doi.org/10.3390/md18090479
Chicago/Turabian StyleGuo, Xing-Chen, Ya-Hui Zhang, Wen-Bin Gao, Li Pan, Hua-Jie Zhu, and Fei Cao. 2020. "Absolute Configurations and Chitinase Inhibitions of Quinazoline-Containing Diketopiperazines from the Marine-Derived Fungus Penicillium polonicum" Marine Drugs 18, no. 9: 479. https://doi.org/10.3390/md18090479
APA StyleGuo, X.-C., Zhang, Y.-H., Gao, W.-B., Pan, L., Zhu, H.-J., & Cao, F. (2020). Absolute Configurations and Chitinase Inhibitions of Quinazoline-Containing Diketopiperazines from the Marine-Derived Fungus Penicillium polonicum. Marine Drugs, 18(9), 479. https://doi.org/10.3390/md18090479