1. Introduction
The marine environment has played an essential role in the discovery of compelling secondary metabolites with fascinating antitumor, immunomodulatory, analgesic, anti-inflammatory, anti-allergic, antimicrobial, and antiviral effects [
1,
2]. Since 1963, more than 30,000 new chemical entities have been identified from marine organisms, including macro- and micro-organisms [
3]. Secondary metabolites obtained from marine invertebrates have received great attention from pharmacologists and chemists due to their remarkable chemical diversity and biological activities [
4,
5,
6]. The fact that 14 marine-derived approved drugs and another 23 drug leads in different phases (I–III) of clinical trials [
7], mostly from marine invertebrates [
7], clearly indicates the role of marine invertebrates as a vigorous source for the drug-discovery process [
7]. Sponges belonging to the genus
Hemimycale are excellent producers of alkaloids with both guanidine [
8,
9] and hydantoin backbones [
10,
11]. Ptilomycalin A, with its exceptional polycyclic guanidine backbone linked with a ω-hydroxyhexadecanoyl-spermidine moiety via an ester linkage, has displayed notable antimicrobial and antiviral activities [
8,
9].
The skeletal muscle relaxant dantrolene and the anticonvulsive drugs phenytoin, norantoin, mephenythoin, ethotoin, methetoin, and fosphenytoin are hydantoin-derived compounds [
11,
12]. Similarly, 5-substituted hydantoins (5,5-dithienylhydantoin, 5,5-dipyridylhydantoin, dithiohydantoins, thiohydantoin, and spirothiohydantoin) have anticonvulsive activity [
13,
14]. Other significant activities for hydantoin derivatives include antimicrobial (nitrofurantoin), antiarrhythmic (azimilide), and nonsteroidal antiandrogens (nilutamide) activities. Allantoin is used as an antacid, antipsoriatic, keratolytic, and astringent, as well as in wound remedy [
12]. Additionally, antiviral, antidepressant, and antithrombotic and enzyme inhibition are additional pharmacological properties of hydantoins [
15]. Finally, the herbicidal effects of spirohydantoin and thioxohydantocidin, as well as the fungicidal properties of clodantoin, are attributed to the hydantoin backbone in their structures [
16,
17]. Recently, the in vitro anti-growth and anti-invasive effects of (
Z)-5-(4-hydroxybenzylidene)imidazolidine-2,4-dione and its analogue (
Z)-5-(4-(ethylthio)benzylidene)-hydantoin against PC-3M prostate cancer were reported [
18]. The compounds reduced the growth of orthotopic tumors and repressed the formation of tumor micrometastases in distant organs without apparent cytotoxic effects at the test doses [
18].
As a continuation of our work to uncover biologically active alkaloids from marine organisms [
19,
20,
21,
22], the cytotoxic fractions of a methanolic extract of the sponge
Hemimycale species were investigated. Three new alkaloids, hemimycalins C–E (
1–
3) with hydantoin and 2-iminoimidazolidin-4-one backbones, were obtained from the active fractions of the extract, and their structures were characterized. Here, we report on the structural determination and the antimicrobial and cytotoxic activities of the compounds.
