
Scalarane-Type Sesterterpenoids from the Marine Sponge Lendenfeldia sp. Alleviate Inflammation in Human Neutrophils

 , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Exploring the Metabolomic Diversity of Marine Sponge Lendenfeldia sp. Using the Molecular Networking (MN) Approach
2.2. Chemical Identification of 24-Homoscalaranes 1–12
2.3. Assessments of O2•− Generation and Elastase Release in fMLF-Activated Human Neutrophils
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Animal Material and Isolation of Compounds
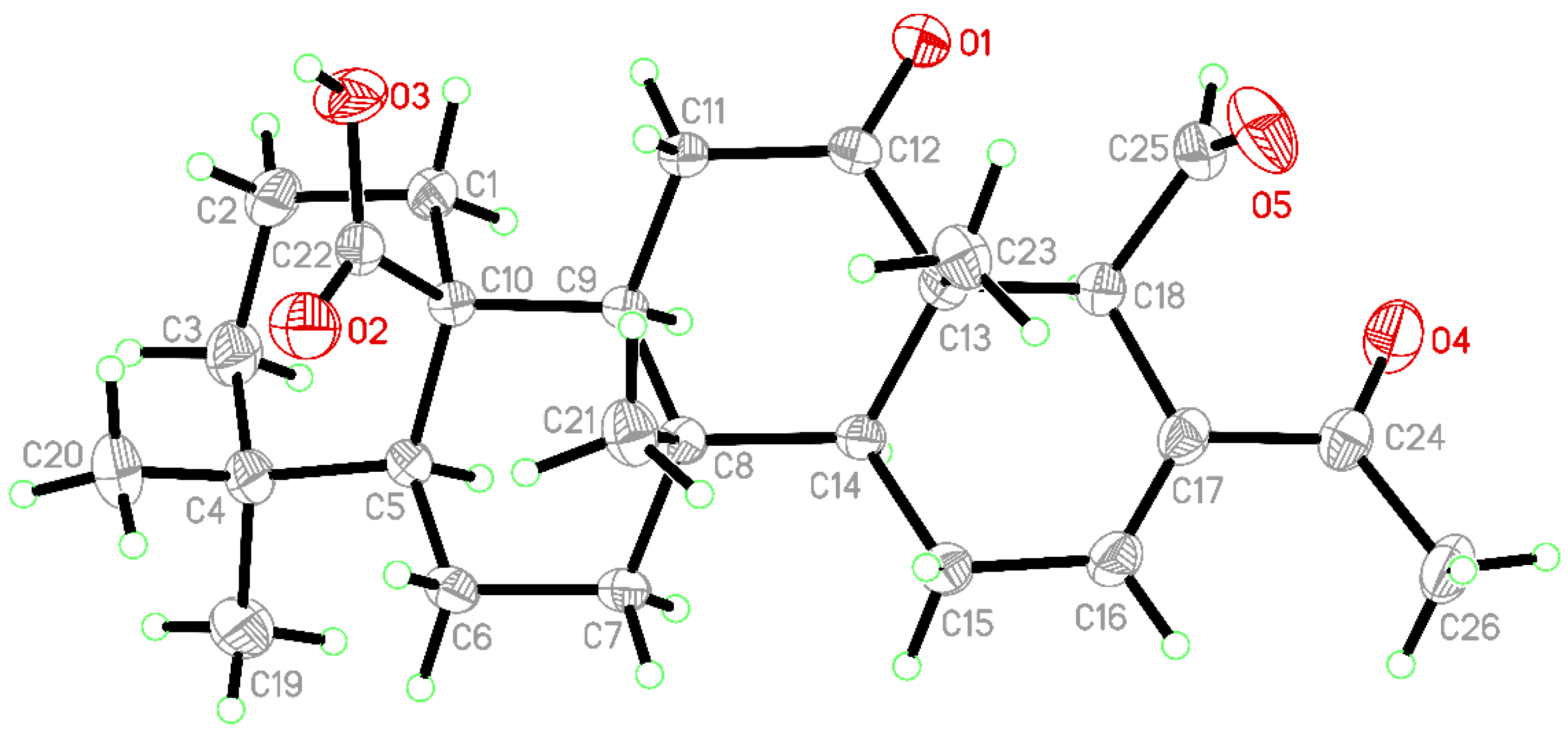
3.3. X-ray Crystallographic Analysis of 24-Methyl-12,24,25-trioxoscalar-16-en-22-oic Acid (10)
3.4. MS/MS Fragmentations Collection Using Ultra-Performance Liquid Chromatography Quadrupole Time-of-Flight (UPLC-QTOF) Mass Spectrometry
3.5. Chromatographic and Spectral Preprocessing Using MZmine
3.6. GNPS Molecular Networking
3.7. Preparation of Human Neutrophils
3.8. Measurement of Superoxide Anion (O2•−) Generation
3.9. Measurement of Elastase Release
3.10. Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Peng, B.-R.; Lai, K.-H.; Chang, Y.-C.; Chen, Y.-Y.; Su, J.-H.; Huang, Y.M.; Chen, P.-J.; Yu, S.S.-F.; Duh, C.-Y.; Sung, P.-J. Sponge-derived 24-homoscalaranes as potent anti-inflammatory agents. Mar. Drugs 2020, 18, 434. [Google Scholar] [CrossRef]
- Peng, B.-R.; Lai, K.-H.; Chen, Y.-Y.; Su, J.-H.; Huang, Y.M.; Chen, Y.-H.; Lu, M.-C.; Yu, S.S.-F.; Duh, C.-Y.; Sung, P.-J. Probing Anti-proliferative 24-homoscalaranes from a sponge Lendenfeldia sp. Mar. Drugs 2020, 18, 76. [Google Scholar] [CrossRef] [Green Version]
- Dai, J.; Liu, Y.; Zhou, Y.-D.; Nagle, D.G. Cytotoxic metabolites from an Indonesian sponge Lendenfeldia sp. J. Nat. Prod. 2007, 70, 1824–1826. [Google Scholar] [CrossRef] [Green Version]
- Oda, Y.; Zhang, Q.; Matsunaga, S.; Fujita, M.J.; Sakai, R. Two new mycosporine-like amino acids LC-343 and mycosporine-ethanolamine from the Micronesian marine sponge Lendenfeldia chondrodes. Chem. Lett. 2017, 46, 1272–1274. [Google Scholar] [CrossRef]
- Radwan, M.M.; Manly, S.P.; Ross, S.A. Two new sulfated sterols from the marine sponge Lendenfeldia dendyi. Nat. Prod. Commun. 2007, 2, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Radwan, M.M.; Wanas, A.S.; Fronczek, F.R.; Jacob, M.R.; Ross, S.A. Polybrominated diphenyl ethers from the marine organisms Lendenfeldia dendyi and Sinularia dura with anti-MRSa activity. Med. Chem. Res. 2015, 24, 3398–3404. [Google Scholar] [CrossRef]
