
Compounds Identified from Marine Mangrove Plant (Avicennia alba) as Potential Antiviral Drug Candidates against WDSV, an In-Silico Approach




Abstract
:1. Introduction
2. Results
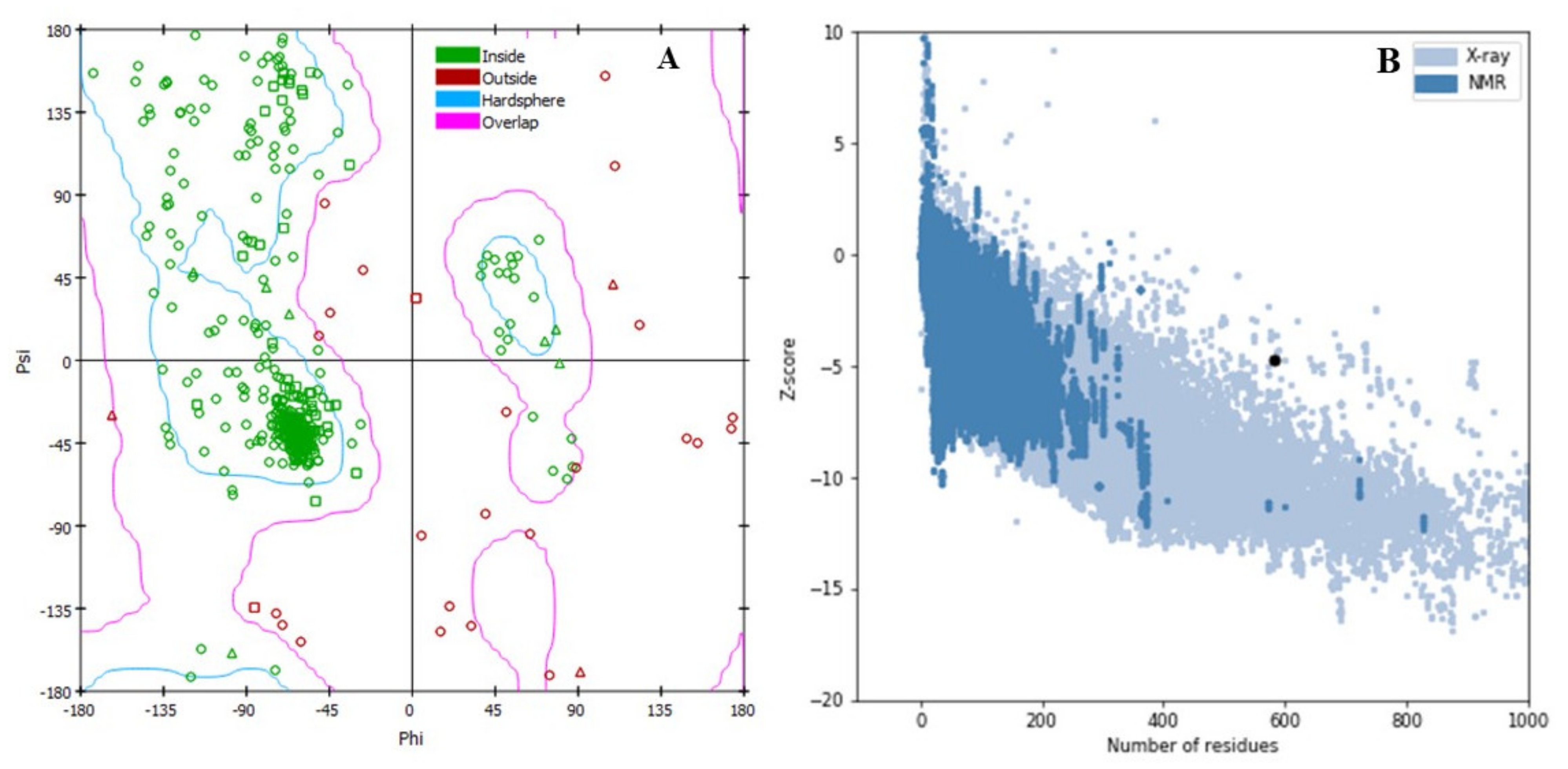
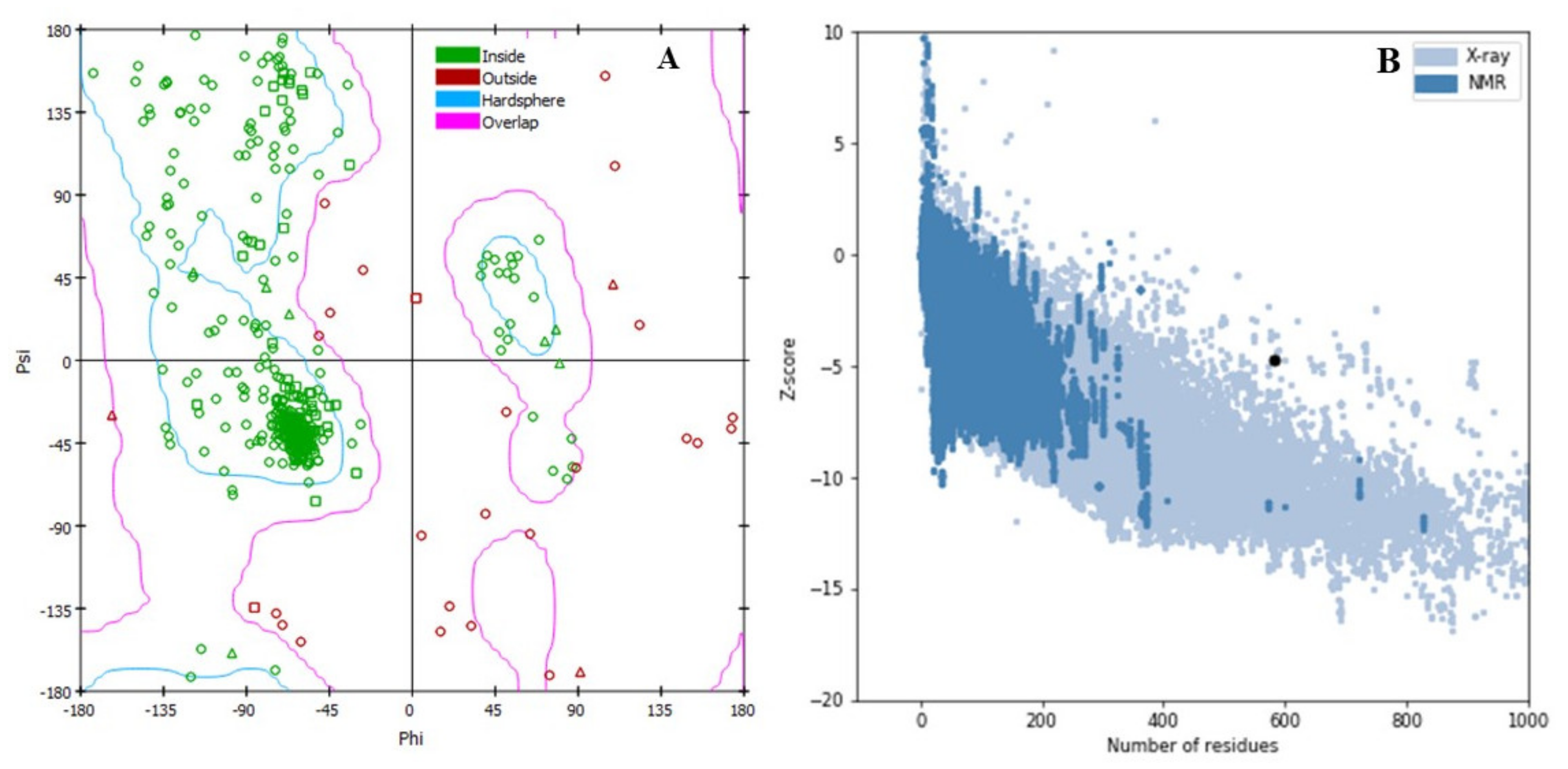
2.1. Protein 3D Structure, Refinement, and Validation
2.2. Phytochemical and Protein Preparation
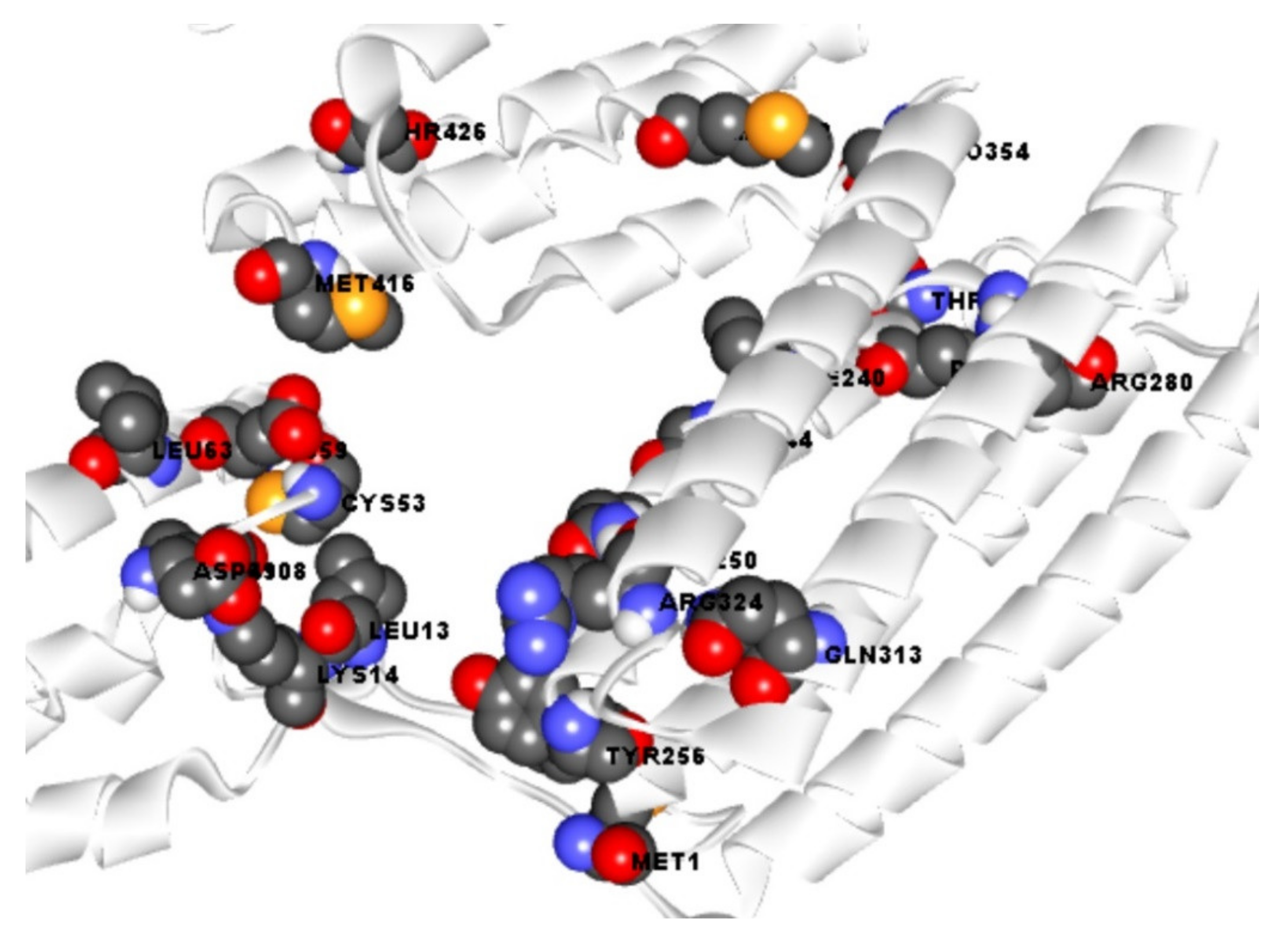
2.3. Active Site Identification and Receptor Grid Generation
2.4. Molecular Docking Simulation
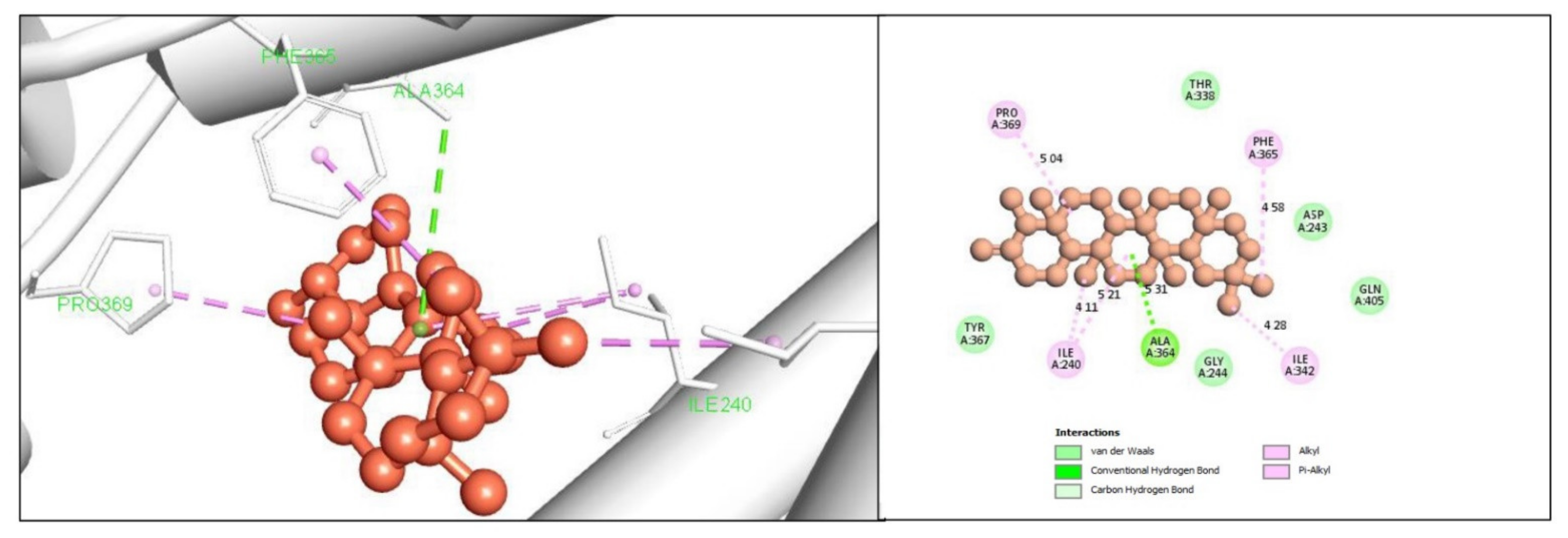
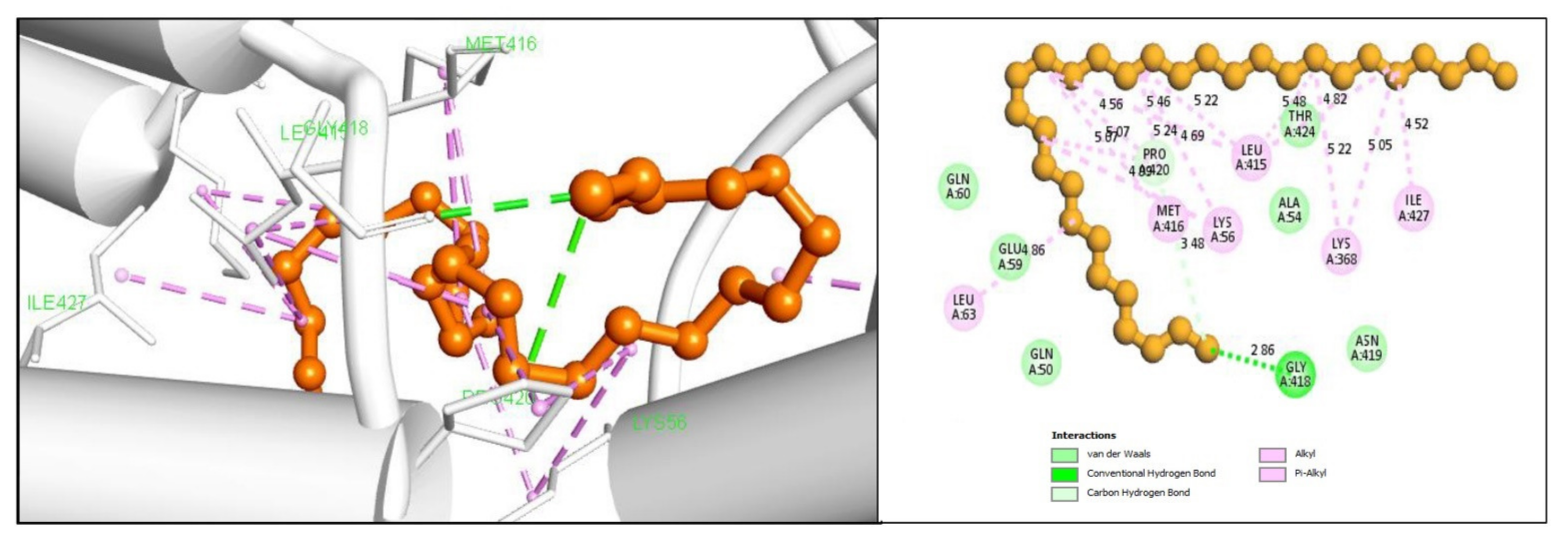
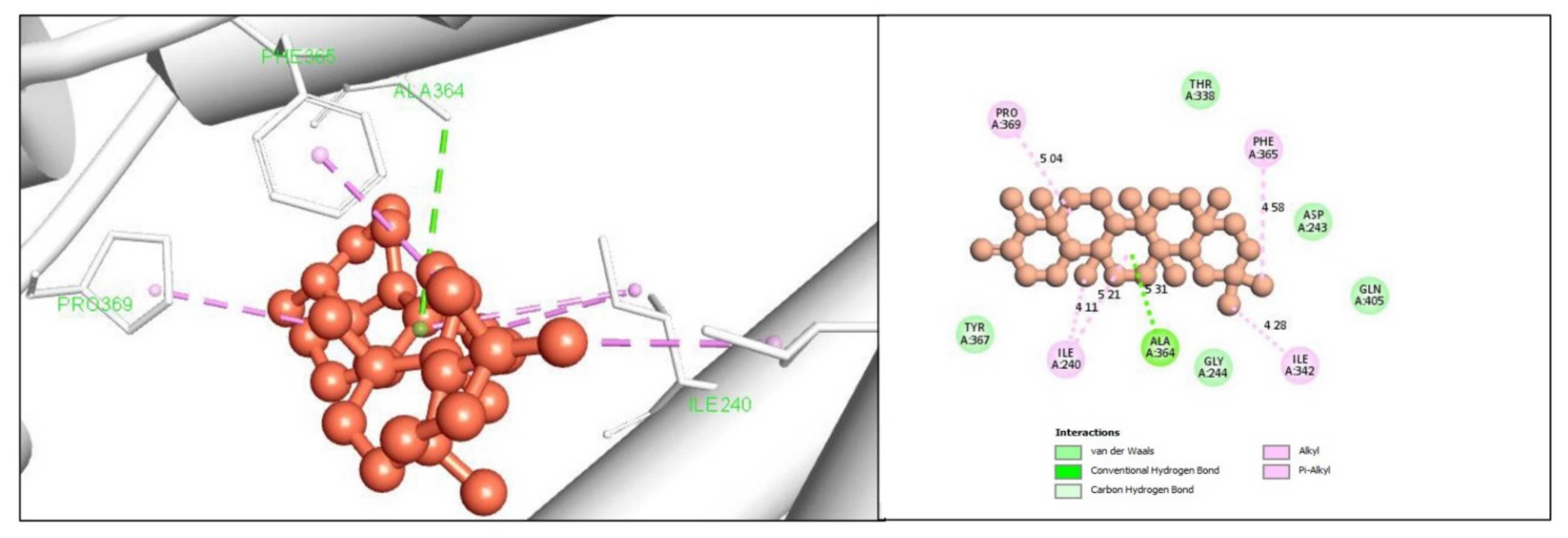
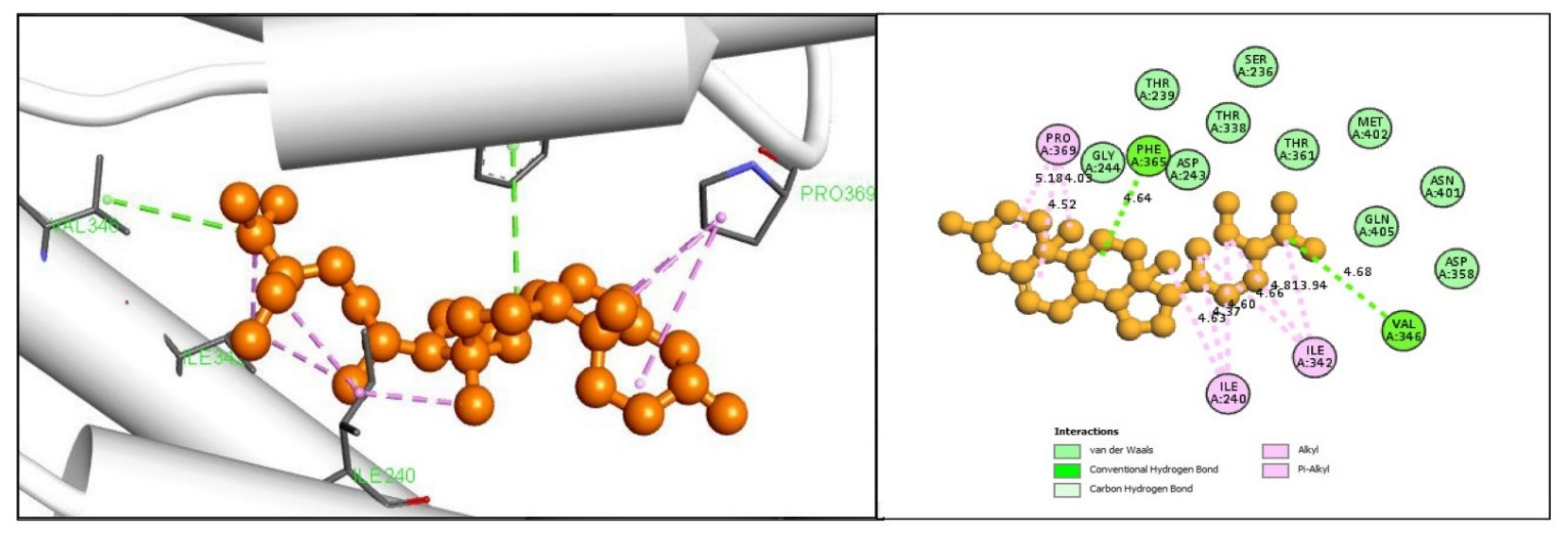
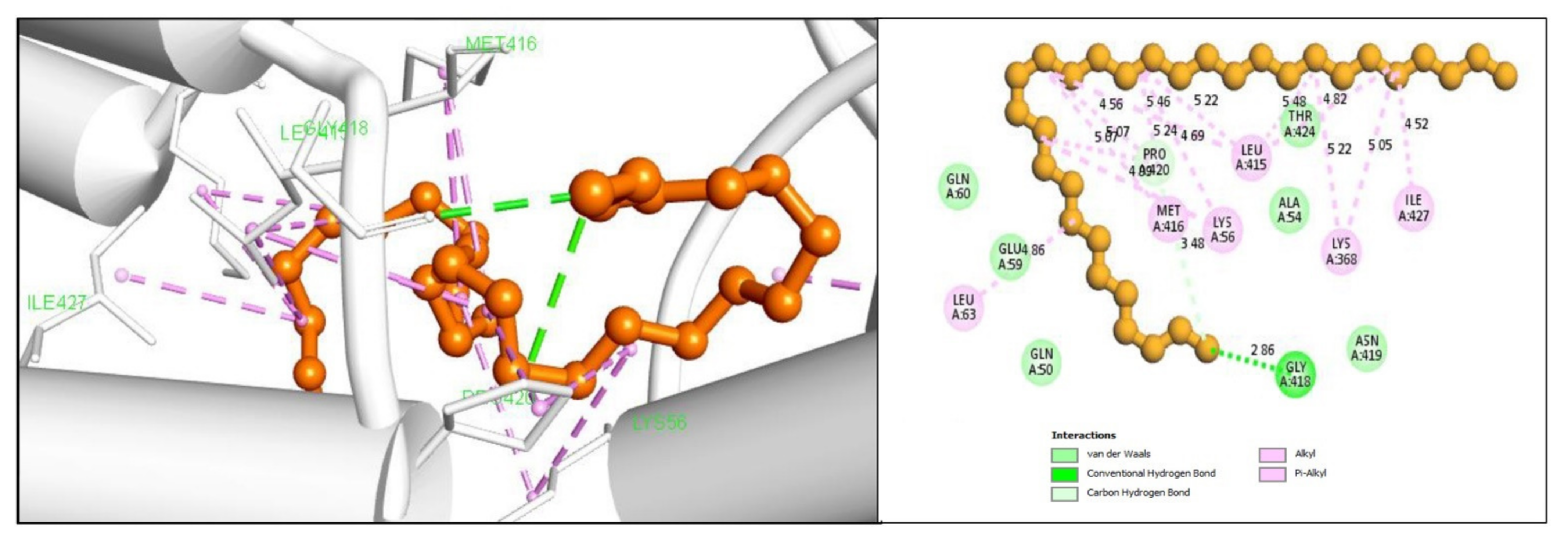
2.5. Protein-Ligand Interaction Analysis
2.6. ADME Analysis
2.7. Toxicity Prediction
2.8. MD Simulation Analysis
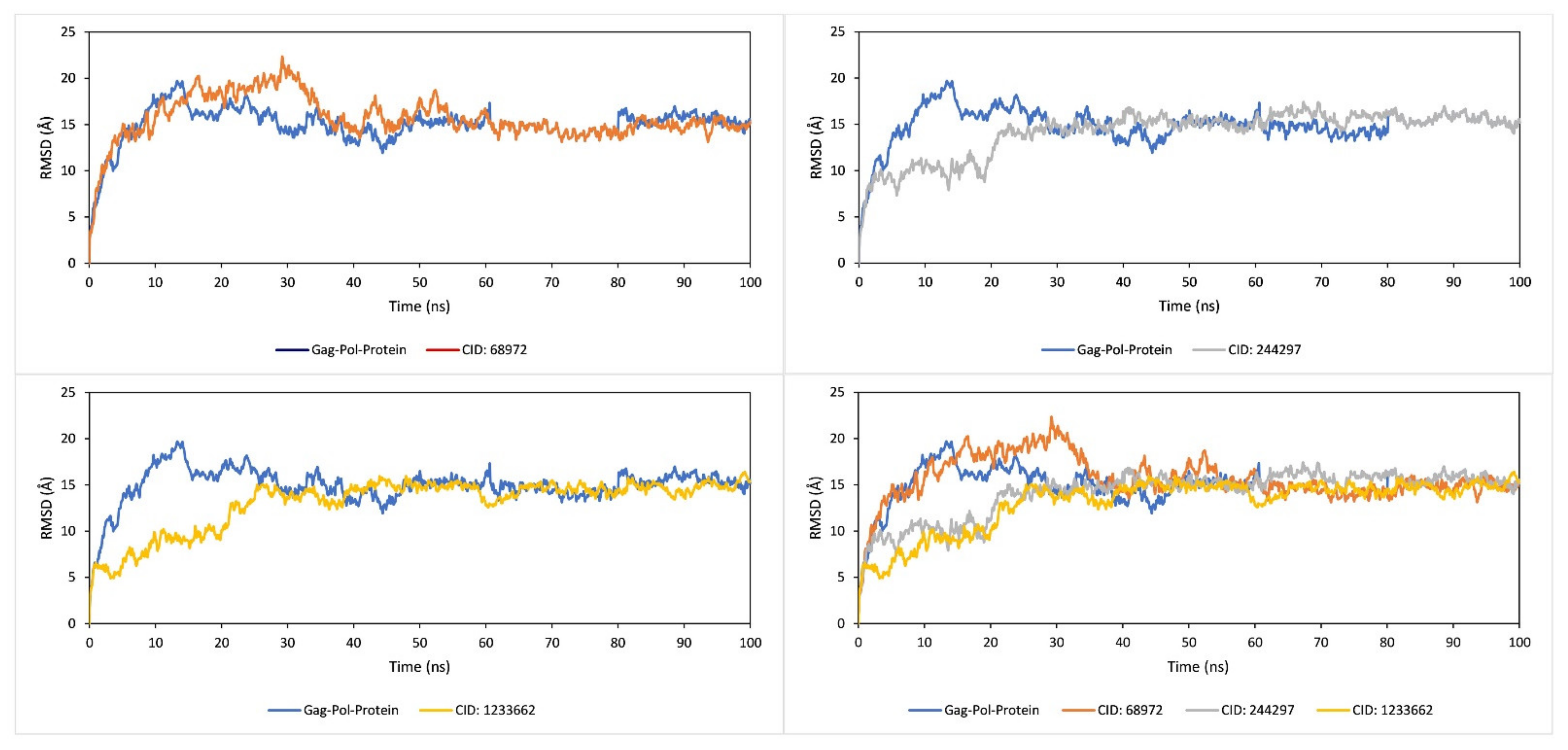
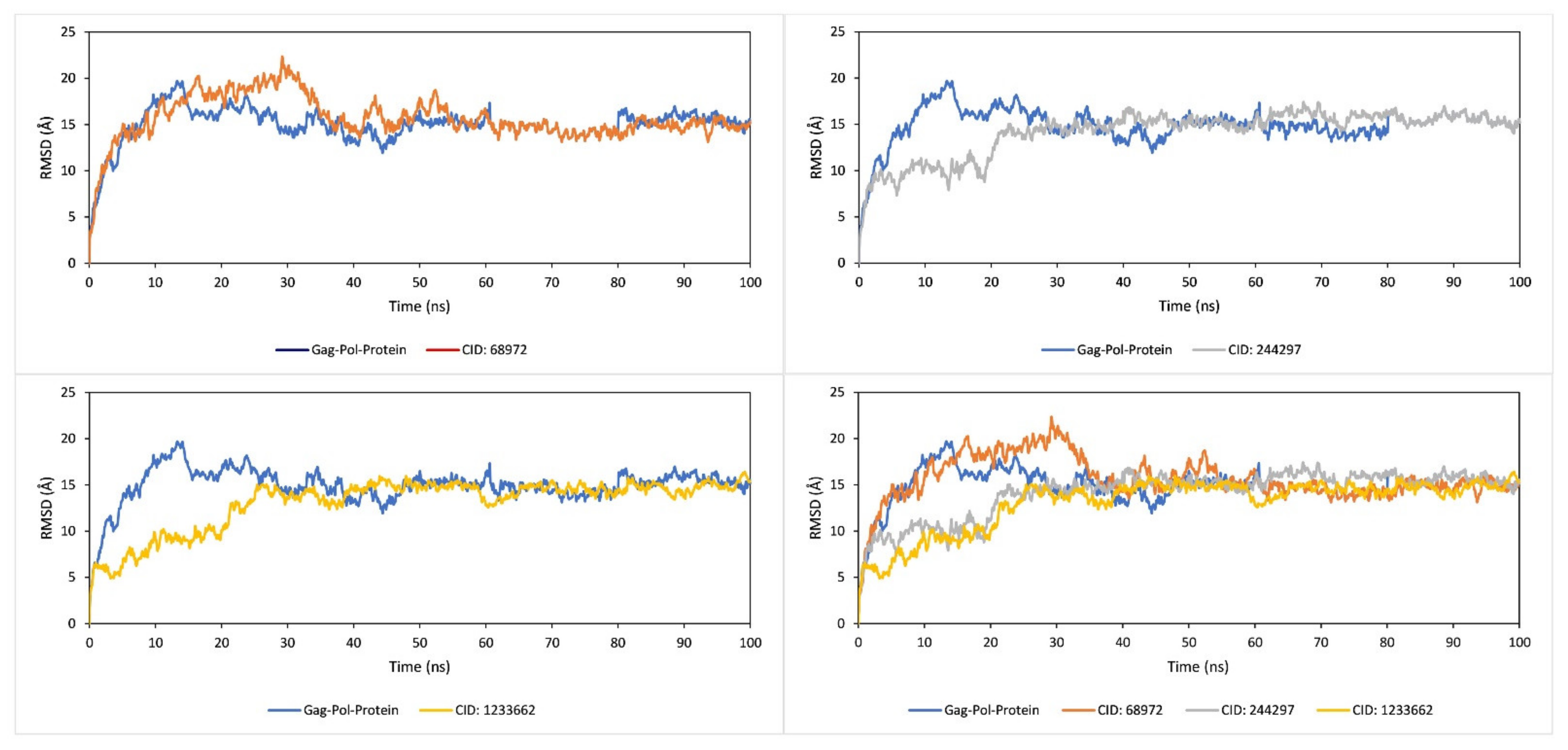
2.9. RMSD Analysis
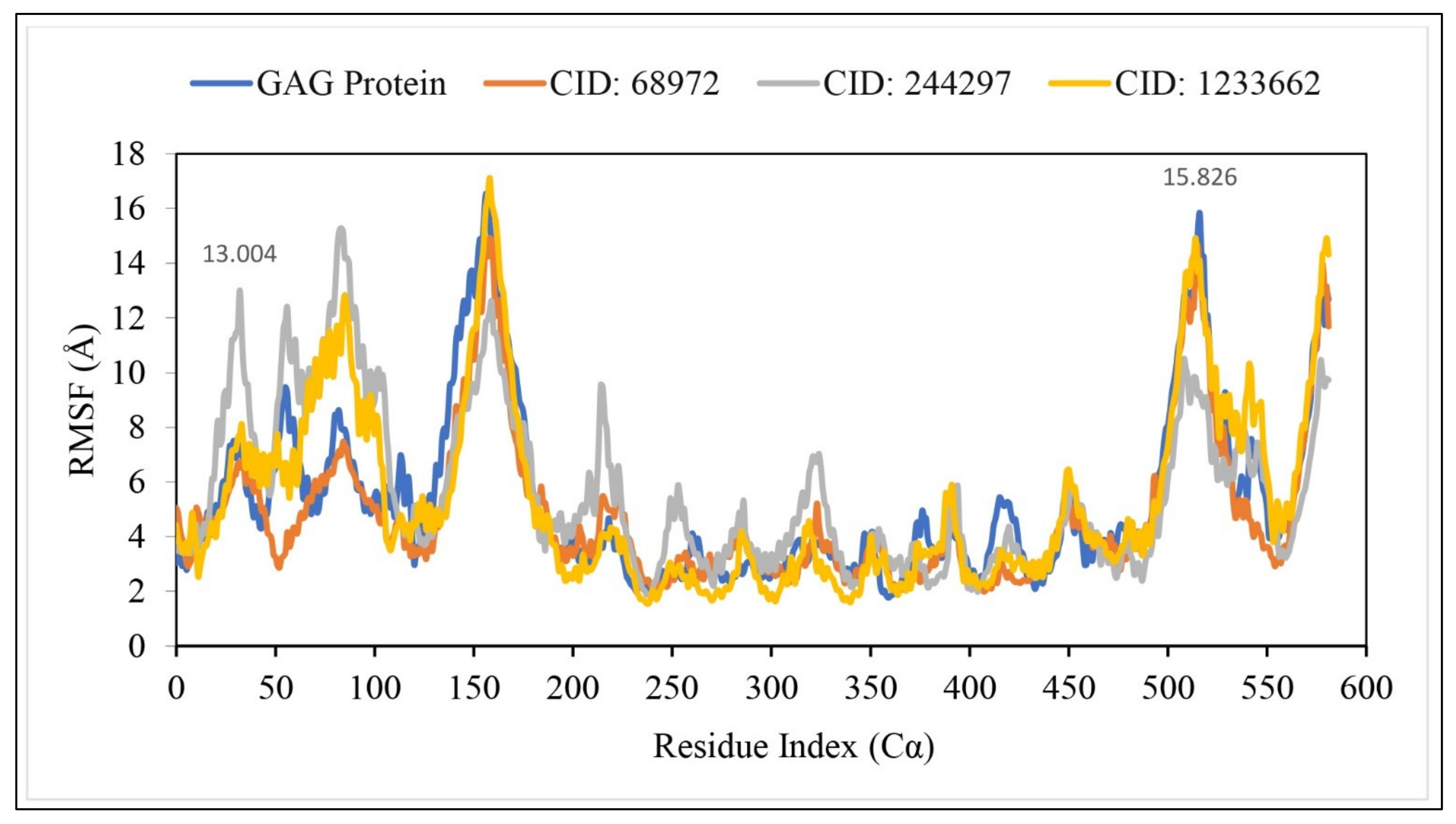
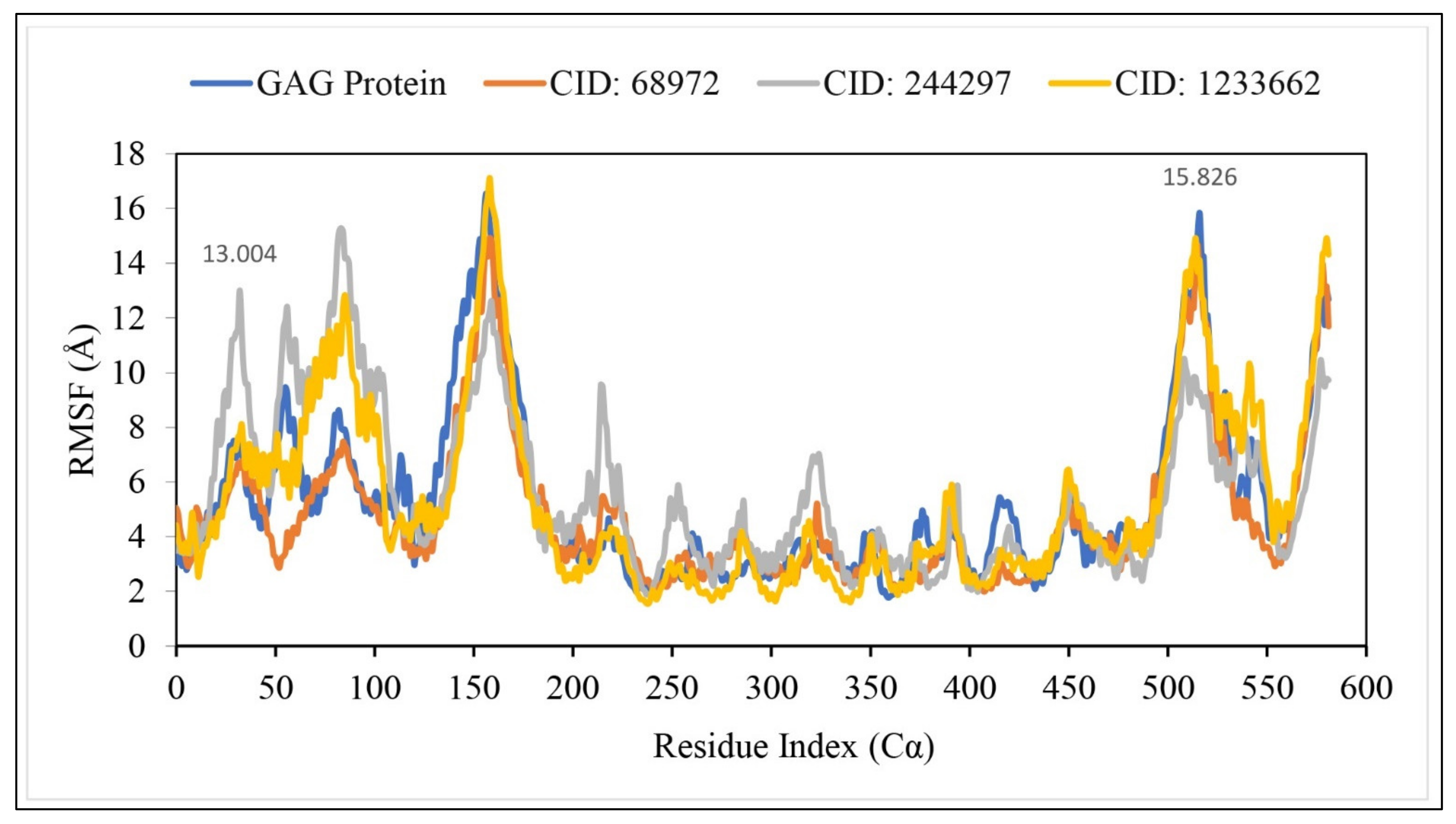
2.10. RMSF Analysis
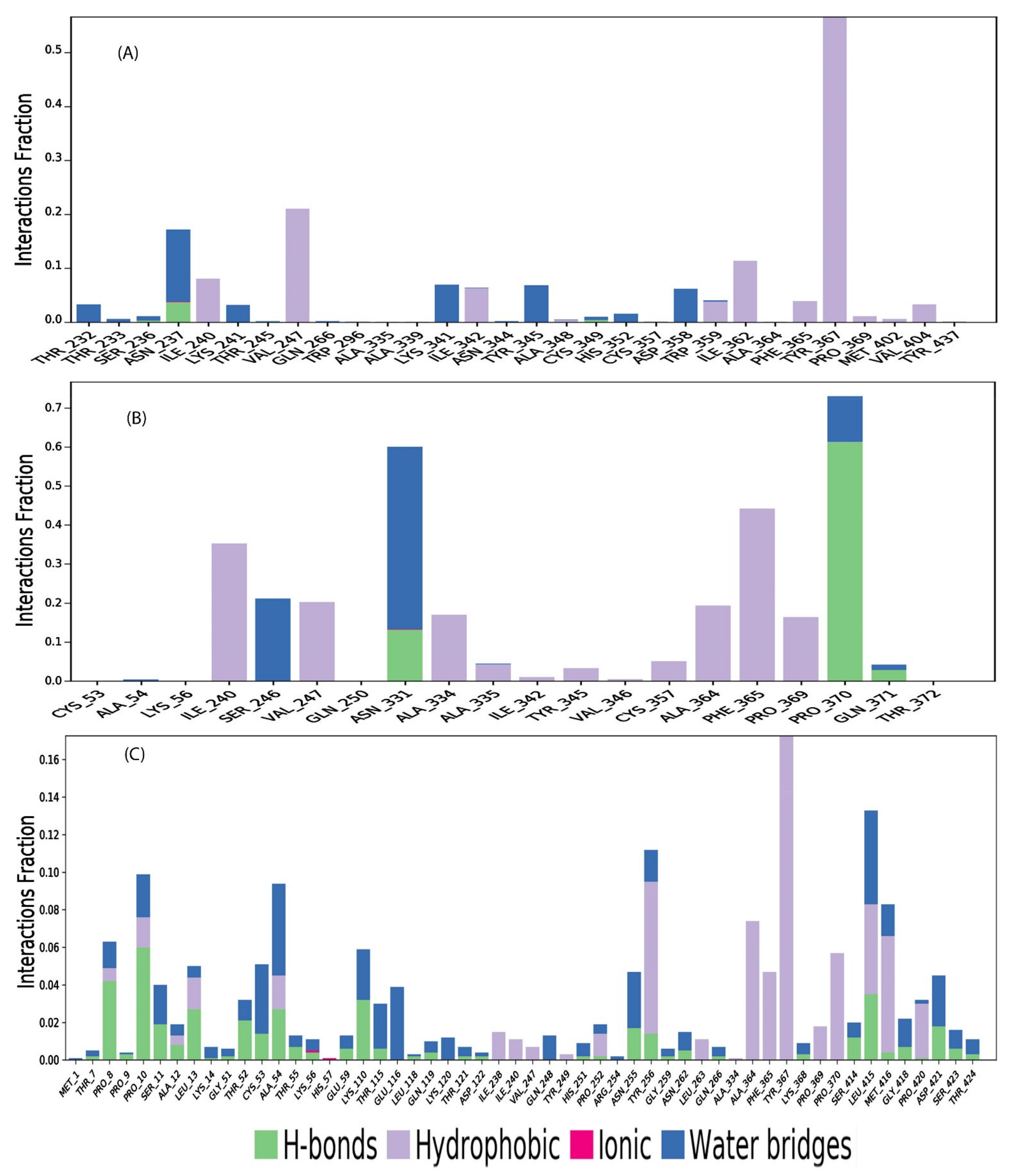
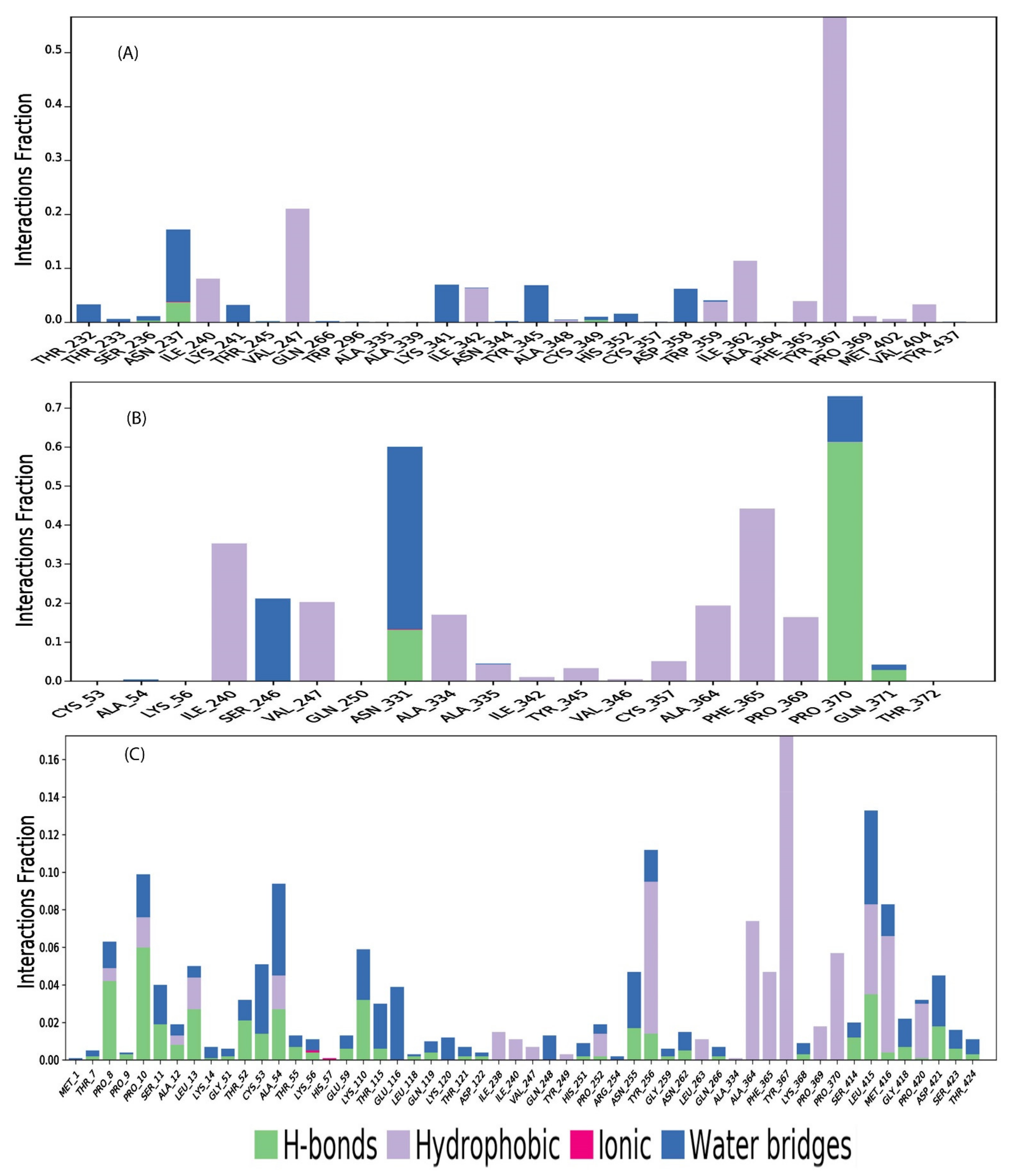
2.11. Protein-Ligand Contact Mapping
3. Discussion
4. Materials and Methods
4.1. Homology Modeling
4.2. Model Refinement and Validation
4.3. Protein and Ligand Preparation
4.4. Protein Active Site Identification and Receptor Grid Generation
4.5. Molecular Docking Simulation
4.6. ADME Analysis
4.7. Toxicity Analysis
4.8. Molecular Dynamic Simulation (MD)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chan, C.Y.; Tran, N.; Pethiyagoda, S.; Crissman, C.C.; Sulser, T.B.; Phillips, M.J. Prospects and challenges of fish for food security in Africa. Glob. Food Secur. 2019, 20, 17–25. [Google Scholar] [CrossRef]