2. Results and Discussion
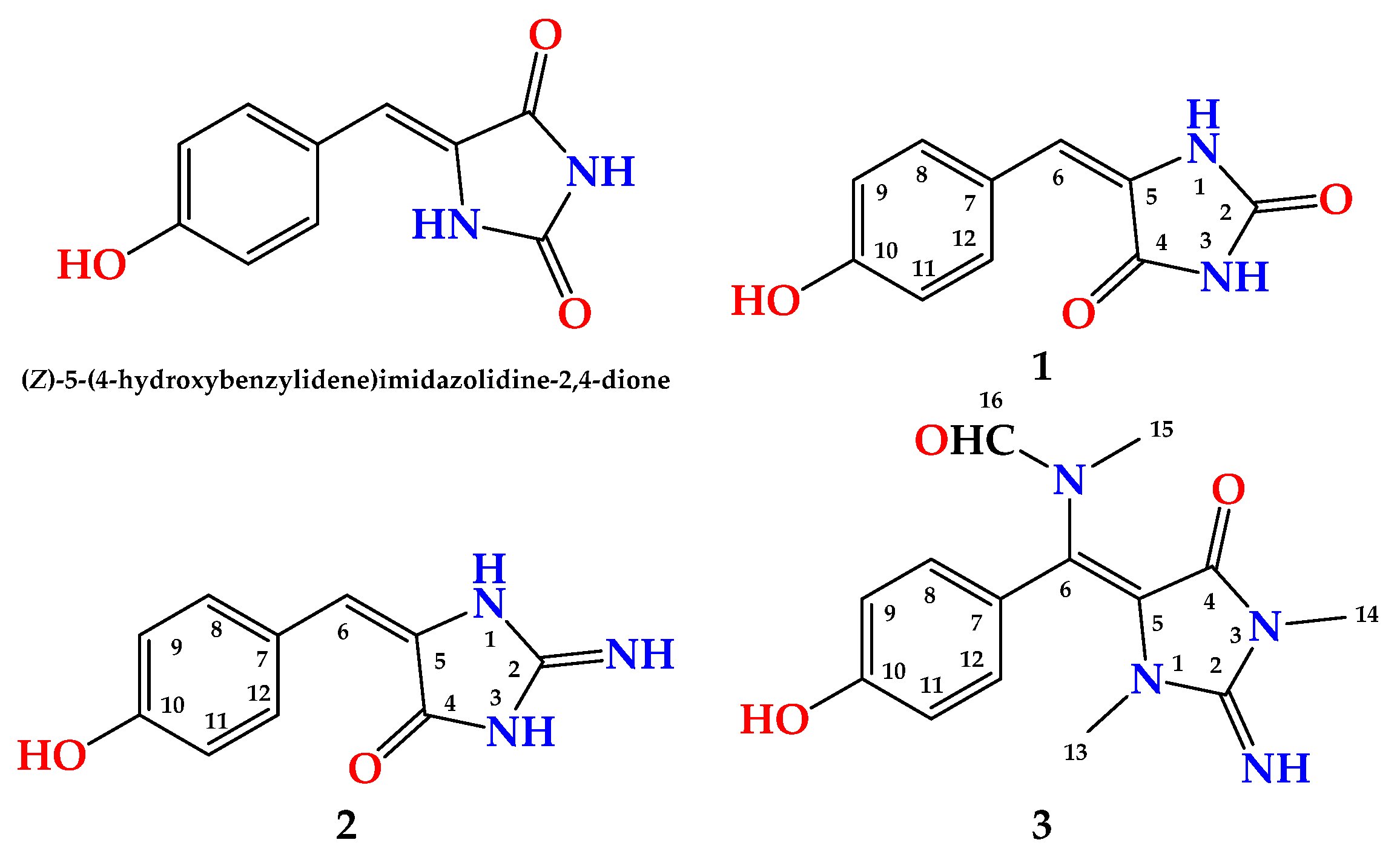
Compound
1 (
Figure 1) was obtained as a yellow powder. The molecular formula was C
10H
8N
2O
3, and it was obtained from the (+)-HRESIMS peak at
m/z 205.0609 [M + H]
+. The interpretation of its NMR spectral data including
1H (
Figures S1 and S2),
13C (
Figure S3), DEPT (
Figure S4), HSQC (
Figure S5) and HMBC (
Figure S6) supported the structure of the compound. The
1H NMR spectra showed two parts: a benzene ring and an imidazolidine-2,4-dione (hydantoin) part connected together via a vinylic carbon (C-6) (
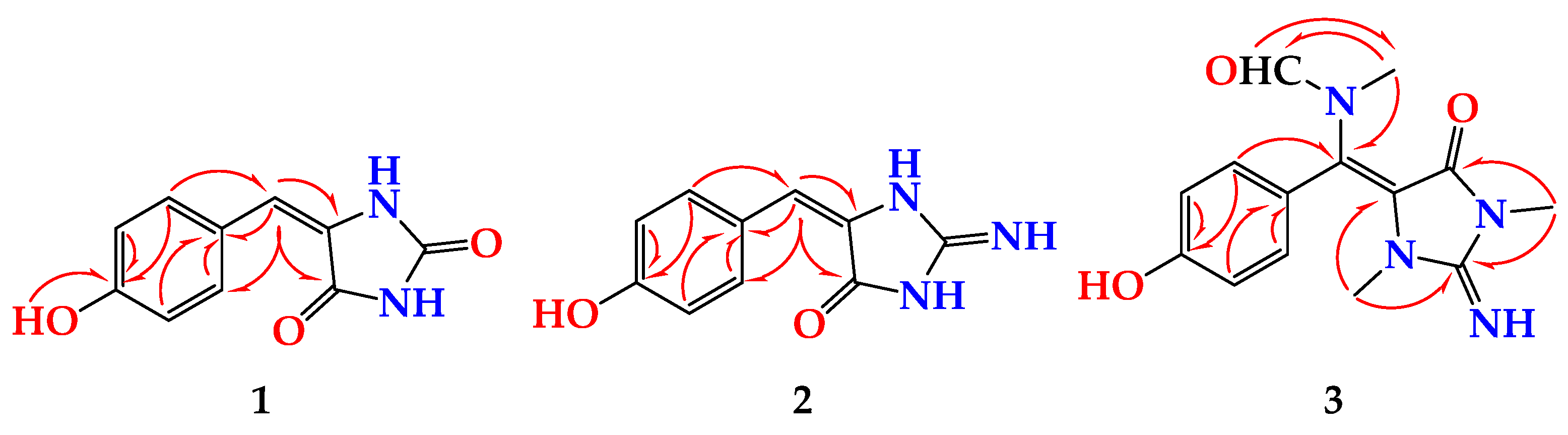
Figure 1). The HMBC cross peaks from H-6 (δ
H 6.23) to C-4 (δ
C 163.6) and C-5 (δ
C 127.0) and from H-8 (δ
H 7.82) and H-12 ((δ
H 7.82) to C-6 (δ
C 116.8) supported the connection of the fragments of
1 through the vinylic C-6 (
Table 1 and
Figure 2). The
1H and
13C NMR signals of
1 were found to be similar to those of (
Z)-5-(4-hydroxybenzylidene)imidazolidine-2,4-dione [
10] with differences in the chemical shifts of some
1H and
13C NMR signals (
Table 2). In a comparison of the NMR data of (
Z)-5-(4-hydroxybenzylidene)imidazolidine-2,4-dione [
10] with those of
