
Synthesis-Driven Stereochemical Assignment of Marine Polycyclic Ether Natural Products


Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
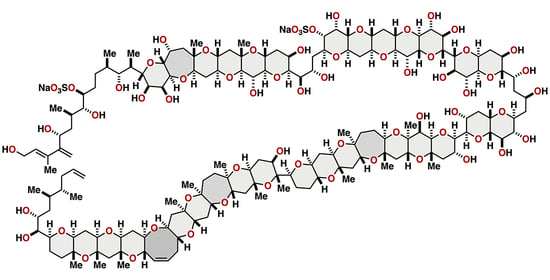
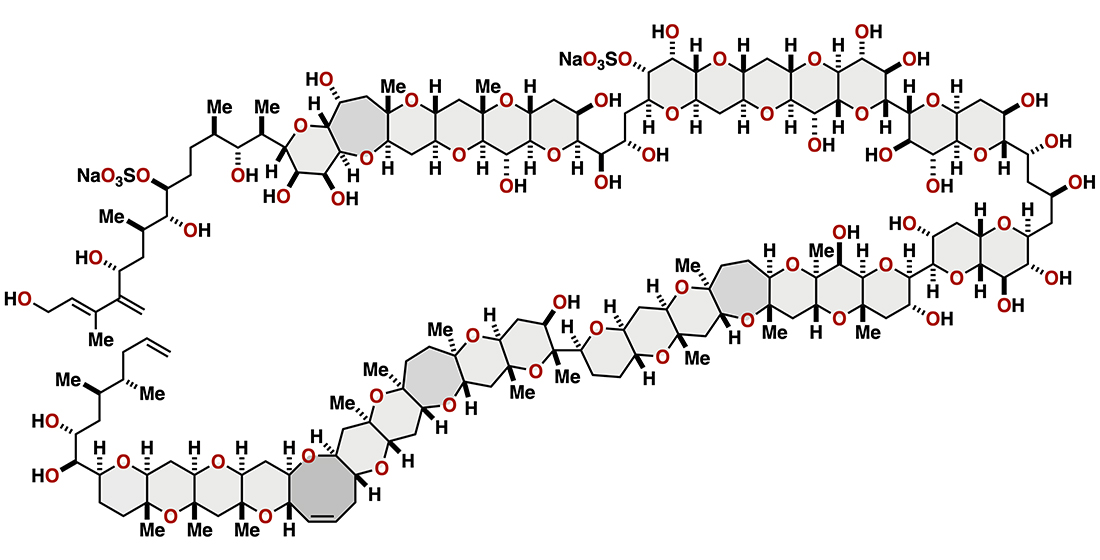
2. Maitotoxin
2.1. Determination of the Absolute Configuration of Maitotoxin by the Tachibana Group and Yasumoto Group
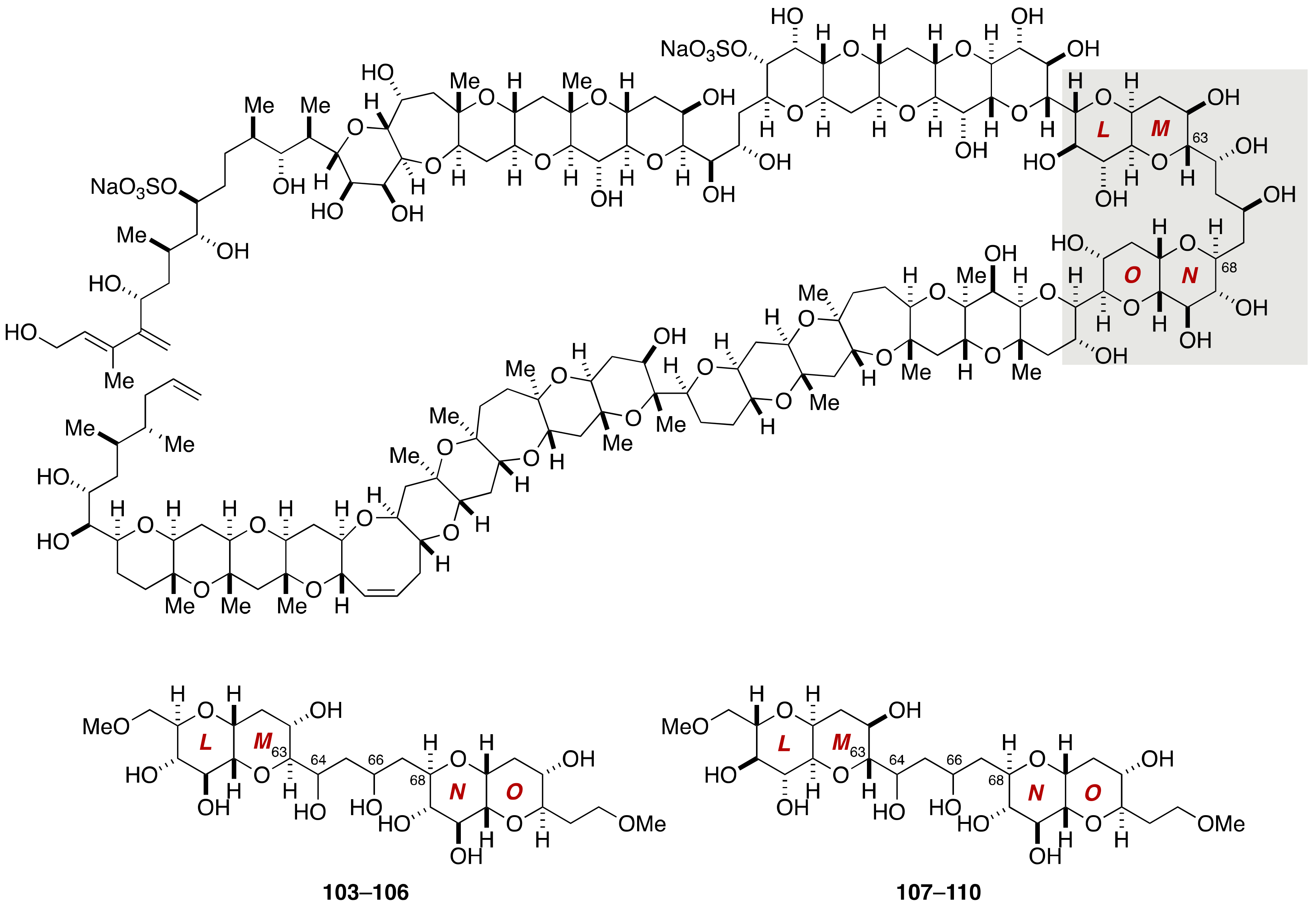