- Walker, P.J.; Winton, J.R. Emerging viral diseases of fish and shrimp. Vet. Res. 2010, 41, 51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chávez, C.; Dresdner, J.; Figueroa, Y.; Quiroga, M. Main issues and challenges for sustainable development of salmon farming in Chile: A socio-economic perspective. Rev. Aquac. 2019, 11, 403–421. [Google Scholar] [CrossRef]
- Rovnak, J.; Quackenbush, S.L. Walleye dermal sarcoma virus: Molecular biology and oncogenesis. Viruses 2010, 2, 1984–1999. [Google Scholar] [CrossRef]
- Galluzzi, L.; Brenner, C.; Morselli, E.; Touat, Z.; Kroemer, G. Viral Control of Mitochondrial Apoptosis. PLoS Pathog. 2008, 4, e1000018. [Google Scholar] [CrossRef] [Green Version]
- Ahammad, F.; Abd Rashid, T.R.T.; Mohamed, M.; Tanbin, S.; Fuad, F.A.A. Contemporary strategies and current trends in designing antiviral drugs against dengue fever via targeting host-based approaches. Microorganisms 2019, 7, 296. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Chen, C. Understanding HIV-1 protease autoprocessing for novel therapeutic development. Future Med. Chem. 2013, 5, 1215–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z. The modification of natural products for medical use. Acta Pharm. Sin. B 2017, 7, 119–136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zandi, K.; Taherzadeh, M.; Yaghoubi, R.; Tajbakhsh, S.; Rastian, Z.; Fouladvand, M.; Sartavi, K. Antiviral activity of Avicennia marina against herpes simplex virus type 1 and vaccine strain of poliovirus (An in vitro study). J. Med. Plants Res. 2009, 3, 771–775. [Google Scholar]