1, a significant downfield shift of C-6 (Δδ
C = + 7.6 ppm) was observed in
1, suggesting a different configuration of Δ
5,6 in
1. Additional
13C NMR chemical shift variations were observed in the imidazolidine-2,4-dione moiety (C-2, C-4 and C-5) ranging from −2.1 to +1.7 ppm (
Table 2).
It is well known that H-6 possesses a higher chemical shift value in
Z-configured double bonds than in the
E-configured ones [
23,
24]. Additionally, the
13C chemical shift of C-6 is more highly downfield in compounds with the
E configuration than those with the Z configuration [
25]. This effect could be a result of both anisotropic and diamagnetic effects on H-6 by the adjacent carbonyl group (C-4) [
23]. In addition, significant downfield shifts (+0.36 ppm) for the signals of H-8 and H-12 in
1 when compared to those reported for (
Z)-5-(4-hydroxybenzylidene)imidazolidine-2,4-dione [
10] were noticed. Finally, the remaining
1H and
13C signals in
1 displayed marginal down- or up-field shifts from those of (
Z)-5-(4-hydroxybenzylidene)imidazolidine-2,4-dione [
10]. Accordingly,
1 was assigned as (
E)-5-(4-hydroxybenzylidene)imidazolidine-2,4-dione and is reported as a new natural compound and named hemimycalin C.