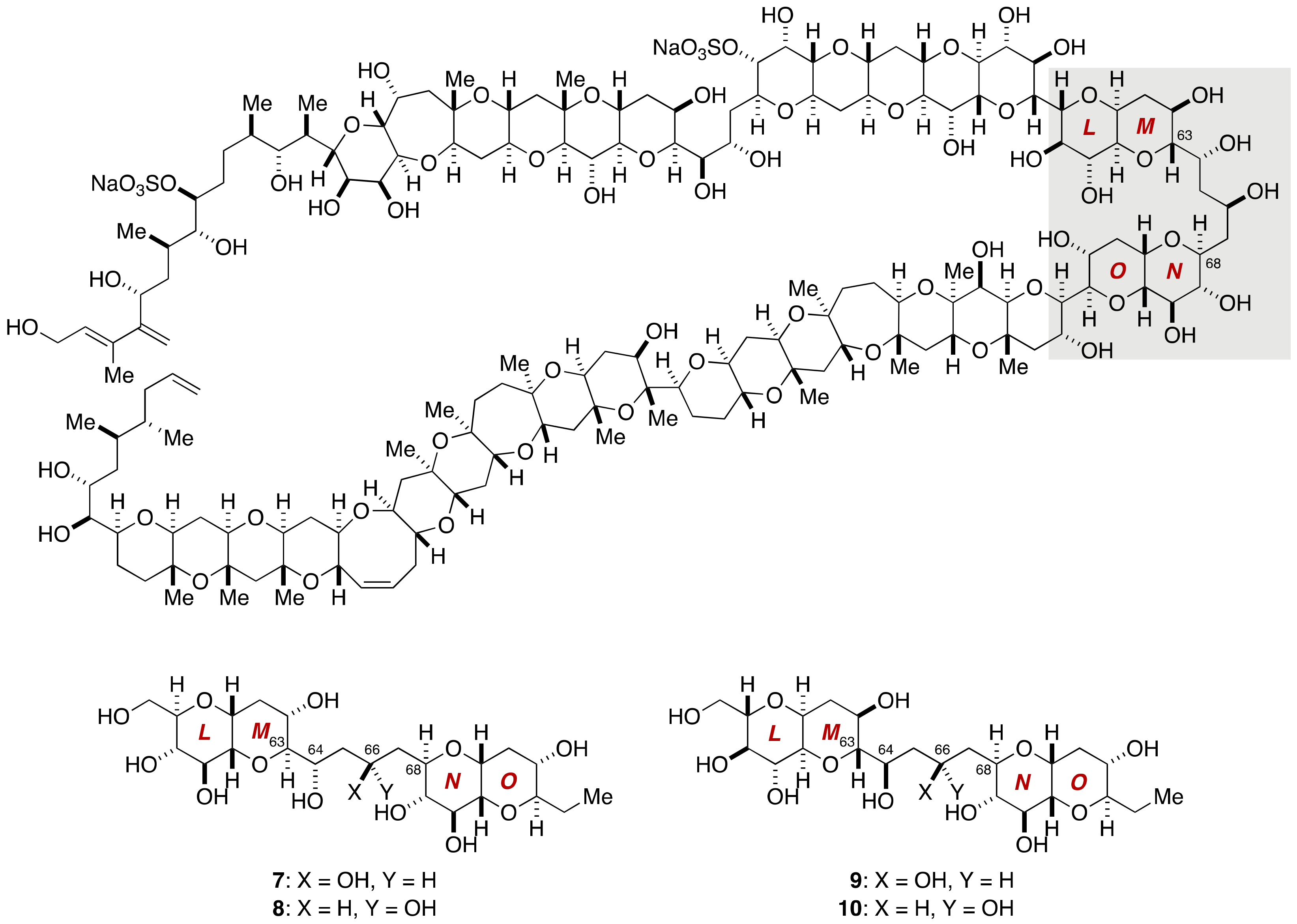
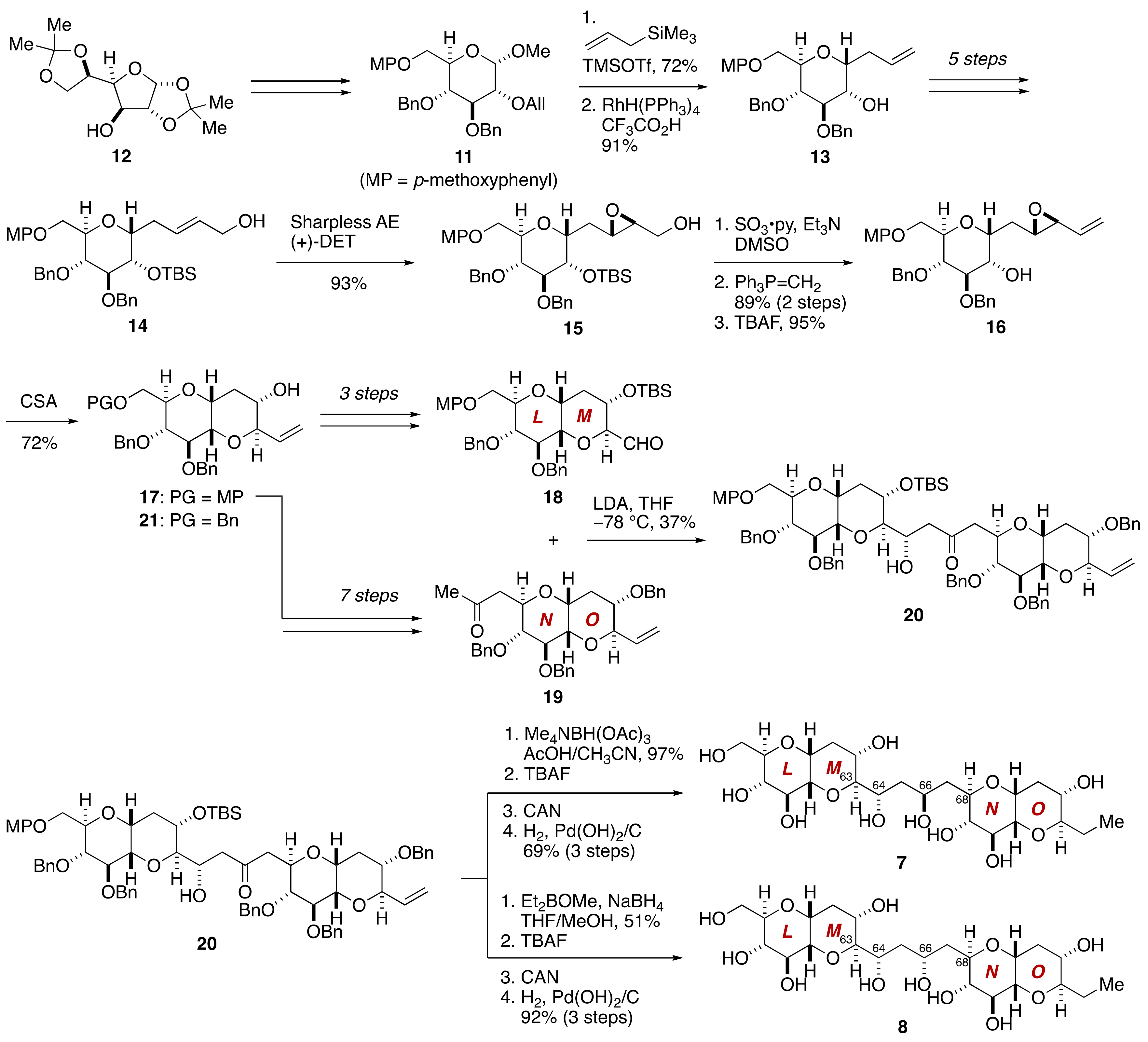
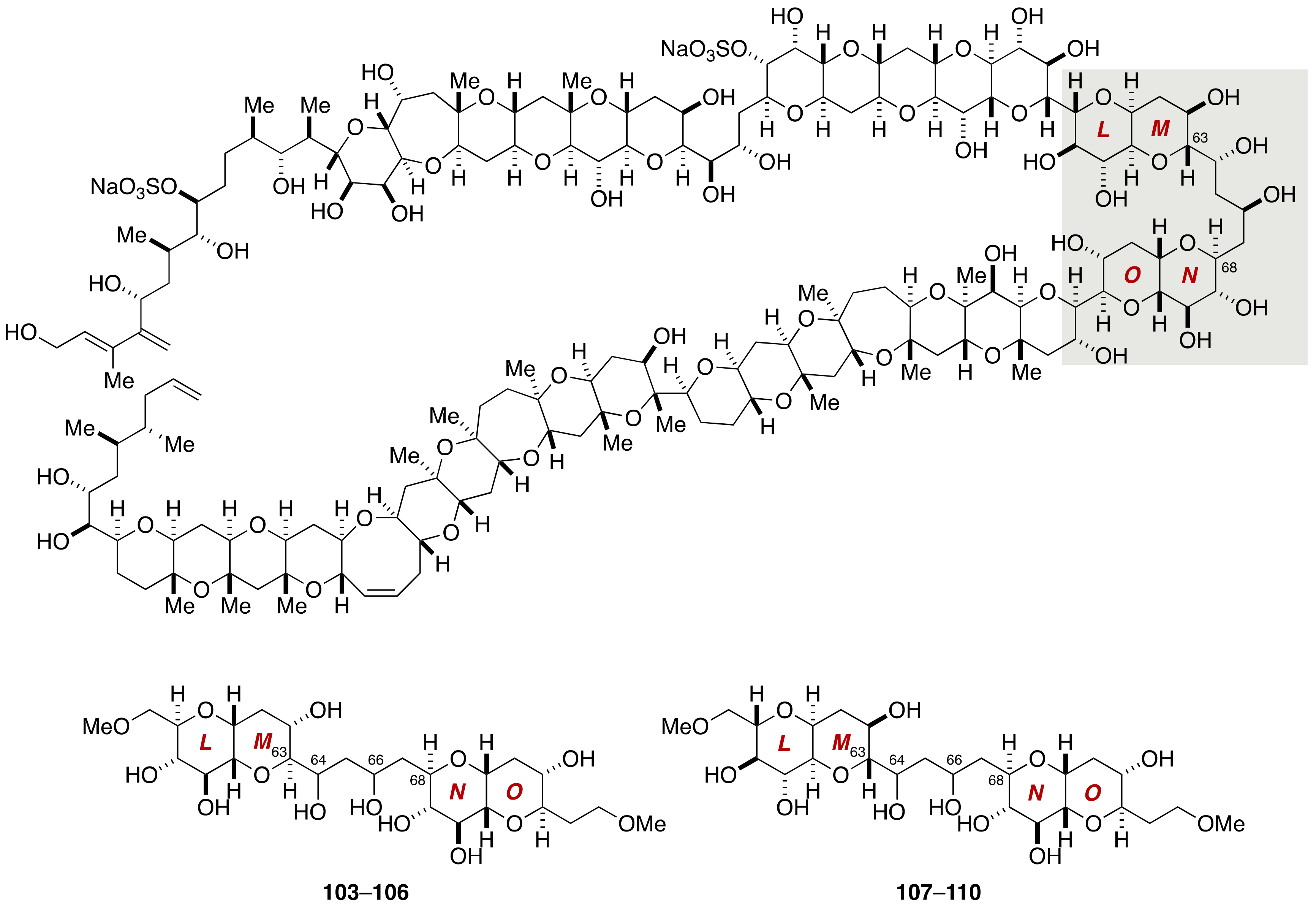
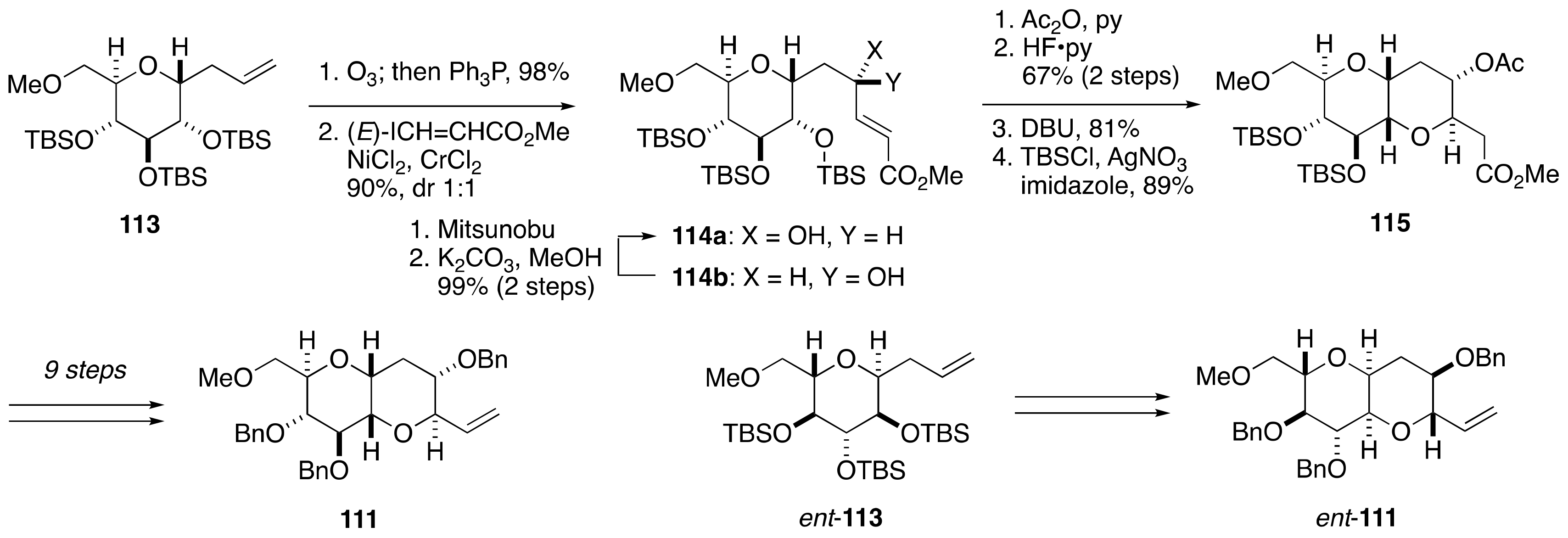
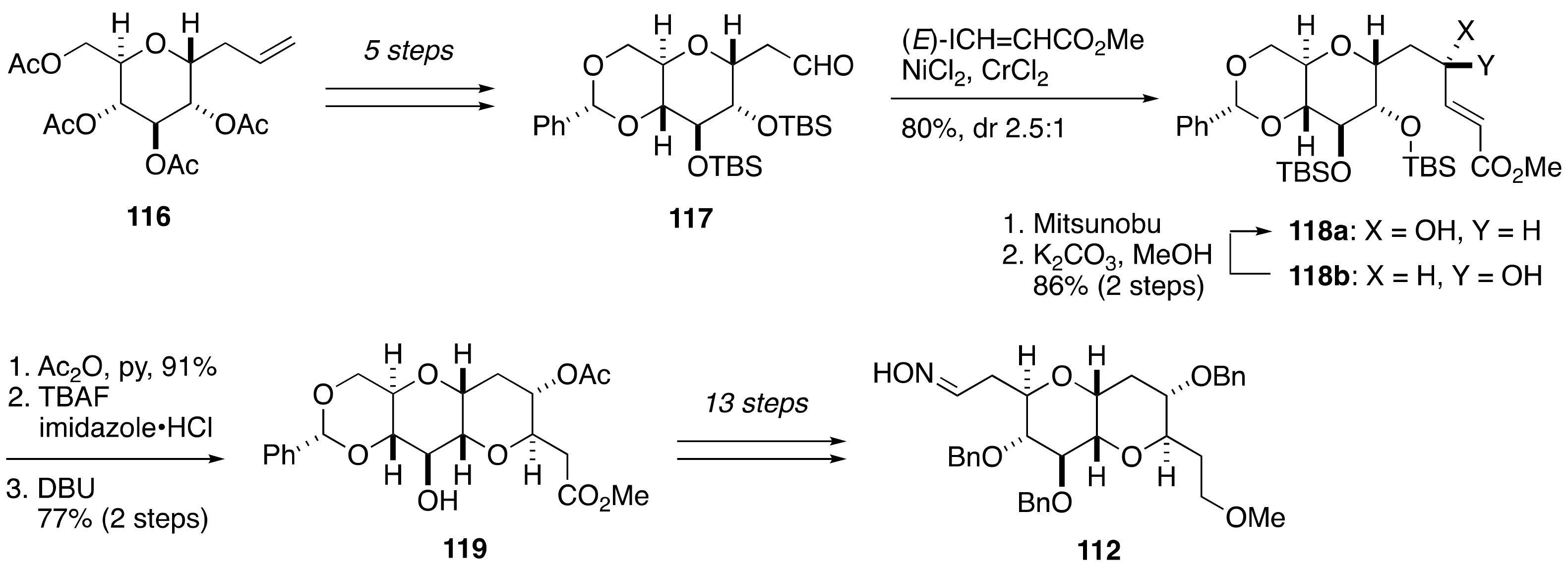