- Ghildiyal, R.; Prakash, V.; Chaudhary, V.K.; Gupta, V.; Gabrani, R. Phytochemicals as antiviral agents: Recent updates. In Plant-Derived Bioactives: Production, Properties and Therapeutic Applications; Springer: Singapore, 2020; pp. 279–295. ISBN 9789811517617. [Google Scholar]
- Rahman, S.M.M.; Atikullah, M.; Islam, M.N.; Mohaimenul, M.; Ahammad, F.; Islam, M.S.; Saha, B.; Rahman, M.H. Anti-inflammatory, antinociceptive and antidiarrhoeal activities of methanol and ethyl acetate extract of Hemigraphis alternata leaves in mice. Clin. Phytosci. 2019, 5, 16. [Google Scholar] [CrossRef]
- Choudhari, A.S.; Mandave, P.C.; Deshpande, M.; Ranjekar, P.; Prakash, O. Phytochemicals in cancer treatment: From preclinical studies to clinical practice. Front. Pharmacol. 2020, 10, 1614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ziegler, S.; Pries, V.; Hedberg, C.; Waldmann, H. Target identification for small bioactive molecules: Finding the needle in the haystack. Angew. Chem. Int. Ed. 2013, 52, 2744–2792. [Google Scholar] [CrossRef]
- Yin, L.; Zheng, L.; Xu, L.; Dong, D.; Han, X.; Qi, Y.; Zhao, Y.; Xu, Y.; Peng, J. In-silico prediction of drug targets, biological activities, signal pathways and regulating networks of dioscin based on bioinformatics. BMC Complement. Altern. Med. 2015, 15, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guedes, I.A.; de Magalhães, C.S.; Dardenne, L.E. Receptor-ligand molecular docking. Biophys. Rev. 2014, 6, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Samad, A.; Ahammad, F.; Nain, Z.; Alam, R.; Imon, R.R.; Hasan, M.; Rahman, M.S. Designing a multi-epitope vaccine against SARS-CoV-2: An immunoinformatics approach. J. Biomol. Struct. Dyn. 2020, 1–17. [Google Scholar] [CrossRef]