Compound
2 (
Figure 1) was obtained as a yellow powder with the molecular formula C
10H
9N
3O
2 obtained from the (+)-HRESIMS ion peak at
m/
z 204.0771 [M + H]
+, being one atomic mass unit less than
1 and thus suggesting the replacement of one of the oxygen atoms in
2 with NH. The
1H (
Figures S7 and S8) and
13C NMR (
Figure S9) data of
2 (
Table 3) were found to be in good agreement with those of
1 (
Table 4). These data were supported also by HSQC (
Figure S10) and HMBC (
Figure S11) experiments. A comparison of the
1H and
13C NMR of
2 with those of
1 revealed marginal chemical shift differences between all NMR signals ranging from –0.39 to 0.0 ppm in the
1H NMR and from –0.1 to –1.8 ppm in the
13C NMR spectra (
Table 3). A noticeable chemical shift difference was observed for C-2 (Δδ = –1.8 ppm) due to the replacement of the urea part (or hydantoin moiety) in
1 with a guanidine part (or 2-iminoimidazolidin-4-one) [
25] in
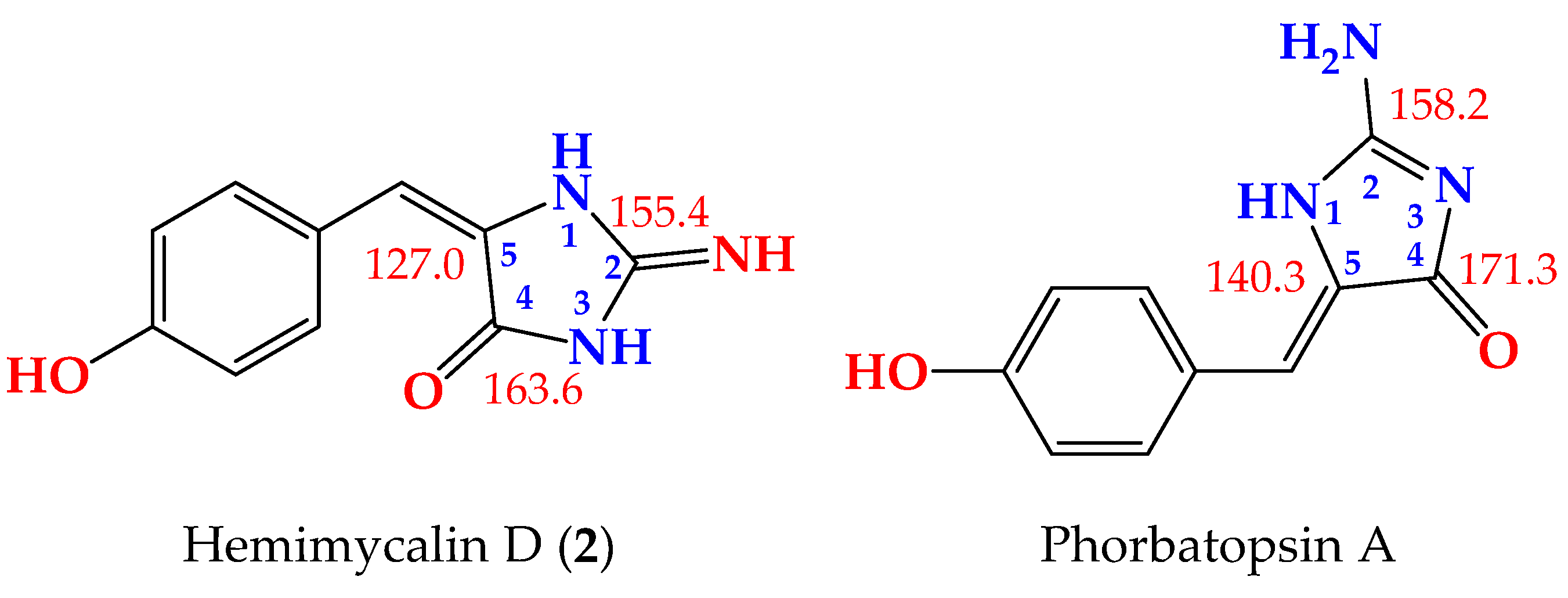
2. Additionally, to exclude the presence of 2-aminoimidazol-4-one moiety in
2, the
13C NMR data of the 2-iminoimidazolidin-4-one moiety in
2 were compared with those reported for 2-aminoimidazol-4-one moiety, both measured in DMSO-
d6 [
23] (
Figure 3). As shown in
Figure 3, the
13C NMR data of 2-f in
2 were completely different from those of 2-aminoimidazol-4-one moiety in phorbatopsin A [
23]. Furthermore, the HMBC correlations supported the assignment of the non-protonated carbons in
2 and the assignment of the 2-iminoimidazolidin-4-one moiety (
Table 2 and
Figure 2). Thus,
2 was assigned as (
E)-5-(4-hydroxybenzylidene)-2-iminoimidazolidin-4-one and named hemimycalin D.