- Sakai, R.; Kamiya, H. 1-Deoxynojirimycin derivatives from the marine sponge Lendenfeldia chondrodes. J. Antibiot. 2006, 59, 507–511. [Google Scholar] [CrossRef] [Green Version]
- Sera, Y.; Adachi, K.; Shizuri, Y. A new epidioxy sterol as an antifouling substance from a Palauan marine sponge, Lendenfeldia chondrodes. J. Nat. Prod. 1999, 62, 152–154. [Google Scholar] [CrossRef]
- Alvi, K.A.; Crews, P. Homoscalarane sesterterpenes from Lendenfeldia frondosa. J. Nat. Prod. 1992, 55, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Chill, L.; Rudi, A.; Aknin, M.; Loya, S.; Hizi, A.; Kashman, Y. New sesterterpenes from Madagascan Lendenfeldia sponges. Tetrahedron 2004, 60, 10619–10626. [Google Scholar] [CrossRef]
- Kazlauskas, R.; Murphy, P.T.; Wells, R.J. Five new C26 tetracyclic terpenes from a sponge (Lendenfeldia sp.). Aust. J. Chem. 1982, 35, 51–59. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, R.; Mao, S.-C.; Morgan, J.B.; Jekabsons, M.B.; Zhou, Y.-D.; Nagle, D.G. Molecular-targeted antitumor agents. 19. Furospongolide from a marine Lendenfeldia sp. sponge inhibits hypoxia-inducible factor-1 activation in breast tumor cells. J. Nat. Prod. 2008, 71, 1854–1860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stessman, C.C.; Ebel, R.; Corvino, A.J.; Crews, P. Employing dereplication and gradient 1D NMR methods to rapidly characterize sponge-derived sesterterpenes. J. Nat. Prod. 2002, 65, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Allard, P.-M.; Péresse, T.; Bisson, J.; Gindro, K.; Marcourt, L.; Pham, V.C.; Roussi, F.; Litaudon, M.; Wolfender, J.-L. Integration of molecular networking and In-Silico MS/MS fragmentation for natural products dereplication. Anal. Chem. 2016, 88, 3317–3323. [Google Scholar] [CrossRef]
- Caesar, L.K.; Kellogg, J.J.; Kvalheim, O.M.; Cech, R.A.; Cech, N.B. Integration of biochemometrics and molecular networking to identify antimicrobials in Angelica keiskei. Planta Med. 2018, 84, 721–728. [Google Scholar] [CrossRef]