- Opo, F.A.D.M.; Rahman, M.M.; Ahammad, F.; Ahmed, I.; Bhuiyan, M.A.; Asiri, A.M. Structure based pharmacophore modeling, virtual screening, molecular docking and ADMET approaches for identification of natural anti-cancer agents targeting XIAP protein. Sci. Rep. 2021, 11, 4049. [Google Scholar] [CrossRef]
- Ahammad, F.; Alam, R.; Mahmud, R.; Akhter, S.; Talukder, E.K.; Tonmoy, A.M.; Fahim, S.; Al-Ghamdi, K.; Samad, A.; Qadri, I. Pharmacoinformatics and molecular dynamics simulation-based phytochemical screening of neem plant (Azadiractha indica) against human cancer by targeting MCM7 protein. Brief. Bioinform. 2021. [Google Scholar] [CrossRef]
- Madhavi Sastry, G.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef]
- Mahmud, S.; Uddin, M.A.R.; Paul, G.K.; Shimu, M.S.S.; Islam, S.; Rahman, E.; Islam, A.; Islam, M.S.; Promi, M.M.; Emran, T.B.; et al. Virtual screening and molecular dynamics simulation study of plant-derived compounds to identify potential inhibitors of main protease from SARS-CoV-2. Brief. Bioinform. 2021, 22, 1402–1414. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhuiyan, M.A.; Quayum, S.T.; Ahammad, F.; Alam, R.; Samad, A.; Nain, Z. Discovery of potential immune epitopes and peptide vaccine design—A prophylactic strategy against Rift Valley fever virus. F1000Research 2020, 9, 999. [Google Scholar] [CrossRef]