2.1.1. Stereochemical Assignment of the C63–C68 Linkage
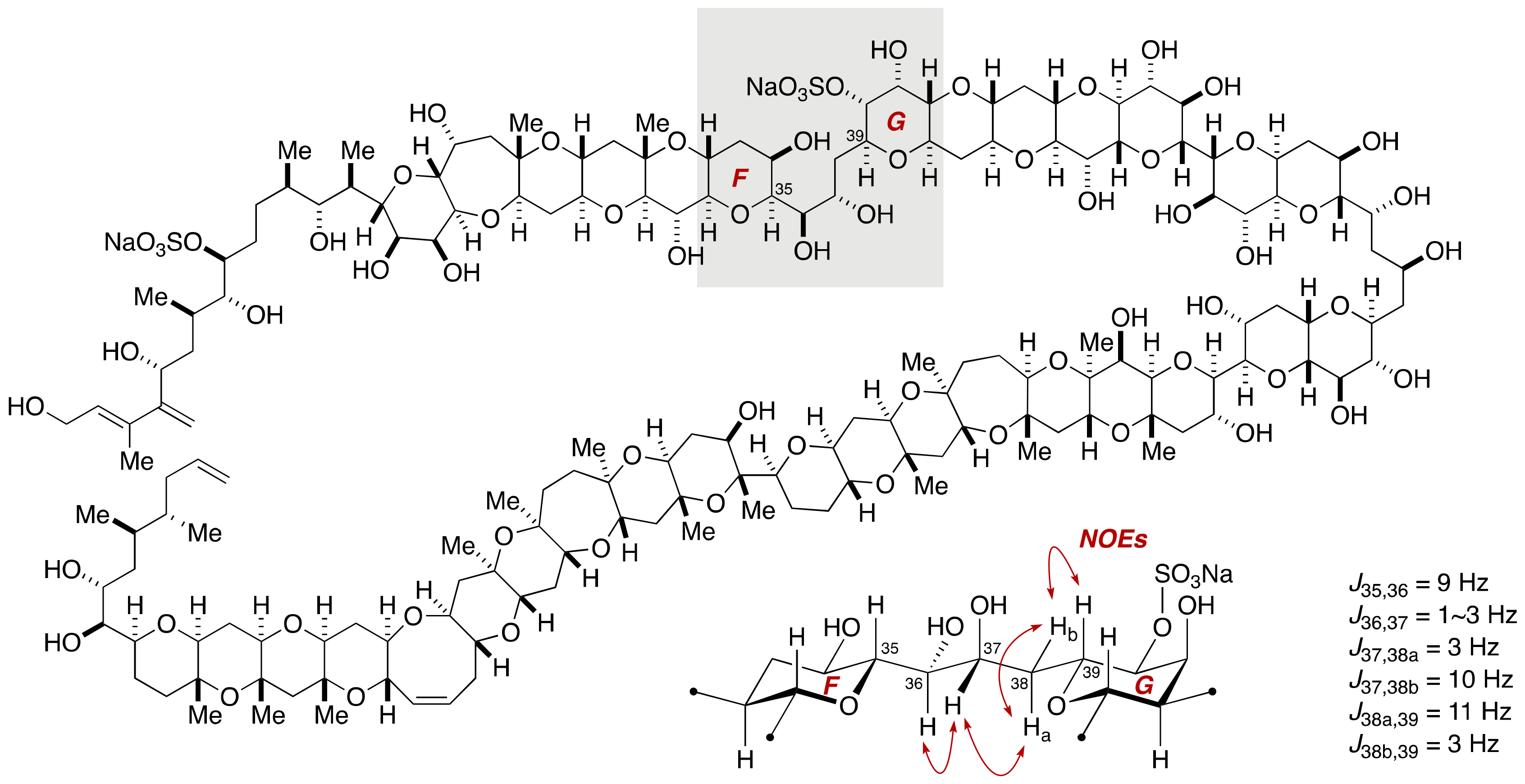
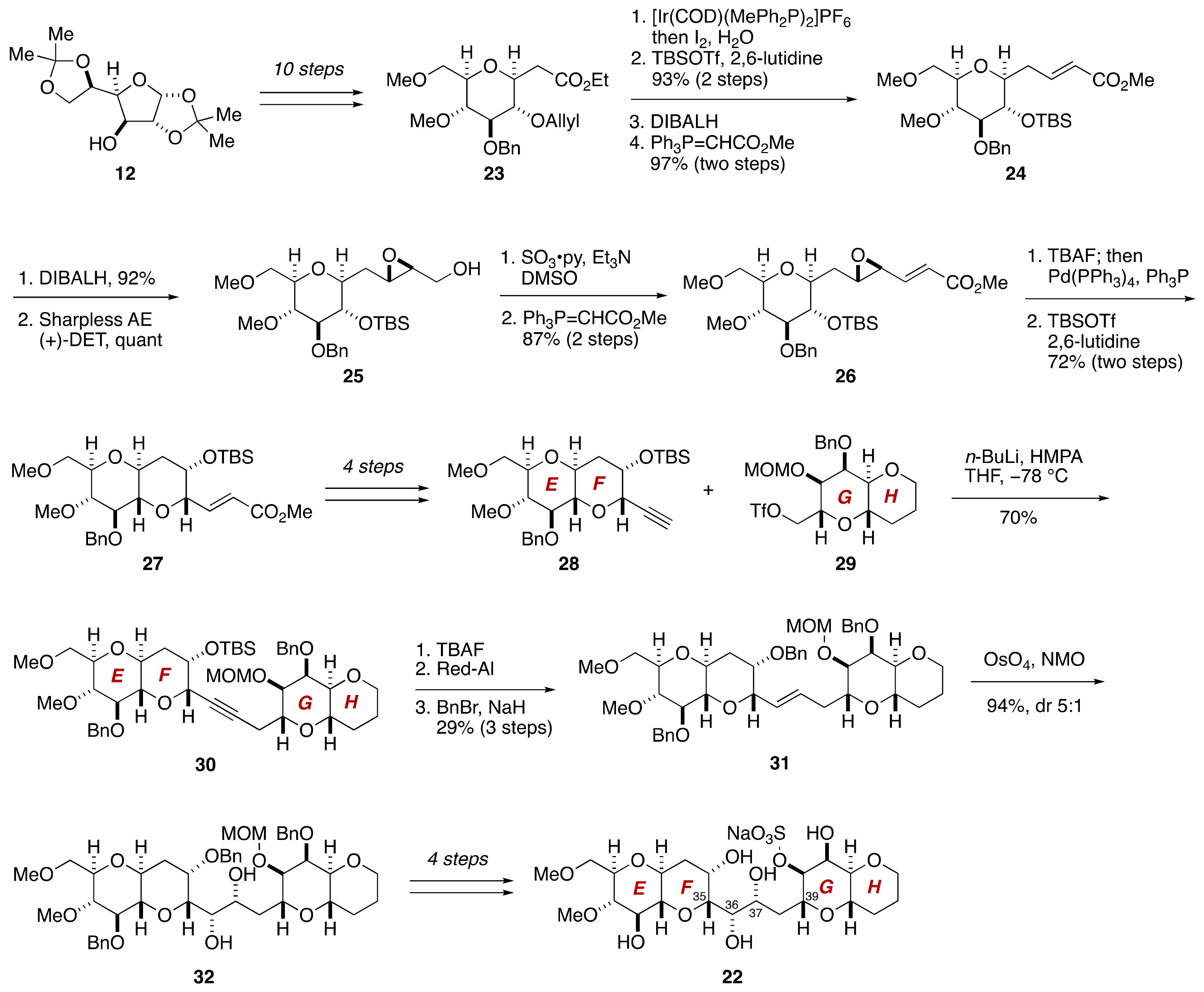
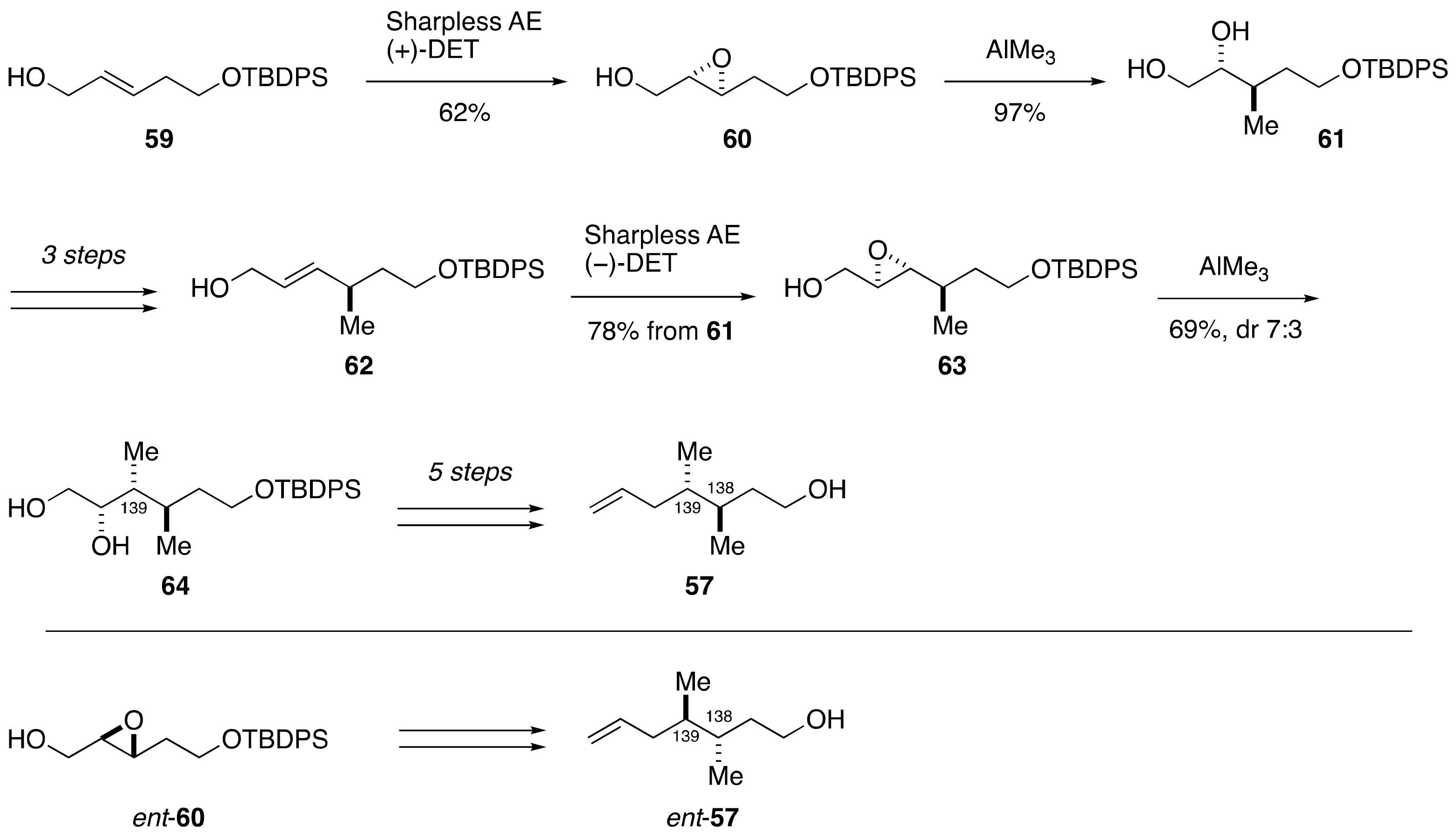
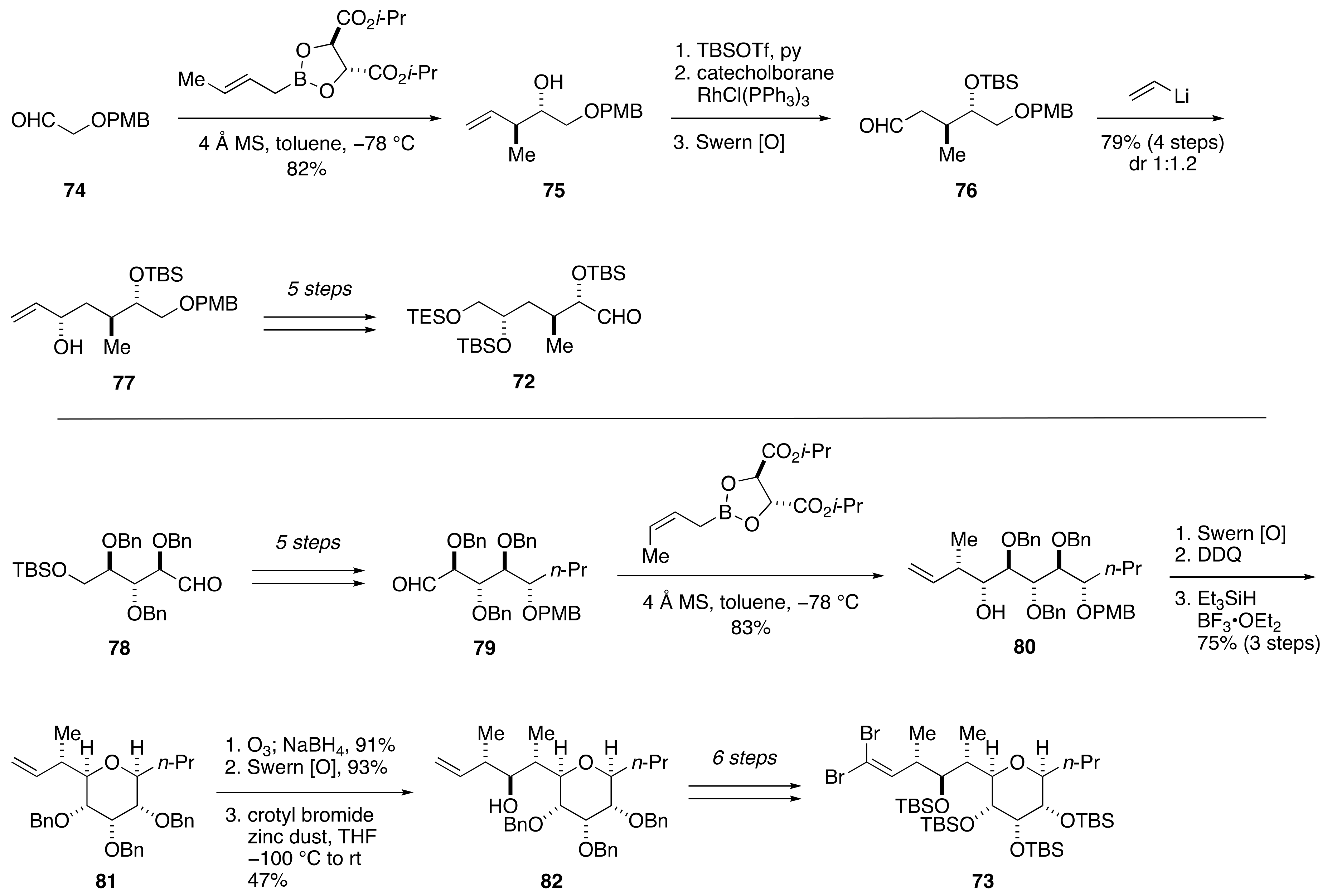