- Watrous, J.; Roach, P.; Alexandrov, T.; Heath, B.S.; Yang, J.Y.; Kersten, R.D.; van der Voort, M.; Pogliano, K.; Gross, H.; Raaijmakers, J.M.; et al. Mass spectral molecular networking of living microbial colonies. Proc. Natl. Acad. Sci. USA 2012, 109, E1743–E1752. [Google Scholar] [CrossRef] [Green Version]
- Woo, S.; Kang, K.B.; Kim, J.; Sung, S.H. Molecular networking reveals the chemical diversity of selaginellin derivatives, natural phosphodiesterase-4 inhibitors from Selaginella tamariscina. J. Nat. Prod. 2019, 82, 1820–1830. [Google Scholar] [CrossRef]
- Nakagawa, M.; Hamamoto, Y.; Ishihama, M.; Hamasaki, S.; Endo, M. Pharmacologically active homosesterterpenes from Palauan sponges. Tetrahedron Lett. 1987, 28, 431–434. [Google Scholar] [CrossRef]
- Lai, Y.-Y.; Lu, M.-C.; Wang, L.-H.; Chen, J.-J.; Fang, L.-S.; Wu, Y.-C.; Sung, P.-J. New scalarane sesterterpenoids from the Formosan sponge Ircinia felix. Mar. Drugs 2015, 13, 4296–4309. [Google Scholar] [CrossRef] [Green Version]
- Hochlowski, J.E.; Faulkner, D.J.; Bass, L.S.; Clardy, J. Metabolites of the dorid nudibranch Chromodoris sedna. J. Org. Chem. 1983, 48, 1738–1740. [Google Scholar] [CrossRef]
- González, M.A. Scalarane sesterterpenoids. Curr. Bioact. Compd. 2010, 6, 178–206. [Google Scholar] [CrossRef] [Green Version]
- Lai, K.-H.; Chen, P.-J.; Chen, C.-C.; Yang, S.-H.; El-Shazly, M.; Chang, Y.-C.; Wu, Y.-H.; Wu, Y.-H.; Wang, Y.-H.; Hsieh, H.-L.; et al. Lophatherum gracile Brongn. attenuates neutrophilic inflammation through inhibition of JNK and calcium. J. Ethnopharmacol. 2021, 264, 113224. [Google Scholar] [CrossRef] [PubMed]
- Flack, H.D.; Bernardinelli, G. Absolute structure and absolute configuration. Acta Cryst. 1999, A55, 908–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Braga, P.C.; Dal Sasso, M.; Culici, M.; Bianchi, T.; Bordoni, L.; Marabini, L. Anti-inflammatory activity of thymol: Inhibitory effect on the release of human neutrophil elastase. Pharmacology 2006, 77, 130–136. [Google Scholar] [CrossRef]
- Chen, X.-B.; Yuan, Q.-J.; Wang, J.; Hua, S.-K.; Ren, J.; Zeng, B.-B. Synthesis of the scalarane sesterterpenoid 16-deacetoxy-12-epi-scalarafuranacetate. J. Org. Chem. 2011, 76, 7216–7221. [Google Scholar] [CrossRef]
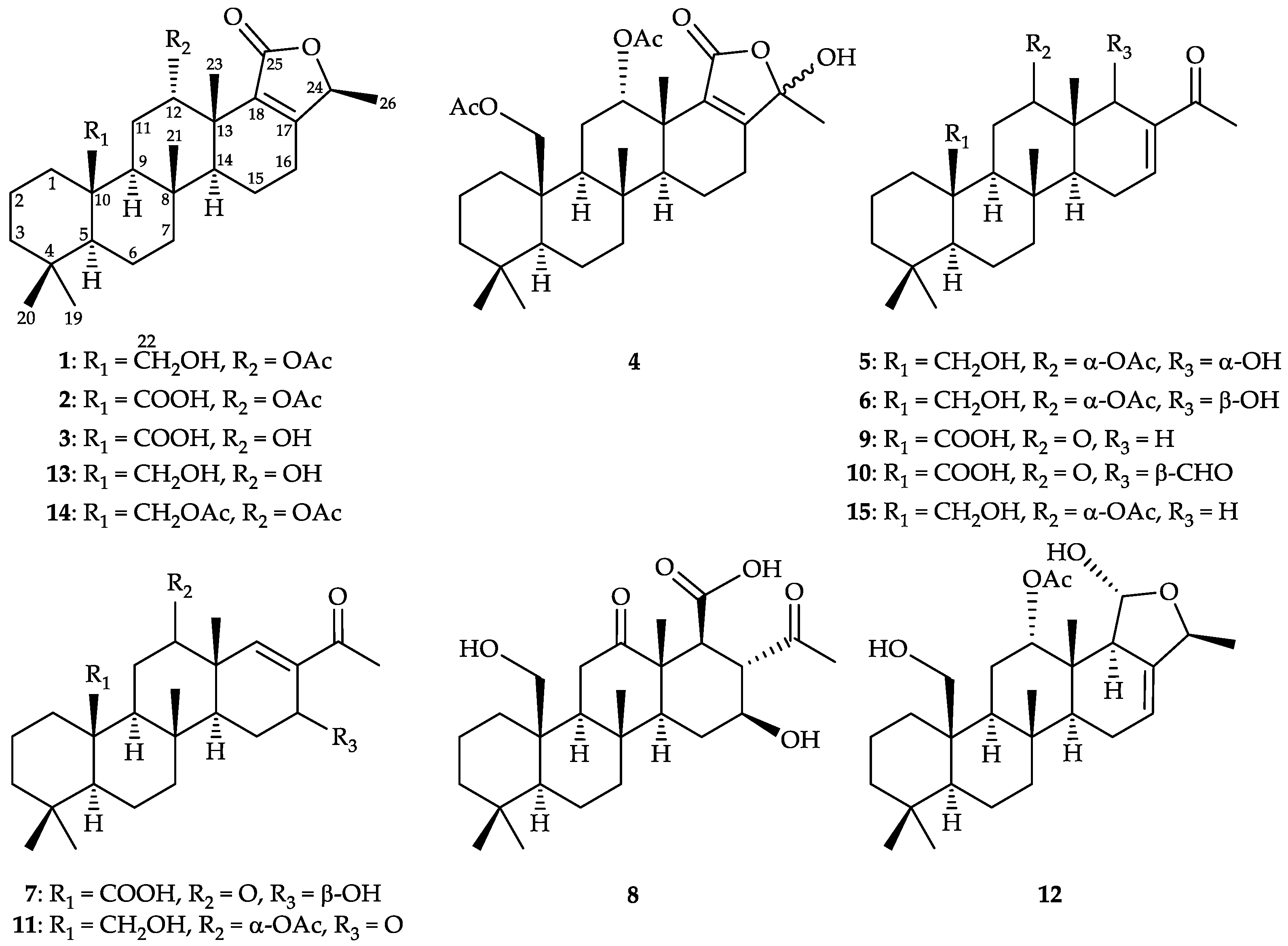

 ), HMBC (
), HMBC (  ), and protons with NOESY (
), and protons with NOESY (  ) correlations of 1.