- Zhou, W.; Wang, Y.; Lu, A.; Zhang, G. Systems pharmacology in small molecular drug discovery. Int. J. Mol. Sci. 2016, 17, 246. [Google Scholar] [CrossRef] [Green Version]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc. 2010, 5, 725–738. [Google Scholar] [CrossRef] [Green Version]
- Heo, L.; Park, H.; Seok, C. GalaxyRefine: Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 2013, 41, W384–W388. [Google Scholar] [CrossRef] [Green Version]
- Ho, B.K.; Thomas, A.; Brasseur, R. Revisiting the Ramachandran plot: Hard-sphere repulsion, electrostatics, and H-bonding in the α-helix. Protein Sci. 2009, 12, 2508–2522. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, 407–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaker, M.S.; Oppenheimer, J.; Grayson, M.; Stukus, D.; Hartog, N.; Hsieh, E.W.Y.; Rider, N.; Dutmer, C.M.; Vander Leek, T.K.; Kim, H.; et al. COVID-19: Pandemic Contingency Planning for the Allergy and Immunology Clinic. J. Allergy Clin. Immunol. Pract. 2020, 8, 1477–1488.e5. [Google Scholar] [CrossRef] [PubMed]
- Ahammad, F.; Fuad, F.A.A. The in silico identification of potent natural bioactive anti-dengue agents by targeting the human hexokinase 2 enzyme. In Proceedings of the 5th International Electronic Conference on Medicinal Chemistry, Basel, Switzerland, 1–30 November 2019; p. 6342. [Google Scholar] [CrossRef]
- Vyas, A.; Saraf, S.; Saraf, S. Cyclodextrin based novel drug delivery systems. J. Incl. Phenom. Macrocycl. Chem. 2008, 62, 23–42. [Google Scholar] [CrossRef]
- Hadni, H.; Elhallaoui, M. 2D and 3D-QSAR, molecular docking and ADMET properties: In silico studies of azaaurones as antimalarial agents. New J. Chem. 2020, 44, 6553–6565. [Google Scholar] [CrossRef]