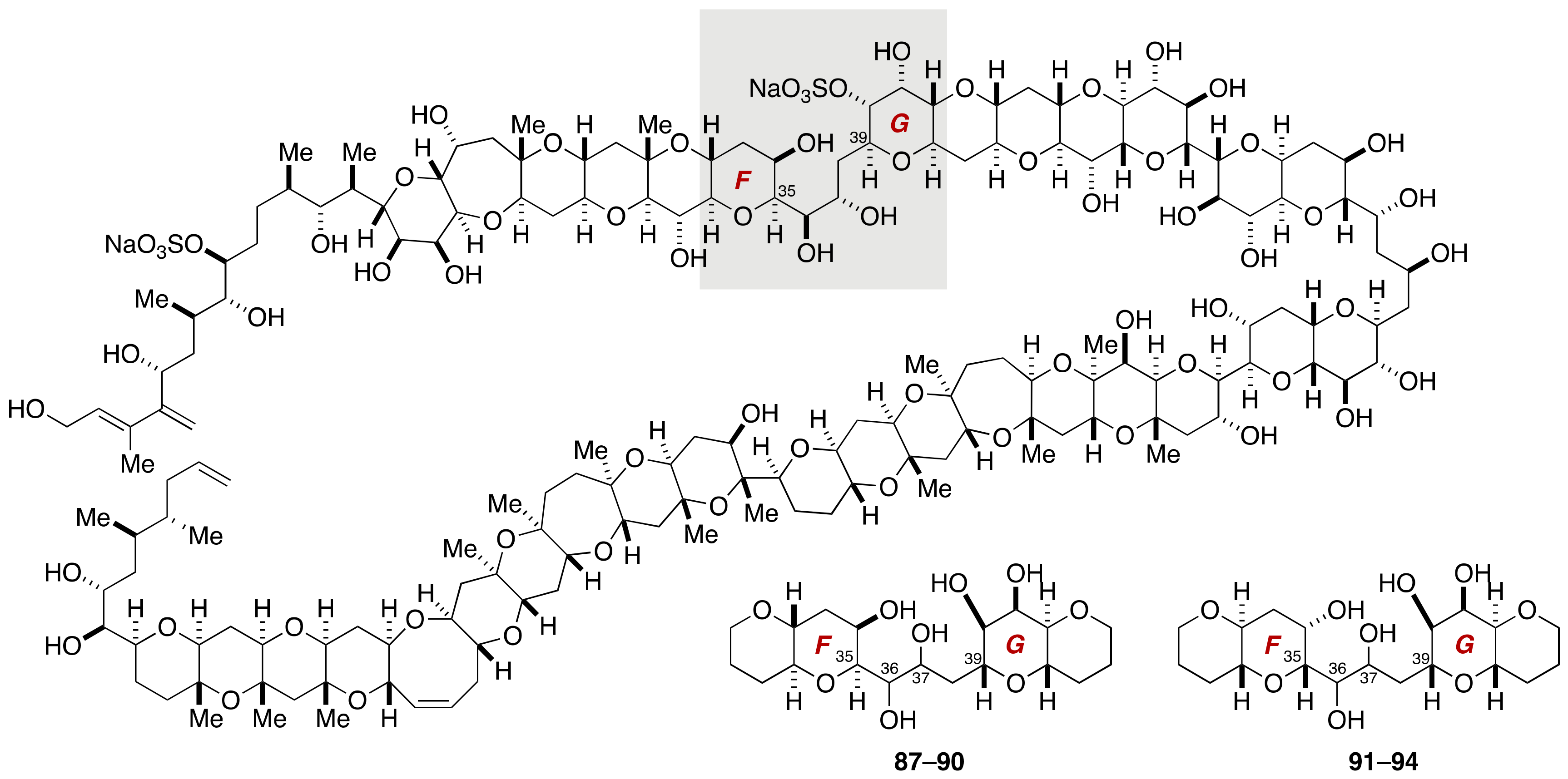
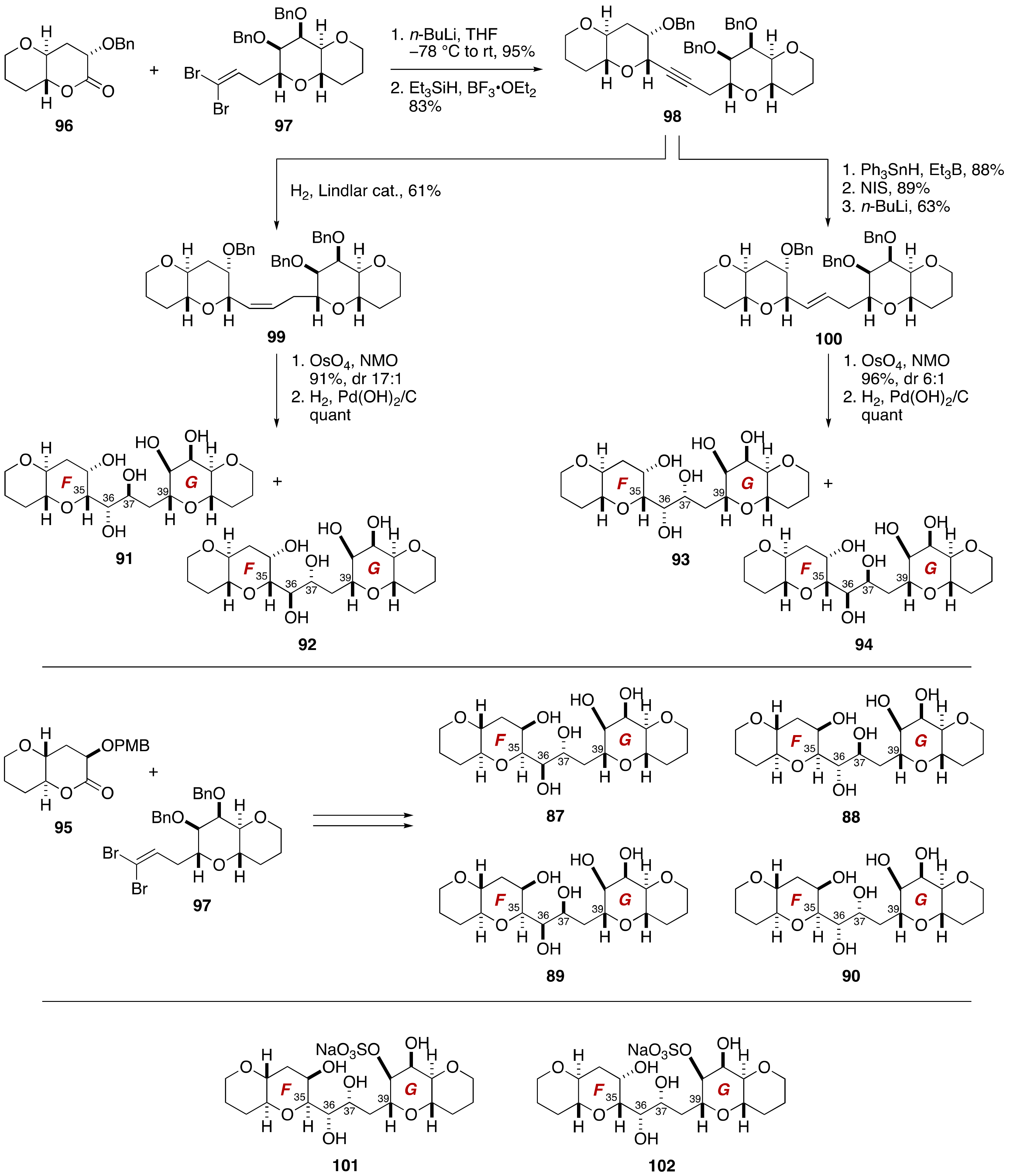
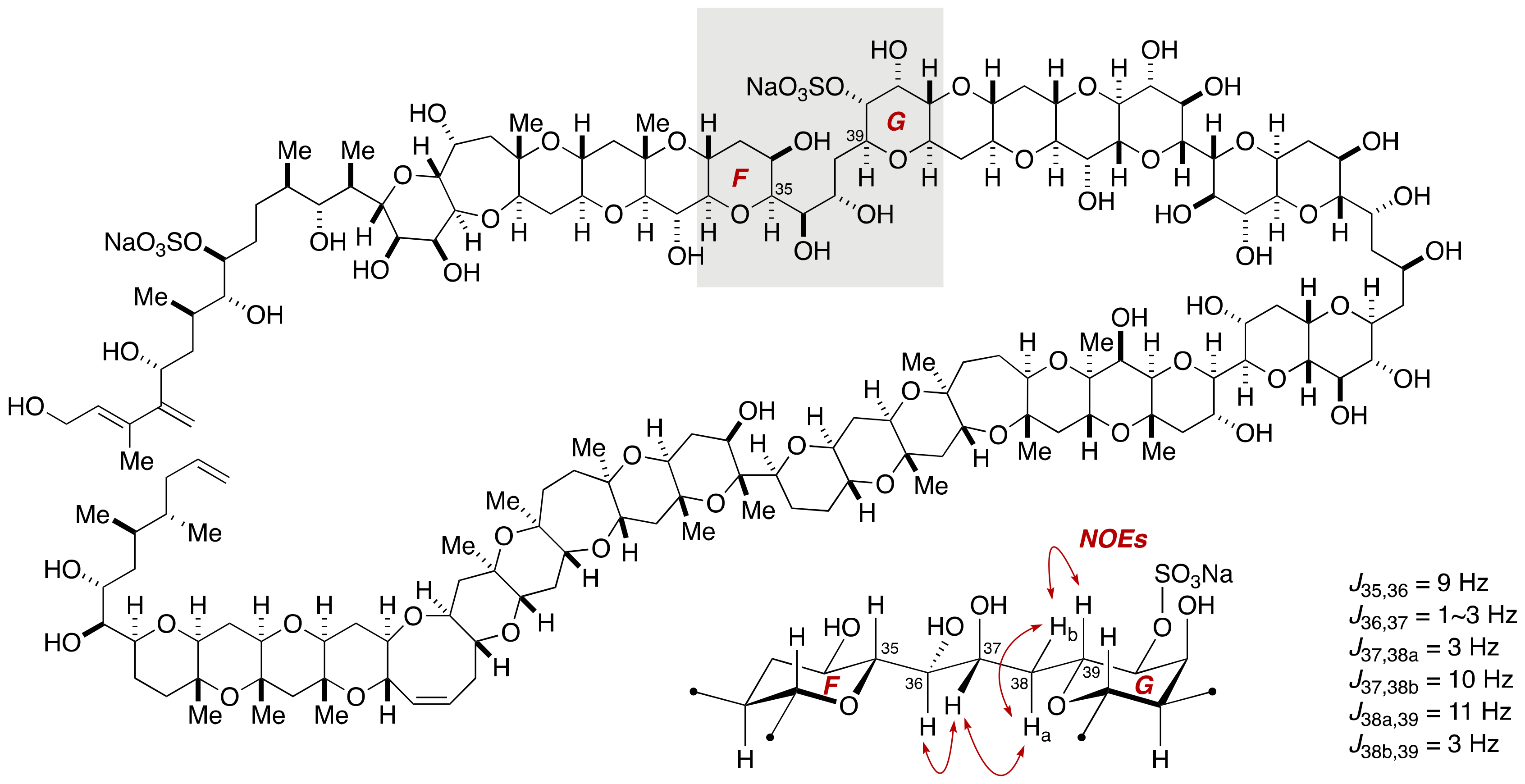
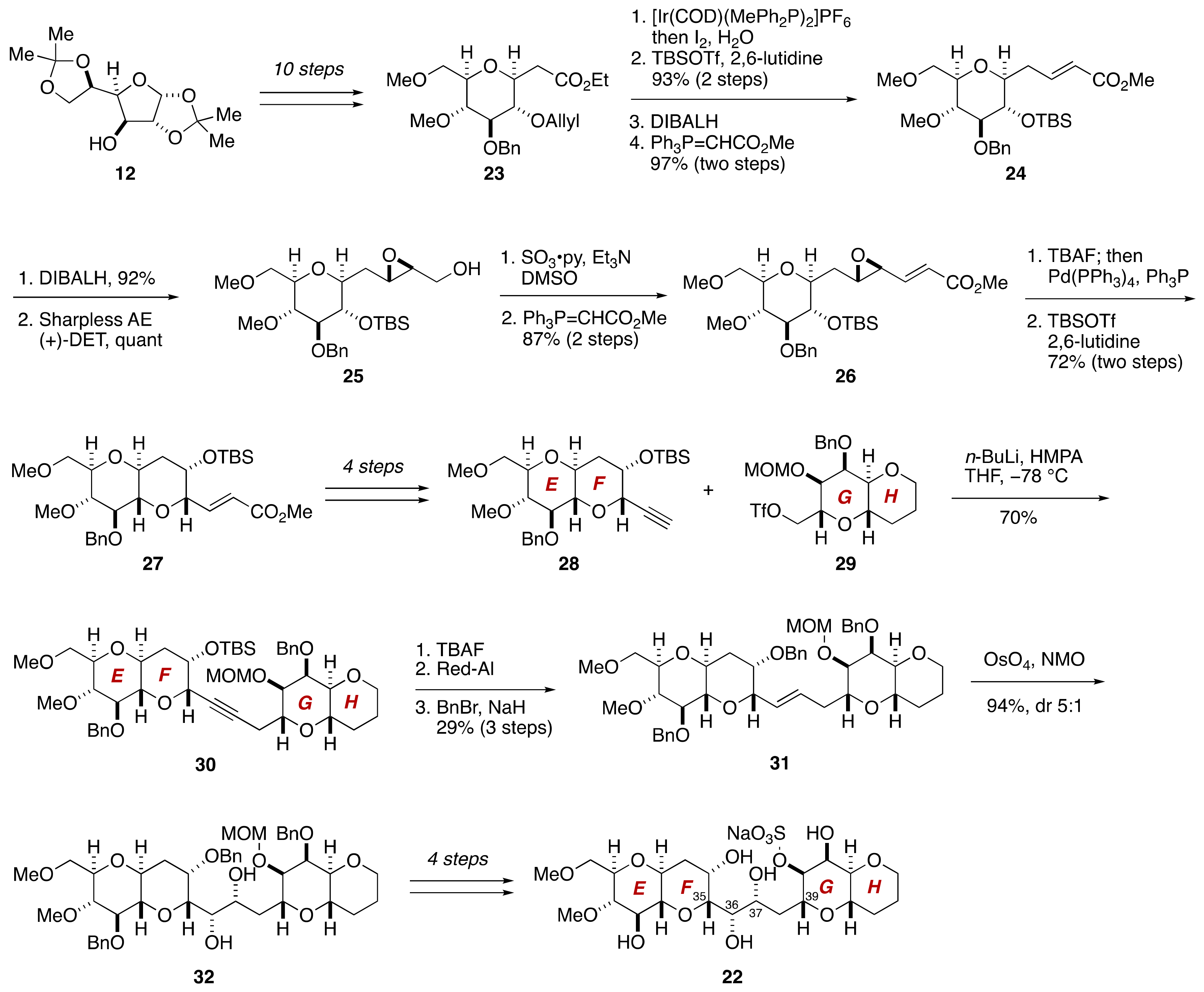