) correlations of 1.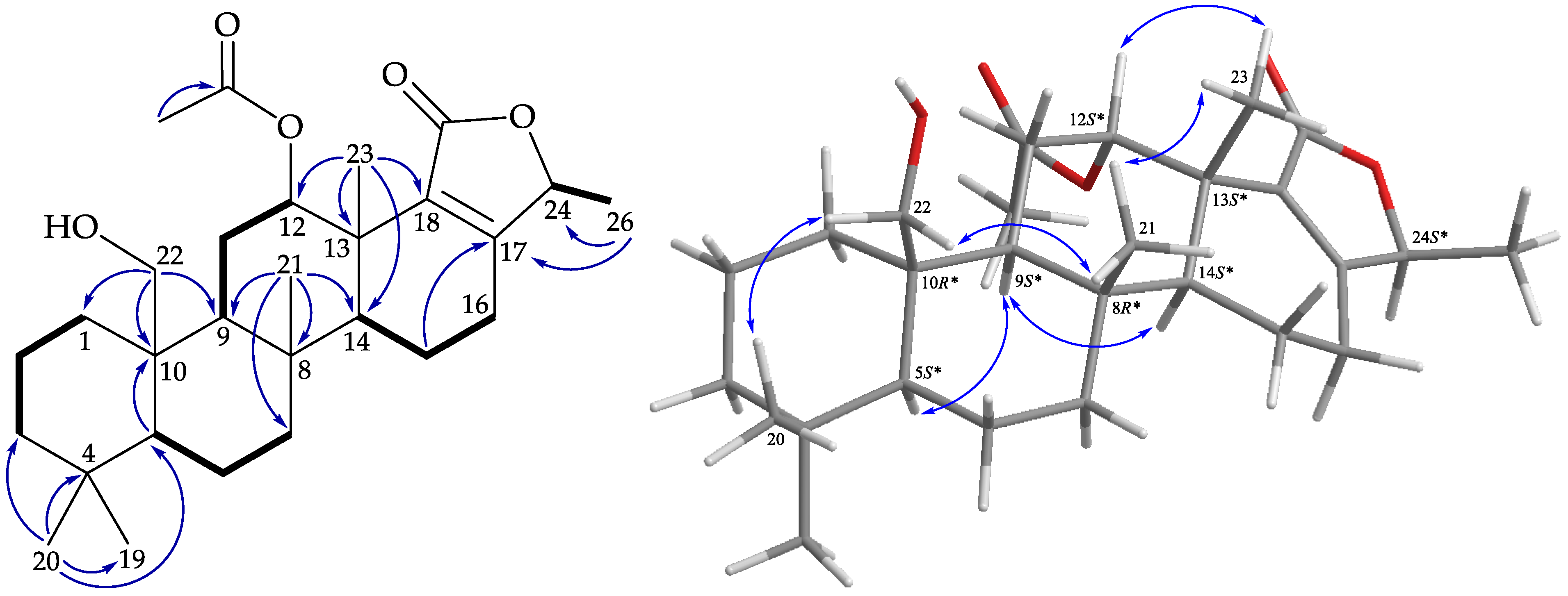 ), HMBC ( ), and protons with NOESY ( ) correlations of 2.