Compound
3 (
Figure 1) was found to possess the formula C
14H
16N
4O
3, as shown by the (+)-HRESIMS ion peak at
m/
z 311.1118 for [M + Na]
+. The
1H (
Figures S12 and S13) and
13C NMR (
Figure S14) spectra of
3 displayed typical resonances for a 1,4-substituted benzene ring, two
N-methyls at δ
H/C 2.79/31.1 and 3.21/29.4 and an
N-methylformamide at δ
H/C 2.83/33.6 (H
3-15/C-15) and 7.91/166.3 (H-16/C-16). The
1H and
13C NMR data of
3 (
Table 4) were found to be comparable with those reported for hemimycalin A [
10], though featuring the replacement of the 1,3-dimethylimidazolidine-2,4-dione moiety in hemimycalin A [
10] with 2-imino-1,3-dimethylimidazolidin-4-one moiety in
3. This assignment was confirmed by HSQC (
Figure S15) experiment and by HMBC (
Figure S16) cross-peaks from H-12 (δ
H 7.48) to C-5 (qc, δ
C = 93.9), from H
3-13 (δ
H 2.79) to C-2 (δ
C 153.6), and from H
3-14 (δ
H 3.21) to C-2 and C-4 (δ
C = 149.7) (
Table 5 and
Figure 2). In addition, the placement of the
N-methylformamide moiety at C-6 was confirmed by the HMBC cross peaks from H
3-15 (δ
H =2.83) to C-6 (δ
C =126.1), from H
3-15 to C-16 (δ
C = 166.3), and from H-16 (δ
H 7.91) to C-15 (δ
C = 33.6). The
E configuration at the olefinic moiety Δ
5,6 in
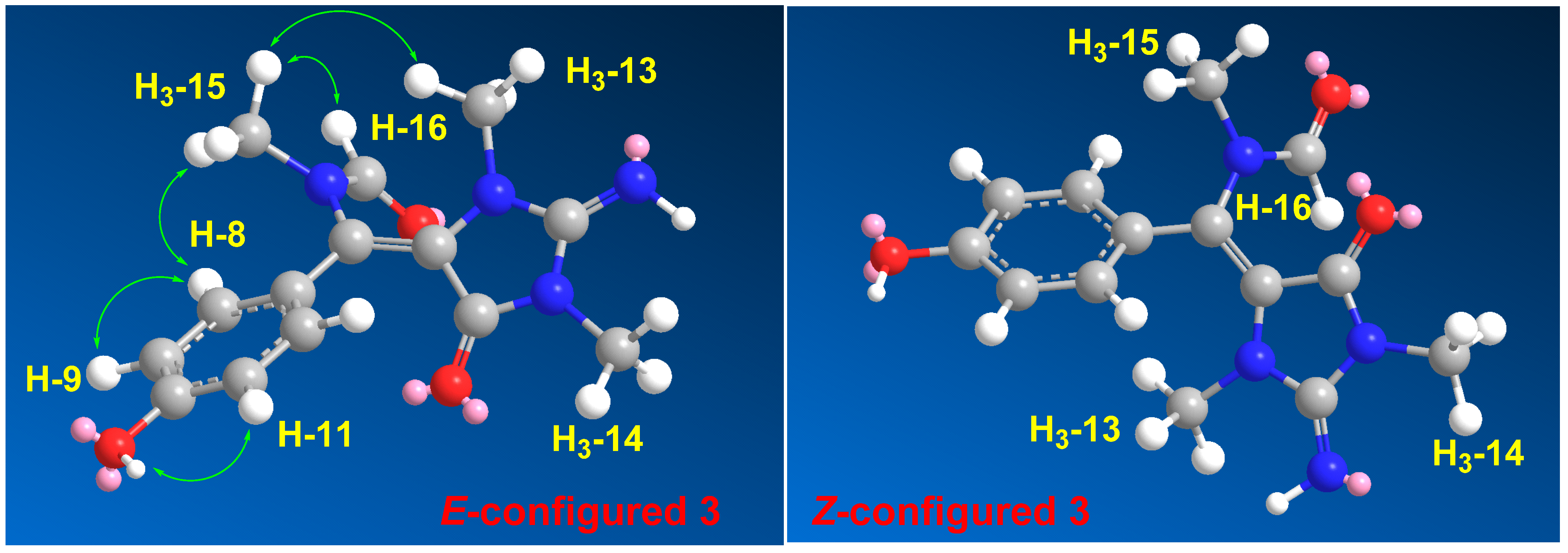
3 was confirmed from NOESY (
Figure S17) correlations between H
3-13 and H
3-15, H
3-15, and H-16, as well as between H
3-15 and H-8,12. The NOESY correlations between H
3-13 and H
3-15 observed in the compound with
E configuration at Δ
5,6 were also confirmed by a comparison of the MM2-minimized drawings of the
E-
3 against Z-
3 (
Figure 4). It is very clear that the compound with the
E configuration at Δ
5,6 displayed significant NOESY between H
3-13 and H
3-15 (
Table 4 and
Figure 4). On the other hand, the isomer with the Z configuration at Δ
5,6 was found to lack any correlation between these two methyl groups. Thus, the
E configuration at Δ
5,6 in
3 was confirmed. Accordingly, compound
3 was assigned as (
E)-
N-((4-hydroxyphenyl)(2-imino-1,3-dimethyl-5-oxoimidazolidin-4-ylidene)methyl)-
N-methylformamide and named hemimycalin E.
An MTT assay showed
1–
3 were mainly active against colorectal carcinoma (HCT 116) cells, with IC