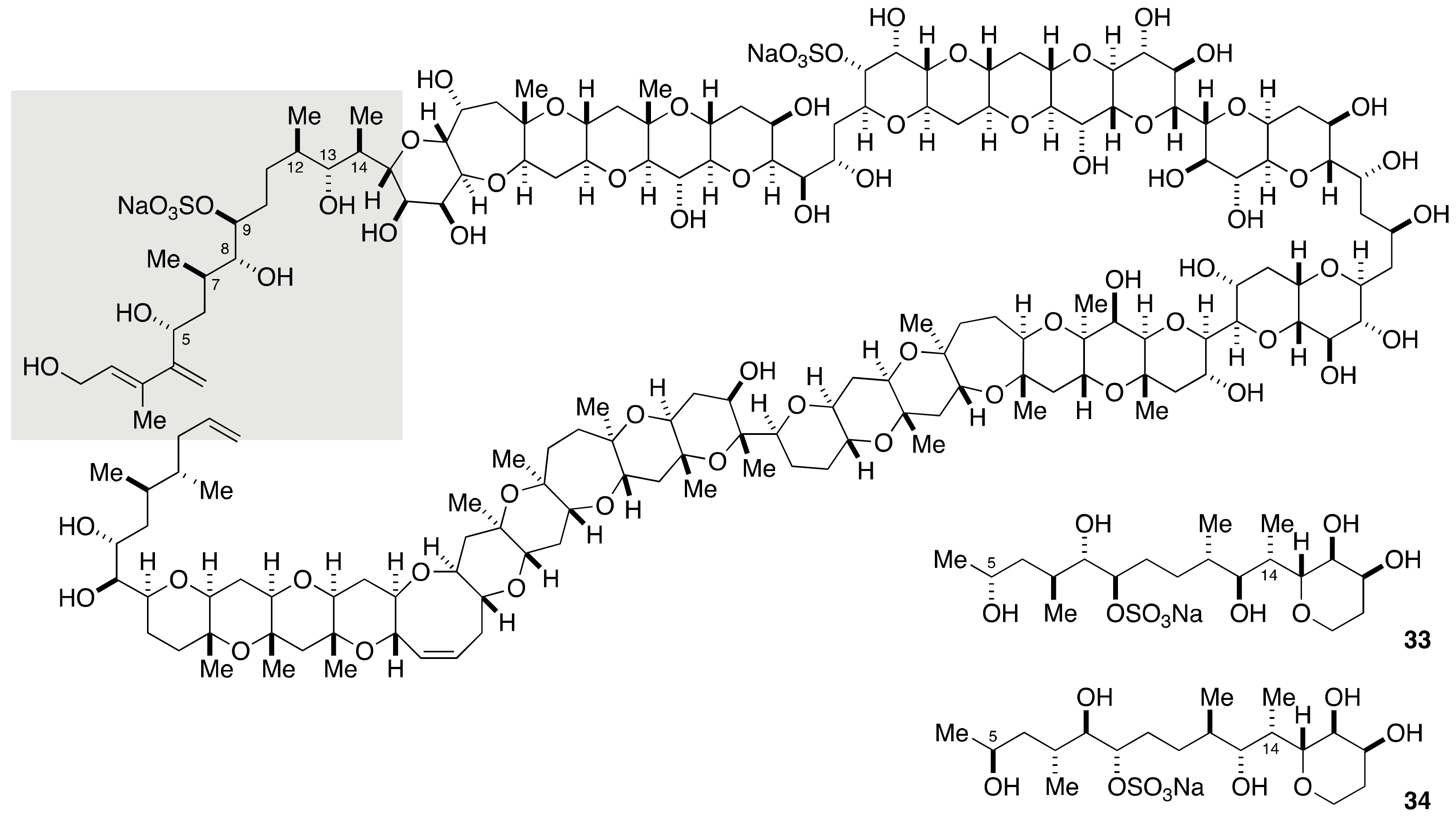
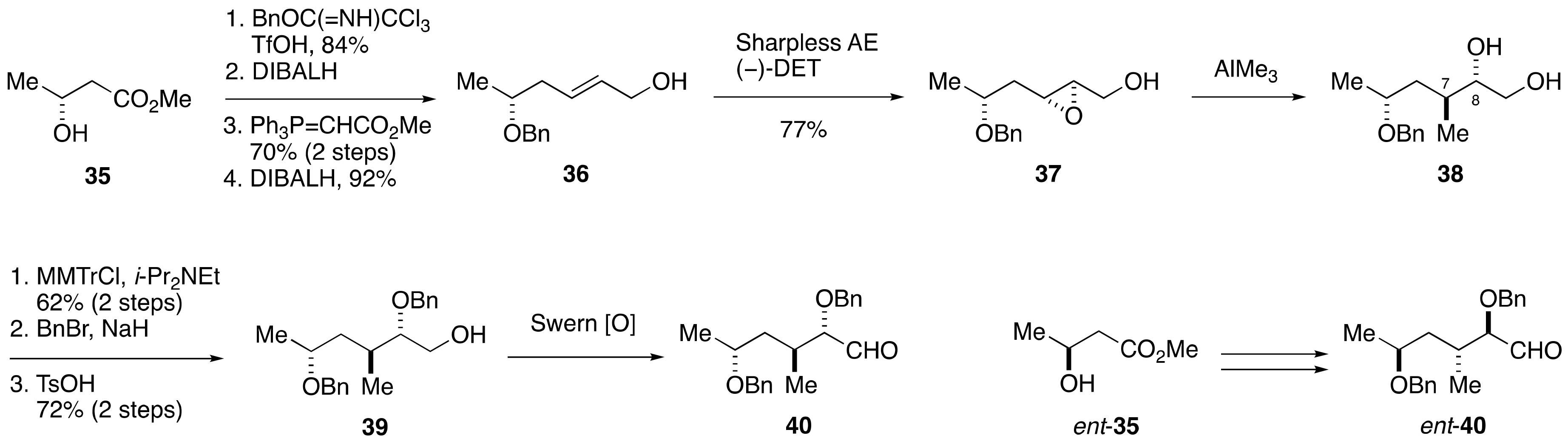
2.1.2. Stereochemical Assignment of the C35–C39 Acyclic Linkage
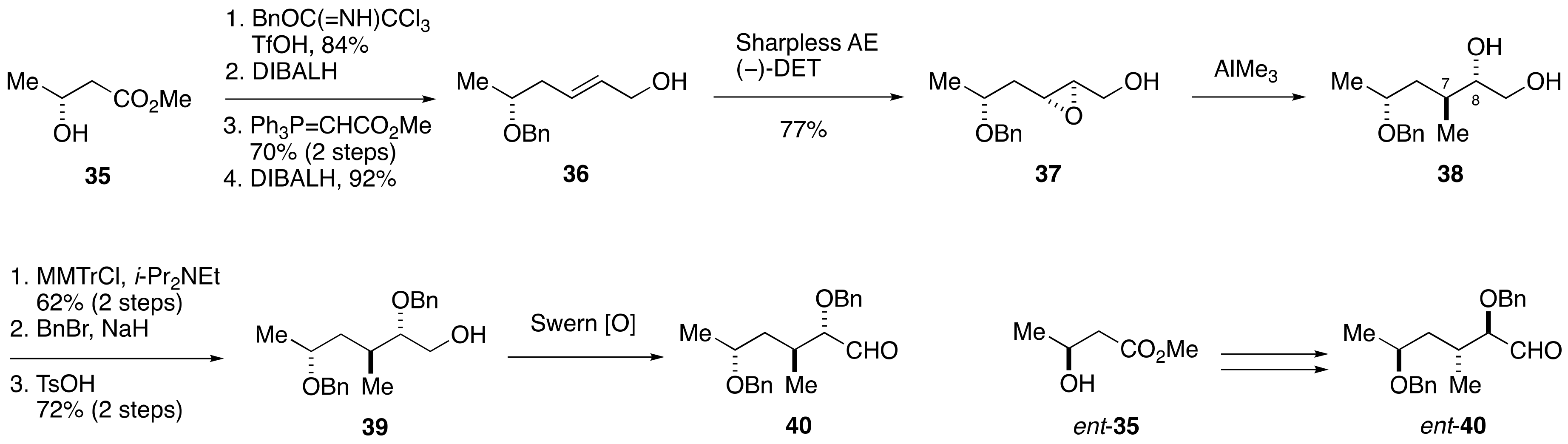
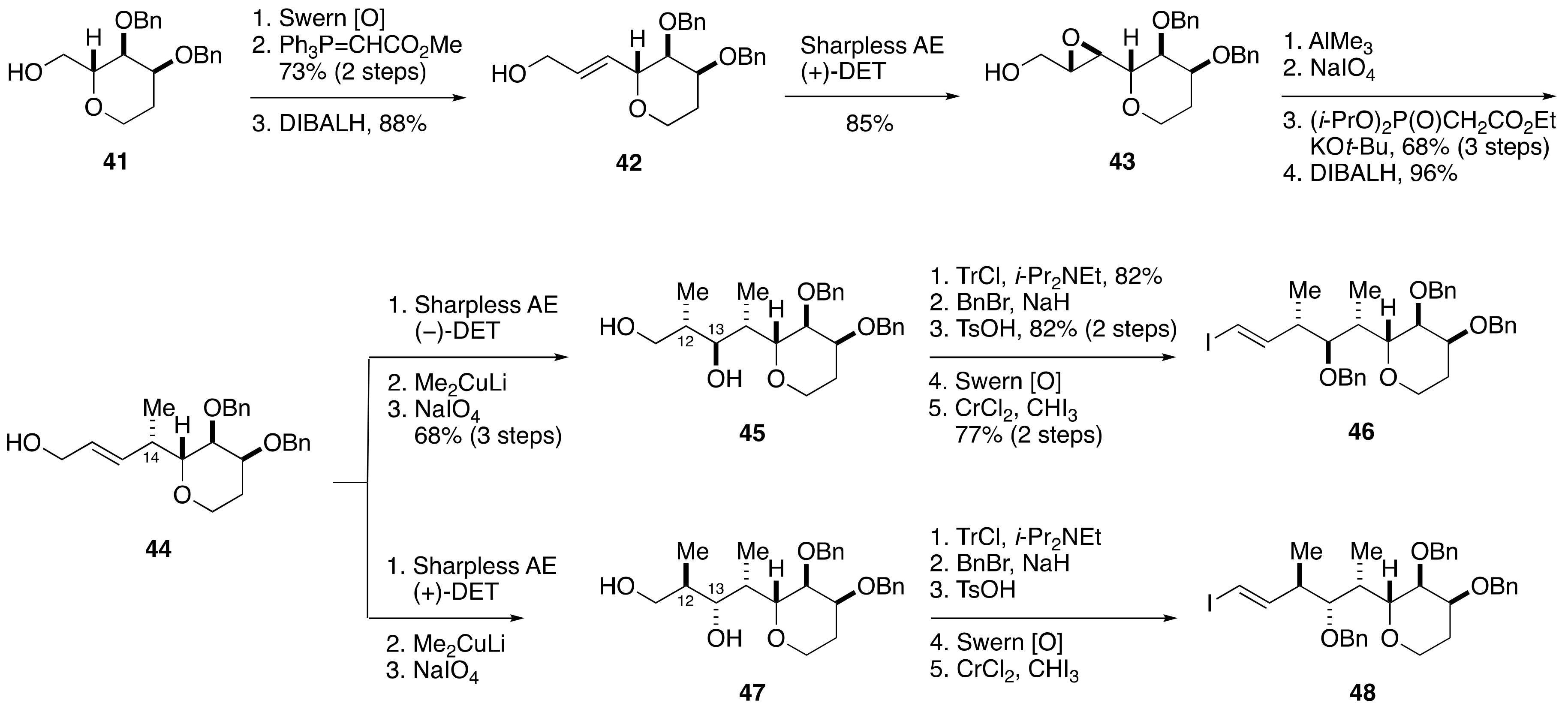