), HMBC ( ), and protons with NOESY ( ) correlations of 2.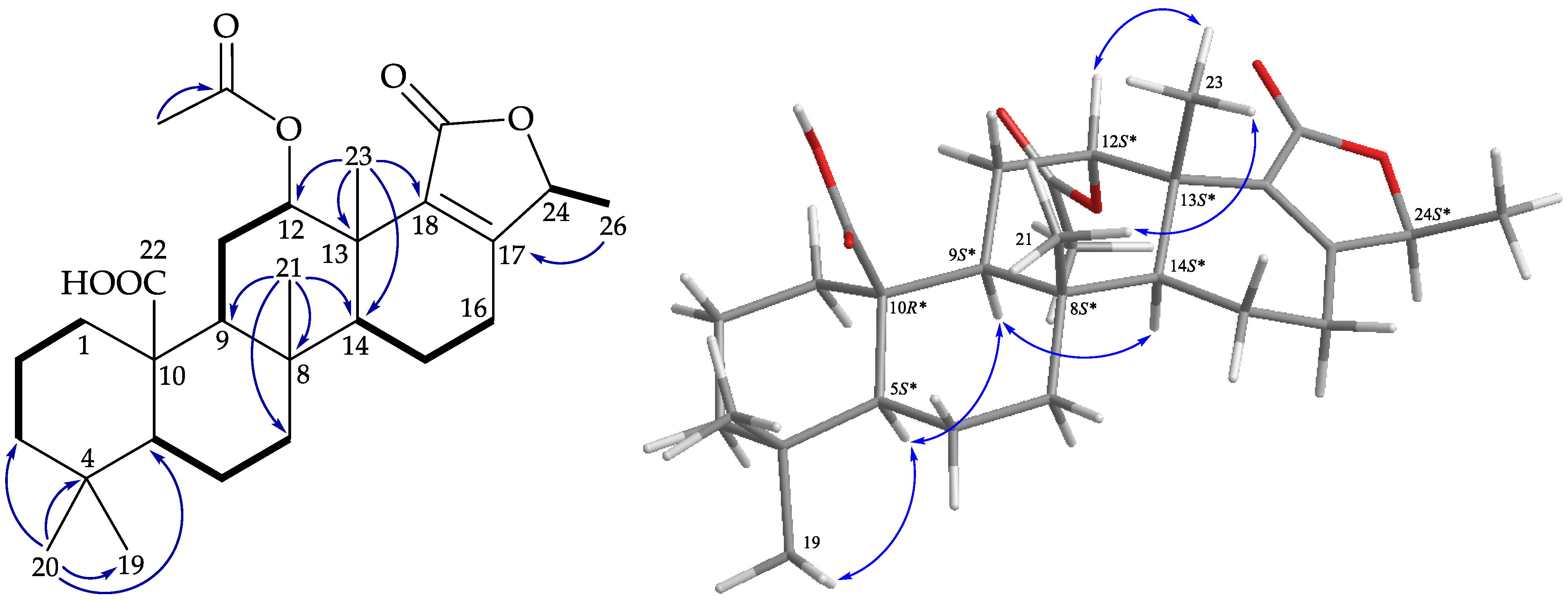 ), HMBC ( ), and protons with NOESY ( ) correlations of 3.
), HMBC ( ), and protons with NOESY ( ) correlations of 3. ), HMBC ( ), and protons with NOESY ( ) correlations of 4.
), HMBC ( ), and protons with NOESY ( ) correlations of 4. ), HMBC ( ), and protons with NOESY ( ) correlations of 5.
), HMBC ( ), and protons with NOESY ( ) correlations of 5.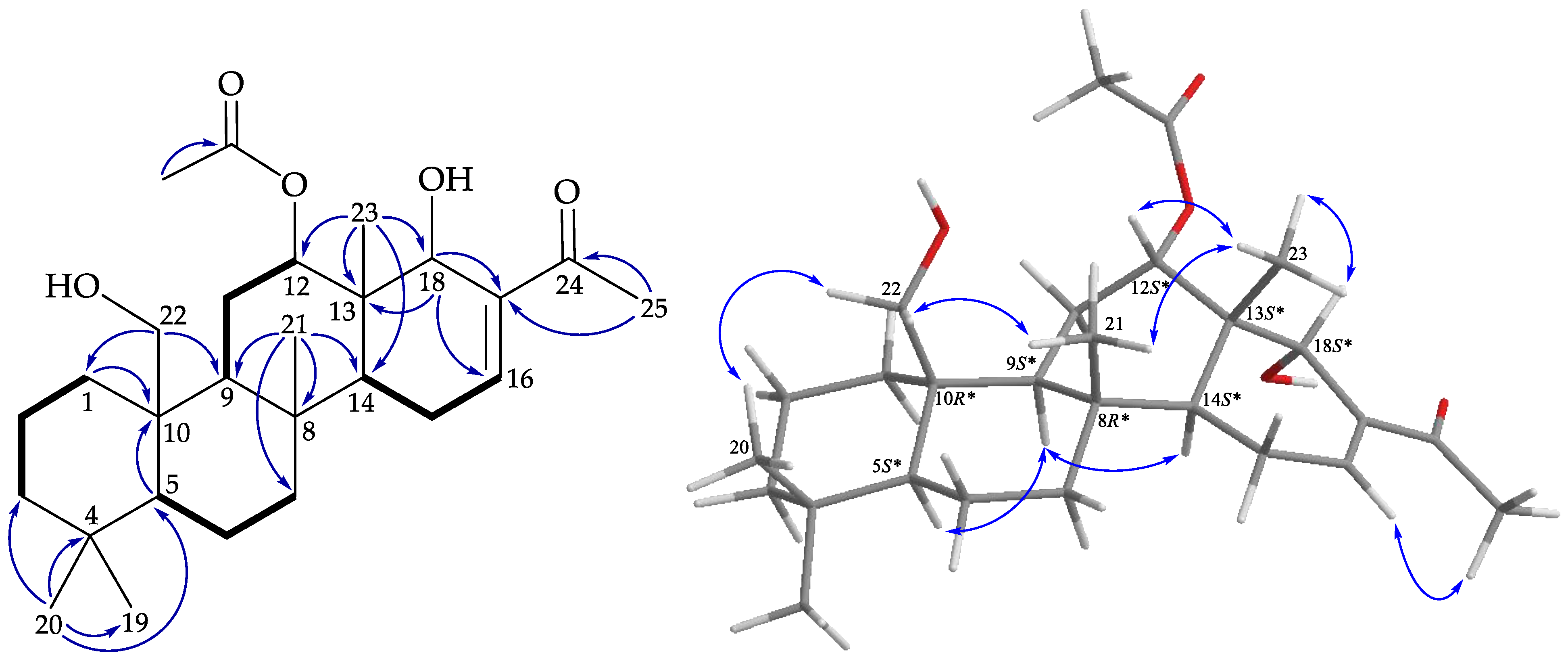 ), HMBC ( ), and protons with NOESY ( ) correlations of 6.