- Yamashita, S.; Inoue, Y.; Maruyama, S.; Murakami, Y.; Yaguchi, H.; Jablonski, M.; Set, S.Y. Saturable absorbers incorporating carbon nanotubes directly synthesized onto substrates and fibers and their application to mode-locked fiber lasers. Opt. Lett. 2004, 29, 1581. [Google Scholar] [CrossRef]
- Raies, A.B.; Bajic, V.B. In silico toxicology: Computational methods for the prediction of chemical toxicity. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2016, 6, 147–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinn, W.E. Emotional facial expression in Parkinson’s disease: A response to Bowers (2006). J. Int. Neuropsychol. Soc. 2007, 13, 721–722. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the opls force field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef]
- Shinoda, W.; Mikami, M. Rigid-body dynamics in the isothermal-isobaric ensemble: A test on the accuracy and computational efficiency. J. Comput. Chem. 2003, 24, 920–930. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PubChem CID | Chemical Name | Formula | Binding Affinity (kcal/mol) |
|---|---|---|---|
| CID244297 | Friedlein | C30H50O | −8.5 |
| CID12303662 | Phytosterols | C29H50O | −8.0 |
| CID68972 | 1-Triacontanol | C30H62O | −7.9 |
| PubChem CID | Residue | Distance | Category | Type |
|---|---|---|---|---|
| CID244297 | ALA364 | 5.30689 | Hydrogen Bond | Conv-H-Bond |
| ILE240 | 5.20546 | Hydrophobic | Alkyl | |
| PRO369 | 5.03758 | Hydrophobic | Alkyl | |
| ILE240 | 4.10592 | Hydrophobic | Alkyl | |
| ILE342 | 4.27502 | Hydrophobic | Alkyl | |
| PHE365 | 4.58263 | Hydrophobic | Pi-Alkyl | |
| CID6897 | GLY418 | 2.86383 | Hydrogen Bond | Conv-H-Bond |