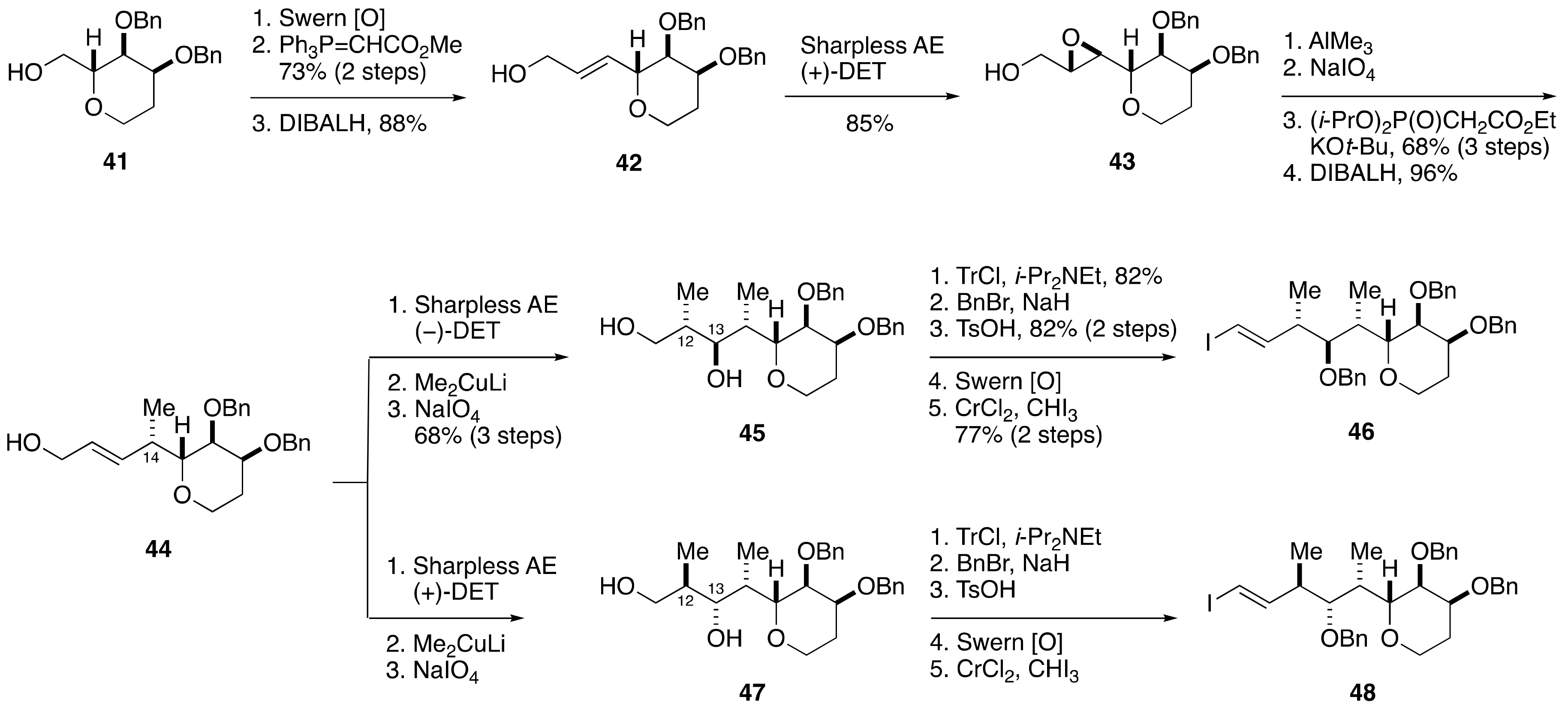
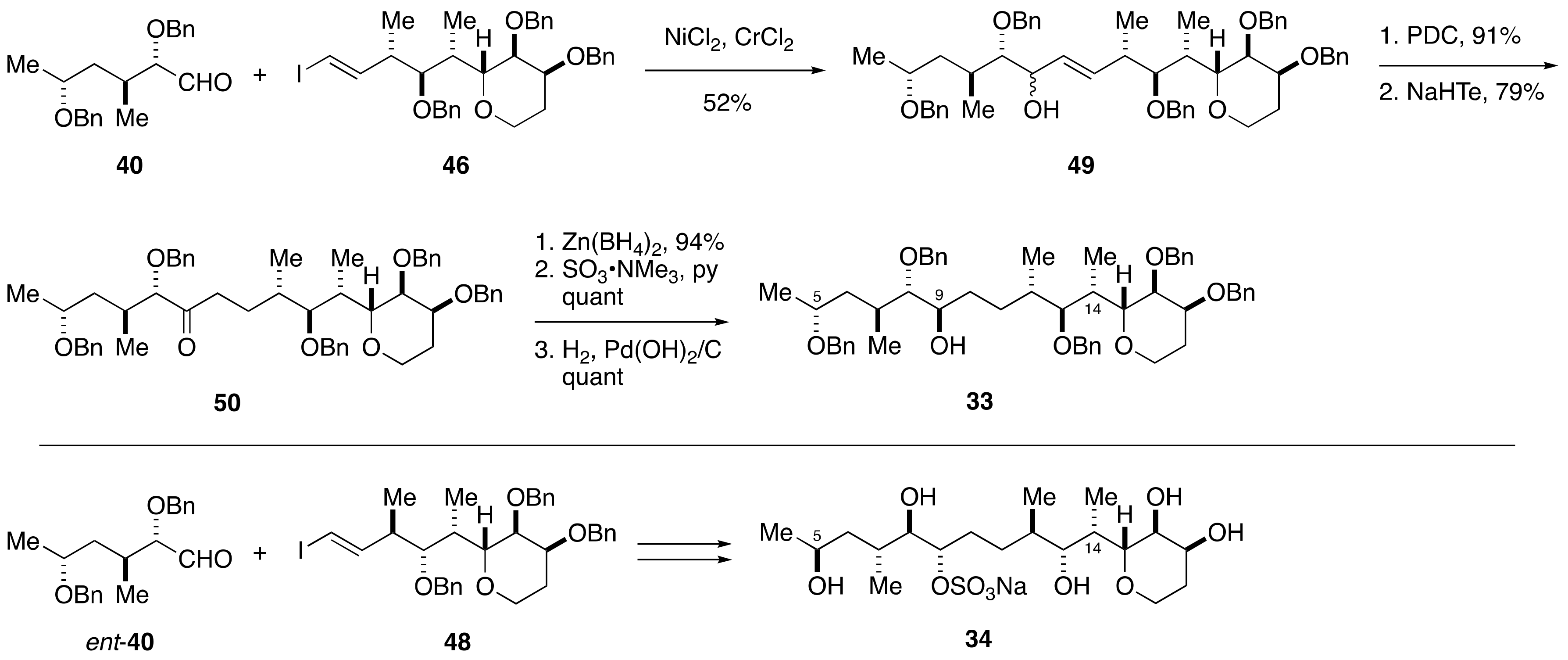
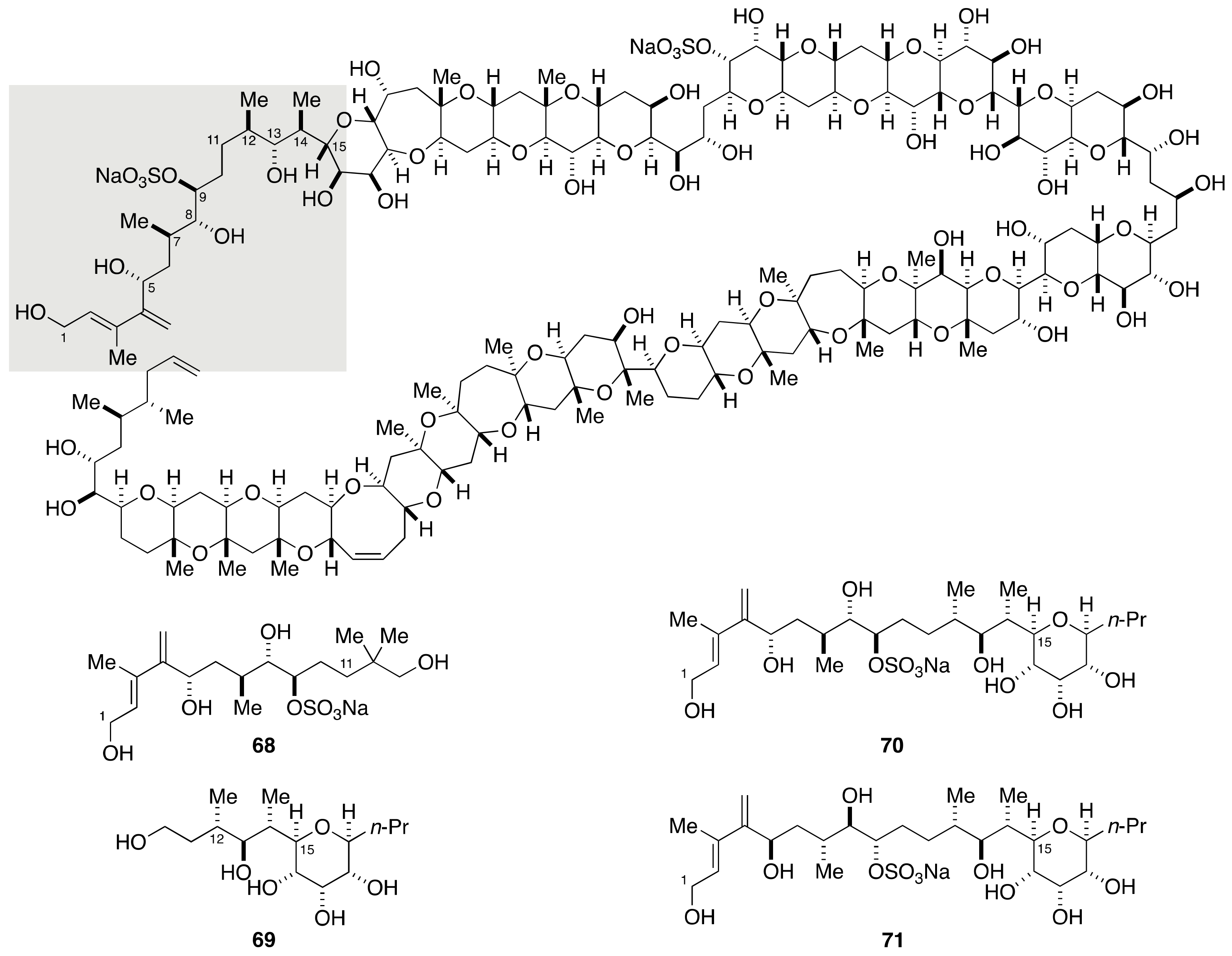
2.1.3. Stereochemical Assignment of the C1–C14 Side Chain