), HMBC ( ), and protons with NOESY ( ) correlations of 6. ), HMBC ( ), and protons with NOESY ( ) correlations of 7.
), HMBC ( ), and protons with NOESY ( ) correlations of 7.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 1 | 2 | |||
|---|---|---|---|---|
| Position | δH (J in Hz) a | δC Mult. b | δH (J in Hz) c | δC Mult. d |
| 1 | 2.12 m; 0.50 ddd (13.2, 13.2, 2.4) | 34.3, CH2 | 2.49 m; 0.67 ddd (12.8, 12.8, 3.2) | 38.4, CH2 |
| 2 | 1.54 m | 18.3, CH2 | 1.60 m | 18.6, CH2 |
| 3 | 1.42 m; 1.15 m | 41.7, CH2 | 1.39 m; 1.14 m | 42.2, CH2 |
| 4 | 33.0, C | 33.3, C | ||
| 5 | 0.96 m | 57.0, CH | 1.01 m | 56.4, CH |
| 6 | 1.94 m; 1.57 m | 17.0, CH2 | 1.93 m; 1.52 m | 16.9, CH2 |
| 7 | 1.93 m; 1.07 m | 42.0, CH2 | 1.94 m; 1.05 ddd (12.8, 12.8, 2.8) | 41.4, CH2 |
| 8 | 37.5, C | 37.7, C | ||
| 9 | 1.25 m | 53.4, CH | 1.45 m | 52.4, CH |
| 10 | 41.8, C | 47.6, C | ||
| 11 | 2.33 m; 2.17 m | 23.9, CH2 | 2.30 m; 1.83 m | 22.7, CH2 |
| 12 | 5.50 dd (3.0, 3.0) | 74.0, CH | 5.56 dd (2.8, 2.8) | 73.6, CH |
| 13 | 38.4, C | 38.2, C | ||
| 14 | 1.56 m | 51.2, CH | 1.51 m | 50.9, CH |
| 15 | 2.35 m | 24.0, CH2 | 2.34 m; 2.20 m | 23.9, CH2 |
| 16 | 1.57 m; 1.44 m | 17.9, CH2 | 1.48 m | 20.2, CH2 |
| 17 | 163.6, C | 163.7, C | ||
| 18 | 132.7, C | 132.5, C | ||
| 19 | 0.87 s | 33.8, CH3 | 0.92 s | 33.8, CH3 |
| 20 | 0.77 s | 21.8, CH3 | 0.86 s | 22.4, CH3 |
| 21 | 1.09 s | 16.3, CH3 | 0.85 s | 14.4, CH3 |
| 22 | 4.03 d (12.0); 3.87 dd (12.0, 1.2) | 62.8, CH2 | 178.8, C | |
| 23 | 1.19 s | 21.1, CH3 | 1.13 s | 21.2, CH3 |
| 24 | 4.77 q (6.6) | 77.6, CH | 4.77 q (6.8) | 77.7, CH |
| 25 | 171.0, C | 170.9, C | ||
| 26 | 1.36 d (6.6) | 18.7, CH3 | 1.35 d (6.8) | 18.6, CH3 |
| OAc-12 | 169.9, C | 169.8, C | ||
| 1.96 s | 21.2, CH3 | 1.96 s | 21.2, CH3 | |
| 3 | 4 | |||
|---|---|---|---|---|
| Position | δH (J in Hz) a | δC Mult. b | δH (J in Hz) a | δC Mult. b |
| 1 | 2.52 m; 0.94 m | 38.3, CH2 | 2.01 m; 0.51 ddd (12.4, 12.4, 4.4) | 34.7, CH2 |
| 2 | 2.30 m; 1.60 m | 16.6, CH2 | 1.58 m | 18.2, CH2 |
| 3 | 1.41 m; 1.16 m | 42.2, CH2 | 1.44 m; 1.13 m | 41.5, CH2 |
| 4 | 33.4, C | 33.0, C | ||
| 5 | 1.12 m | 56.1, CH | 1.01 m | 57.1, CH |
| 6 | 1.91 m; 1.54 m | 18.6, CH2 | 1.58 m; 1.45 m | 18.0, CH2 |
| 7 | 1.92 m; 1.10 m | 41.3, CH2 | 1.93 m; 1.11 m | 41.8, CH2 |
| 8 | 37.8, C | 37.4, C | ||
| 9 | 1.77 m | 51.5, CH | 1.31 m | 53.2, CH |
| 10 | 47.8, C | 40.1, C | ||
| 11 | 1.99 m; 1.89 m | 25.8, CH2 | 2.20 m; 2.03 m | 23.3, CH2 |