50 values of 8.6–18.8 μM (
Table 5). On the contrary,
1–
3 were moderately active towards triple-negative breast cancer (MDA-MB-231), with IC
50 values of 21.5–31.7 μM, and inactive against human cervical carcinoma (Hela) cells. These data suggest that HCT 116 cells have higher sensitivity towards compound 3 than the other cell lines.
In a disk diffusion assay,
1–
3 were evaluated for their effects on three pathogens at a concentration of 50 µg/disc. The compounds displayed high activities against
Candida albicans (inhibition zones = 20–22 mm) and
Escherichia coli (inhibition zones = 17–18 mm) but no effects on
Staphylococcus aureus (
Table 6). Finally,
1–
3 displayed a minimum inhibitory concentration (MIC) value of 8 µM against
C. albicans and
E. coli (
Table 6).
4. Conclusions
The bioassay-directed partition and purification of the cytotoxic fraction of the Red Sea sponge
Hemimycale sp. provided three new alkaloids: hemimycalins C–E (
1–
3). The structures of the compounds were assigned via analyses of their spectral data. Interestingly, hemimycalin C (
1) was found to possess an
E configuration [
25] at Δ
5,6 instead of the previously reported
Z configuration of Δ
5,6. In addition, hemimycalins D and E (
2 and
3) were found to possess the 2-iminoimidazolidin-4-one [
25] backbone instead of hydantoin (imidazolidine-2,4-dione) moiety in previously reported alkaloids from the genus
Hemimycale. Furthermore, hemimycalin D (
2) was found to share the
E configuration at Δ
5,6 with hemimycalin C (
1). Consequently, the
E-configured
1 and
2 were shown to possess higher chemical shift values for C-6 than the Z-configured compounds, while H-6 [
23,
24,
25] in the
E-configured compounds [
23,
24,
25] was found to resonate at lower chemical shift values than in the
Z-configured ones.
Hemimycalins C–E showed significant cytotoxic effects and selective antimicrobial effects against E. coli and C. albicans, making them potential scaffolds for the development of drug leads.
The current findings provide a deeper insight and understanding of the chemical diversity and biological activities of the secondary metabolites of the Red Sea sponge Hemimycale sp.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}