| PRO420 | 3.47712 | Hydrogen Bond | C-H Bond | |
| LYS56 | 4.6933 | Hydrophobic | Alkyl | |
| LYS56 | 4.99136 | Hydrophobic | Alkyl | |
| LYS368 | 5.21921 | Hydrophobic | Alkyl | |
| LYS368 | 5.05134 | Hydrophobic | Alkyl | |
| LEU415 | 5.22063 | Hydrophobic | Alkyl | |
| MET416 | 5.23747 | Hydrophobic | Alkyl | |
| PRO420 | 4.55835 | Hydrophobic | Alkyl | |
| PRO420 | 5.075 | Hydrophobic | Alkyl | |
| LEU415 | 5.47725 | Hydrophobic | Alkyl | |
| LEU415 | 4.82065 | Hydrophobic | Alkyl | |
| ILE427 | 4.51538 | Hydrophobic | Alkyl | |
| LEU415 | 5.46433 | Hydrophobic | Alkyl | |
| MET416 | 5.07033 | Hydrophobic | Alkyl | |
| LEU63 | 4.86074 | Hydrophobic | Alkyl | |
| CID12303662 | VAL346 | 4.68285 | Hydrogen Bond | Conv-H-Bond |
| PHE365 | 4.64493 | Hydrogen Bond | Conv-H-Bond | |
| PRO369 | 4.52399 | Hydrophobic | Alkyl | |
| PRO369 | 5.17641 | Hydrophobic | Alkyl | |
| ILE240 | 4.62631 | Hydrophobic | Alkyl | |
| PRO369 | 4.03084 | Hydrophobic | Alkyl | |
| ILE240 | 4.36608 | Hydrophobic | Alkyl | |
| ILE342 | 4.65569 | Hydrophobic | Alkyl | |
| ILE342 | 3.93691 | Hydrophobic | Alkyl | |
| ILE240 | 4.59804 | Hydrophobic | Alkyl | |
| ILE342 | 4.80551 | Hydrophobic | Alkyl |
| Properties | CID244297 | CID12230662 | CID68972 | |
|---|---|---|---|---|
| Physico-chemical attribute | MW (g/mol) | 426.73 g/mol | 414.72 g/mol | 438.8 g/mol |
| Heavy atoms | 31 | 30 | 31 | |
| Aromatic heavy atoms | 0 | 1 | 0 | |
| Rotatable bonds | 0 | 6 | 28 | |
| H-bond acceptors | 1 | 1 | 1 | |
| H-bond donors | 0 | 1 | 4 | |
| Lipophilicity | Log Po/w | 8.46 | 8.02 | 7.67 |
| Water solubility | Log S (ESOL) | Soluble | Soluble | Soluble |