| 12 | 4.65 br s | 69.7, CH | 5.49 br s | 73.9, CH |
| 13 | 40.1, C | 38.5, C | ||
| 14 | 1.55 m | 49.8, CH | 1.53 m | 51.3, CH |
| 15 | 2.26 m | 24.3, CH2 | 2.48 m; 2.35 m | 22.8, CH2 |
| 16 | 1.53 m; 1.42 m | 20.2, CH2 | 1.42 m | 18.1, CH2 |
| 17 | 165.4, C | 161.6, C | ||
| 18 | 133.1, C | 134.3, C | ||
| 19 | 0.92 s | 33.7, CH3 | 0.88 s | 33.7, CH3 |
| 20 | 0.88 s | 22.4, CH3 | 0.83 s | 21.8, CH3 |
| 21 | 0.85 s | 14.3, CH3 | 0.97 s | 16.4, CH3 |
| 22 | 179.3, C | 4.58 d (12.0); 4.14 d (12.0) | 64.7, CH2 | |
| 23 | 1.09 s | 21.5, CH3 | 1.21 s | 21.1, CH3 |
| 24 | 4.78 q (6.8) | 78.6, CH | 103.4, C | |
| 25 | 172.5, C | 178.1, C | ||
| 26 | 1.36 d (6.8) | 18.4, CH3 | 1.59 s | 24.3, CH3 |
| OAc-12 | 169.6, C | |||
| 1.95 s | 21.2, CH3 | |||
| OAc-22 | 171.0, C | |||
| 2.06 s | 21.2, CH3 | |||
| 5 | 6 | |||
|---|---|---|---|---|
| Position | δH (J in Hz) a | δC Mult. b | δH (J in Hz) a | δC Mult. b |
| 1 | 2.11 m; 0.53 ddd (14.4, 14.4, 4.8) | 34.3, CH2 | 2.12 m; 0.53 ddd (13.2, 13.2, 2.4) | 34.4, CH2 |
| 2 | 1.54 m; 1.39 m | 18.0, CH2 | 1.53 m | 17.8, CH2 |
| 3 | 1.44 m; 1.20 m | 41.7, CH2 | 1.45 m; 1.18 m | 41.7, CH2 |
| 4 | 33.0, C | 33.0, C | ||
| 5 | 1.02 dd (12.6, 2.4) | 56.7, CH | 0.98 dd (12.6, 2.4) | 56.9, CH |
| 6 | 1.47 m | 18.2, CH2 | 1.55 m; 1.48 m | 18.4, CH2 |
| 7 | 1.82 ddd (12.6, 3.0, 3.0); 1.20 m | 41.9, CH2 | 1.80 ddd (12.6, 3.0, 3.0); 1.06 m | 41.9, CH2 |
| 8 | 37.2, C | 37.4, C | ||
| 9 | 1.35 br d (12.6) | 53.0, CH | 1.34 m | 52.5, CH |
| 10 | 41.7, C | 41.7, C | ||
| 11 | 2.20 m; 1.98 m | 26.0, CH2 | 2.10 m; 2.00 m | 25.0, CH2 |
| 12 | 4.92 dd (3.0, 3.0) | 79.7, CH | 5.01 dd (3.6, 2.4) | 73.6, CH |
| 13 | 39.2, C | 40.1, C | ||
| 14 | 2.23 m | 42.0, CH | 1.52 m | 47.8, CH |
| 15 | 1.25 m | 29.6, CH2 | 2.26 m | 24.3, CH2 |
| 16 | 6.99 br s | 143.6, CH | 6.88 br s | 141.8, CH |
| 17 | 132.2, C | 140.0, C | ||
| 18 | 4.38 s | 70.9, CH | 4.57 s | 69.7, CH |
| 19 | 0.89 s | 33.8, CH3 | 0.87 s | 33.8, CH3 |
| 20 | 0.77 s | 22.0, CH3 | 0.77 s | 21.9, CH3 |
| 21 | 1.11 s | 15.5, CH3 | 1.15 s | 15.8, CH3 |
| 22 | 4.05 d (10.8); 3.91 dd (10.8, 4.8) | 62.9, CH2 | 4.04 d (12.0); 3.89 dd (12.0, 1.2) | 63.0, CH2 |
| 23 | 0.70 s | 19.7, CH3 | 0.94 s | 12.6, CH3 |
| 24 | 197.8, C | 202.2, C | ||
| 25 | 2.32 s | 24.8, CH3 | 2.32 s | 26.1, CH3 |
| OAc-12 | 169.2, C | 170.1, C | ||
| 2.14 s | 22.0, CH3 | 2.11 s | 21.5, CH3 | |
| 18-OH | 4.11 d (1.8) | |||
| Position | δH (J in Hz) a | δC Mult. b |
|---|---|---|
| 1 | 2.47 m; 0.91 m | 38.3, CH2 |
| 2 | 1.54 m | 20.1, CH2 |
| 3 | 1.40 m; 1.16 m | 42.0, CH2 |
| 4 | 33.4, C | |
| 5 | 1.06 m | 56.2, CH |
| 6 | 2.31 m; 1.65 m | 18.4, CH2 |
| 7 | 2.00 m; 1.13 m | 40.6, CH2 |
| 8 | 37.3, C | |
| 9 | 1.61 m | 58.5, CH |
| 10 | 48.7, C | |
| 11 | 2.87 dd (14.4, 14.4); 2.67 dd (14.4, 2.8) | 36.5, CH2 |
| 12 | 210.3, C | |
| 13 | 50.3, C | |
| 14 | 1.70 m | 48.7, CH |
| 15 | 1.96 m; 1.73 m | 25.1, CH2 |
| 16 | 4.57 dd (4.0, 1.6) | 63.2, CH |
| 17 | 137.7, C | |
| 18 | 7.40 s | 147.2, CH |
| 19 | 0.91 s | 33.7, CH3 |
| 20 | 0.88 s | 22.5, CH3 |
| 21 | 1.03 s | 14.3, CH3 |
| 22 | 179.3, C | |
| 23 | 1.20 s | 19.8, CH3 |
| 24 | 201.8, C | |
| 25 | 2.36 s | 25.7, CH3 |
| Compound | Superoxide Anion generation | Elastase Release | ||
|---|---|---|---|---|
| IC50 (μM) a | Inh % | IC50 (μM) a | Inh % | |
| 1 | 0.87 ± 0.14 | 98.90 ± 0.79 *** | 1.12 ± 0.37 | 101.69 ± 2.91 *** |
| 2 | 1.11 ± 0.10 | 101.77 ± 1.05 *** | 1.65 ± 0.31 | 91.46 ± 5.24 *** |
| 3 | 6.57 ± 0.67 | 70.78 ± 5.80 *** | 48.78 ± 3.17 *** | |
| 4 | 34.63 ± 6.30 ** | 42.52 ± 5.88 ** | ||
| 6 | 1.75 ± 0.02 | 97.57 ± 1.42 *** | 1.59 ± 0.41 | 102.08 ± 0.44 *** |
| 7 | 6.25 ± 1.17 | 71.74 ± 8.89 *** | 45.82 ± 6.54 ** | |
| 8 | 1.47 ± 0.24 | 101.35 ± 0.77 *** | 2.78 ± 0.78 | 101.83 ± 2.28 *** |
| 9 | 1.50 ± 0.08 | 97.27 ± 1.59 *** | 1.74 ± 0.15 | 101.80 ± 3.06 *** |
| 10 | 2.83 ± 0.63 | 98.94 ± 0.60 *** | 1.66 ± 0.09 | 93.47 ± 4.73 *** |
| 11 | 6.33 ± 0.89 | 69.61 ± 6.49 *** | 48.06 ± 5.55 *** | |
| 12 | 44.48 ± 6.95 ** | 6.97 ± 1.01 | 101.69 ± 8.13 *** | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, B.-R.; Lai, K.-H.; Lee, G.-H.; Yu, S.S.-F.; Duh, C.-Y.; Su, J.-H.; Zheng, L.-G.; Hwang, T.-L.; Sung, P.-J. Scalarane-Type Sesterterpenoids from the Marine Sponge Lendenfeldia sp. Alleviate Inflammation in Human Neutrophils. Mar. Drugs 2021, 19, 561. https://doi.org/10.3390/md19100561
Peng B-R, Lai K-H, Lee G-H, Yu SS-F, Duh C-Y, Su J-H, Zheng L-G, Hwang T-L, Sung P-J. Scalarane-Type Sesterterpenoids from the Marine Sponge Lendenfeldia sp. Alleviate Inflammation in Human Neutrophils. Marine Drugs. 2021; 19(10):561. https://doi.org/10.3390/md19100561
Chicago/Turabian StylePeng, Bo-Rong, Kuei-Hung Lai, Gene-Hsiang Lee, Steve Sheng-Fa Yu, Chang-Yih Duh, Jui-Hsin Su, Li-Guo Zheng, Tsong-Long Hwang, and Ping-Jyun Sung. 2021. "Scalarane-Type Sesterterpenoids from the Marine Sponge Lendenfeldia sp. Alleviate Inflammation in Human Neutrophils" Marine Drugs 19, no. 10: 561. https://doi.org/10.3390/md19100561