| Pharmacokinetics | GI absorption | Low | Low | Low |
| Drug-likeness | Lipinski | Yes | Yes | Yes |
| Medi. Chemistry | Synth. accessibility | Easy | Easy | Easy |
| PubChem ID | Hepatotoxicity | Carcinogenicity | Immunotoxicity | Mutagenicity | Cytotoxicity |
|---|---|---|---|---|---|
| CID244297 | Inactive | No | Inactive | Inactive | Inactive |
| CID12230662 | Inactive | No | Light active | Inactive | Inactive |
| CID68972 | Inactive | No | Inactive | Inactive | Inactive |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aljahdali, M.O.; Molla, M.H.R.; Ahammad, F. Compounds Identified from Marine Mangrove Plant (Avicennia alba) as Potential Antiviral Drug Candidates against WDSV, an In-Silico Approach. Mar. Drugs 2021, 19, 253. https://doi.org/10.3390/md19050253
Aljahdali MO, Molla MHR, Ahammad F. Compounds Identified from Marine Mangrove Plant (Avicennia alba) as Potential Antiviral Drug Candidates against WDSV, an In-Silico Approach. Marine Drugs. 2021; 19(5):253. https://doi.org/10.3390/md19050253
Chicago/Turabian StyleAljahdali, Mohammed Othman, Mohammad Habibur Rahman Molla, and Foysal Ahammad. 2021. "Compounds Identified from Marine Mangrove Plant (Avicennia alba) as Potential Antiviral Drug Candidates against WDSV, an In-Silico Approach" Marine Drugs 19, no. 5: 253. https://doi.org/10.3390/md19050253
APA StyleAljahdali, M. O., Molla, M. H. R., & Ahammad, F. (2021). Compounds Identified from Marine Mangrove Plant (Avicennia alba) as Potential Antiviral Drug Candidates against WDSV, an In-Silico Approach. Marine Drugs, 19(5), 253. https://doi.org/10.3390/md19050253