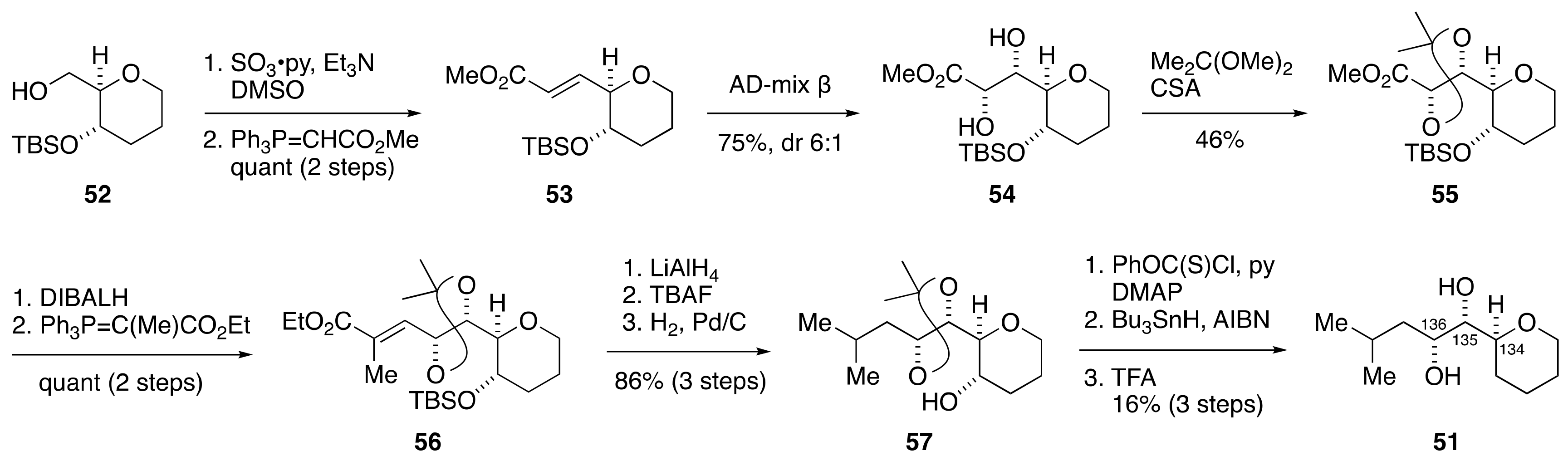
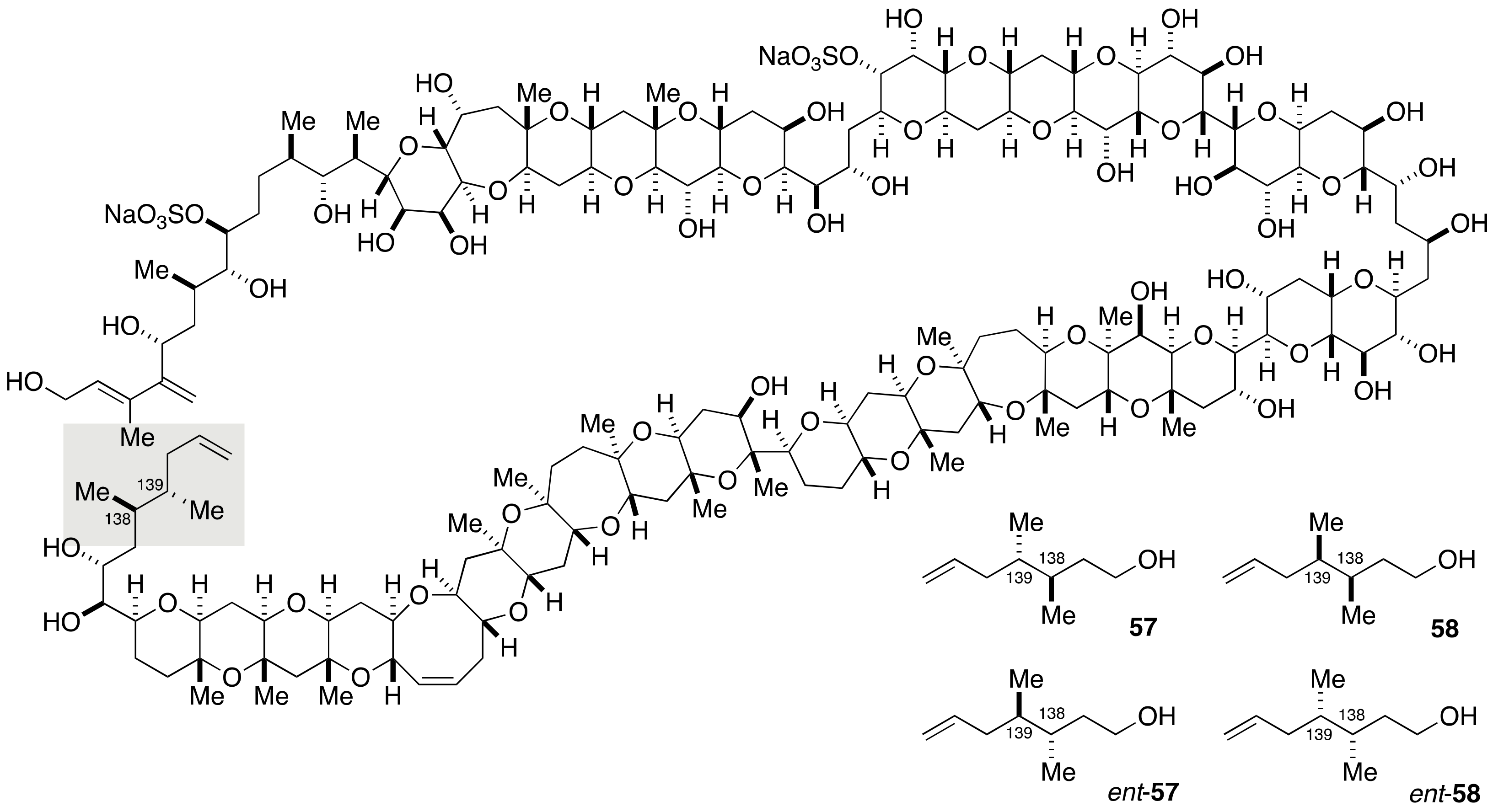
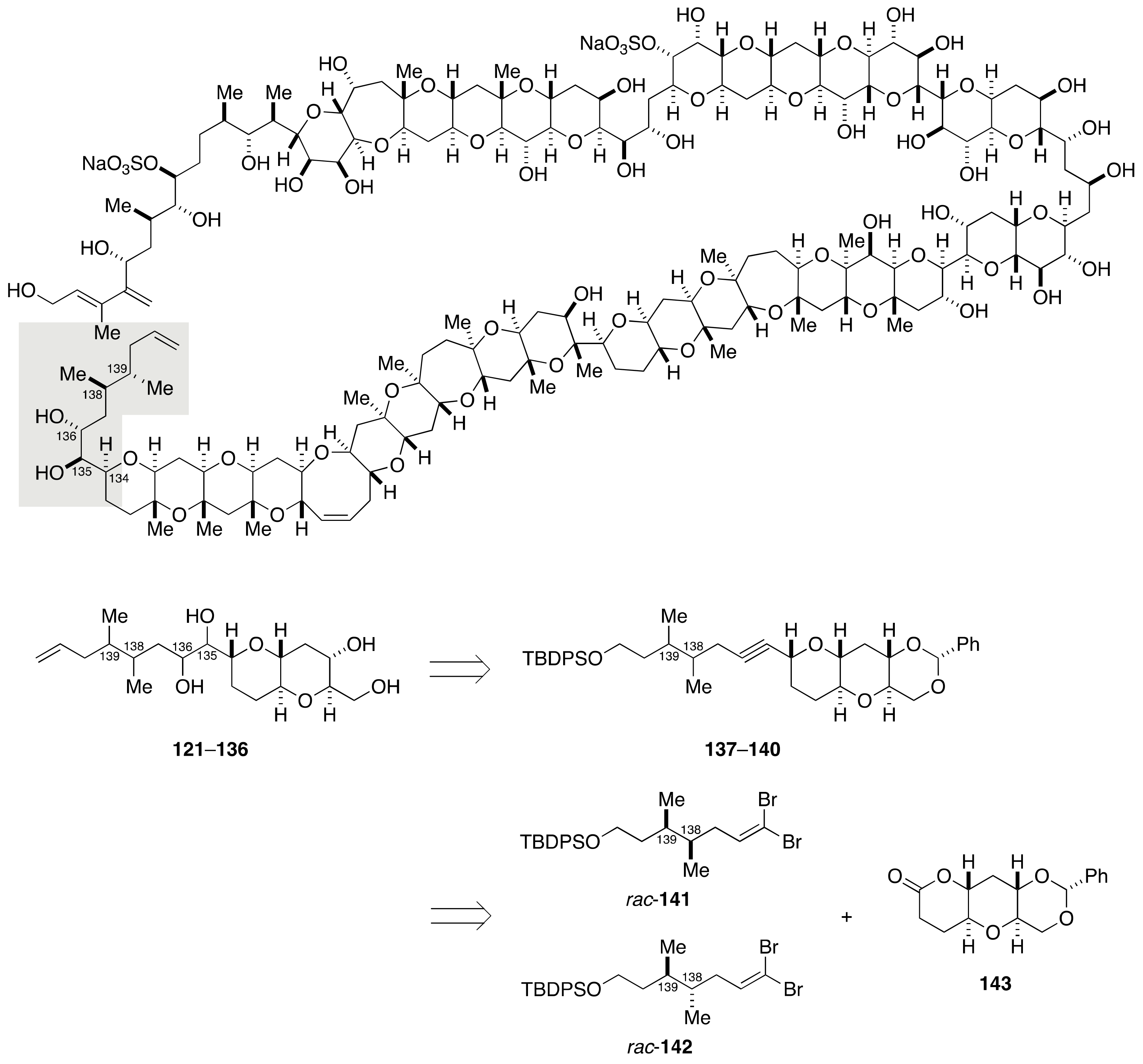
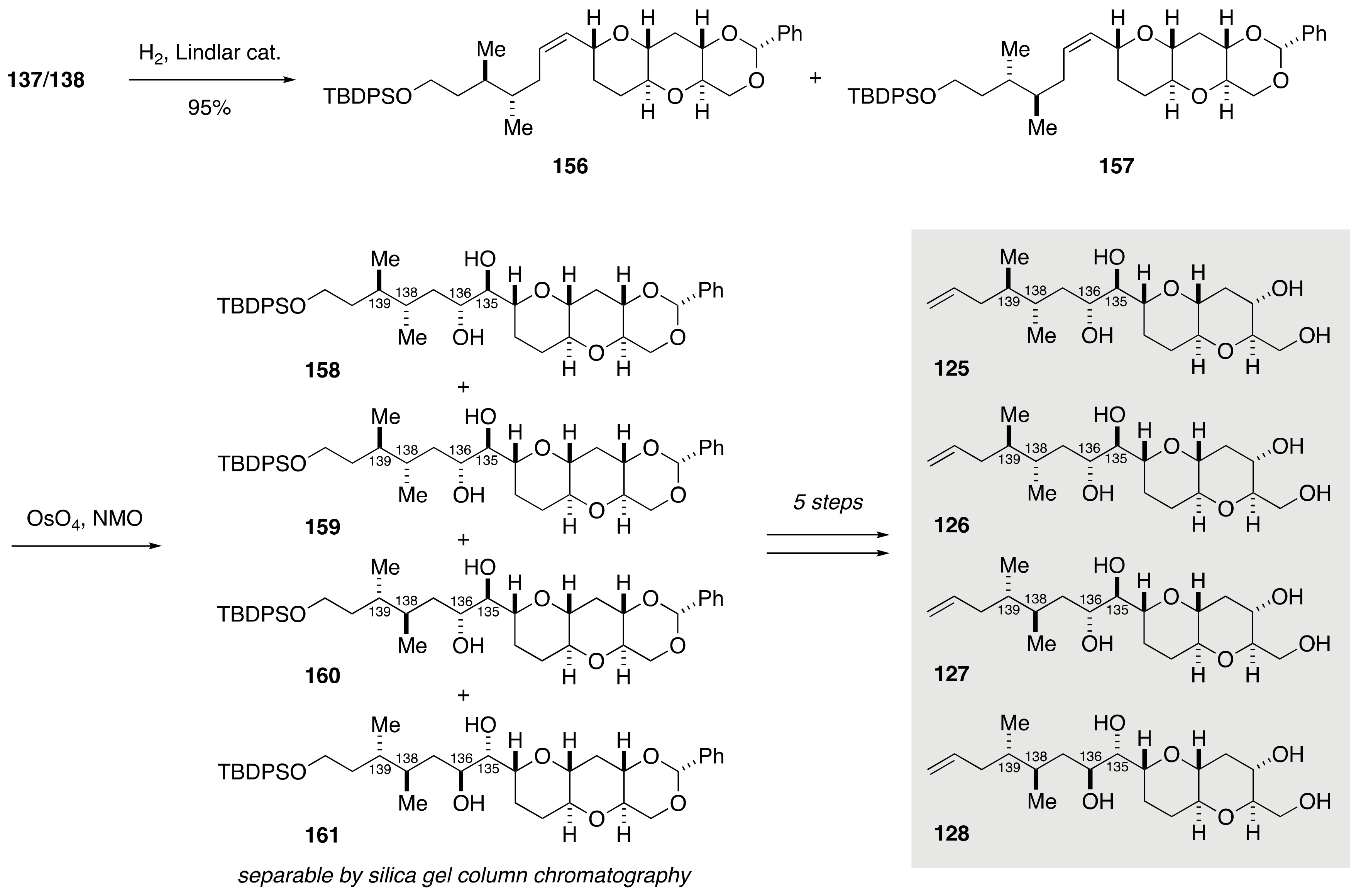
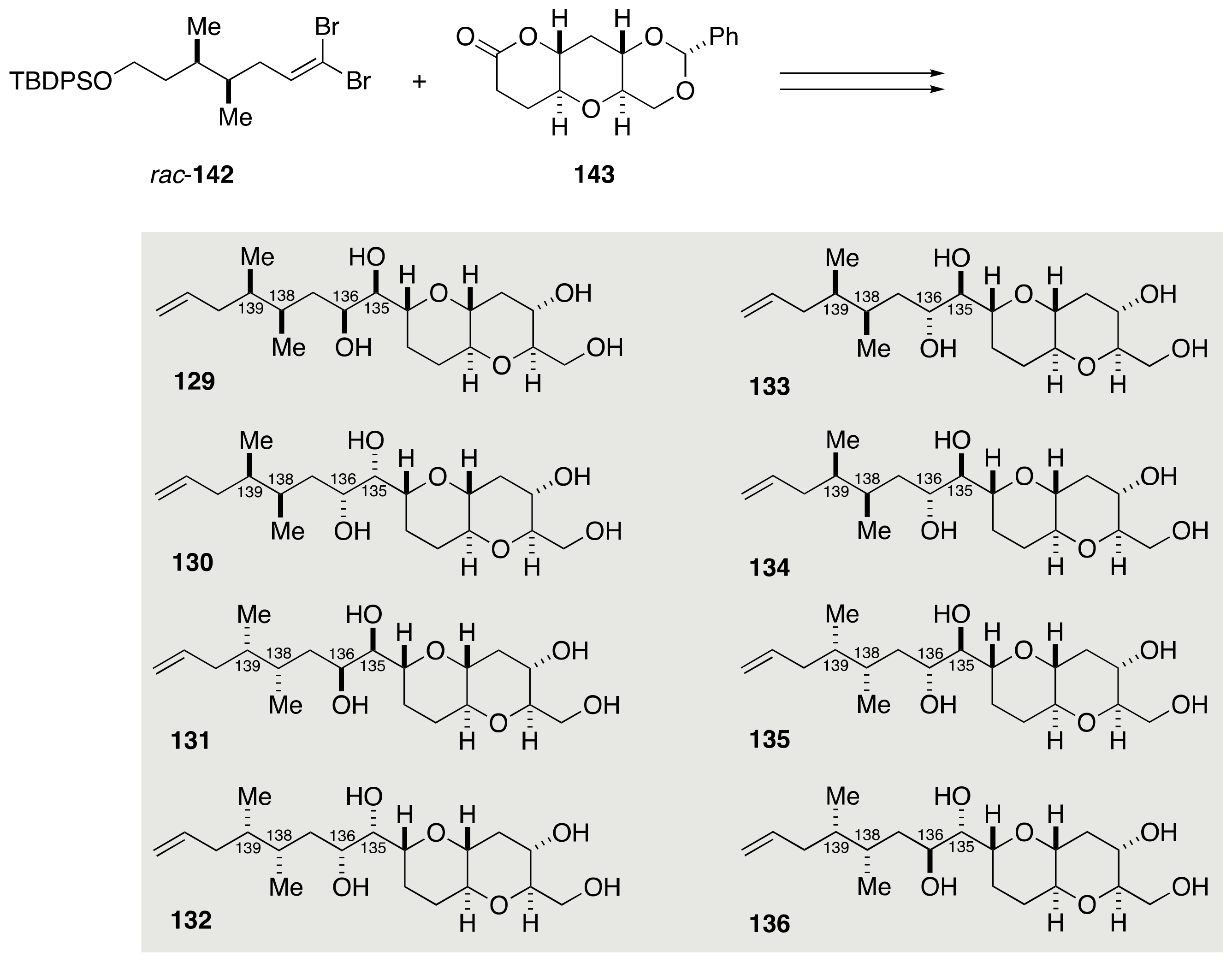
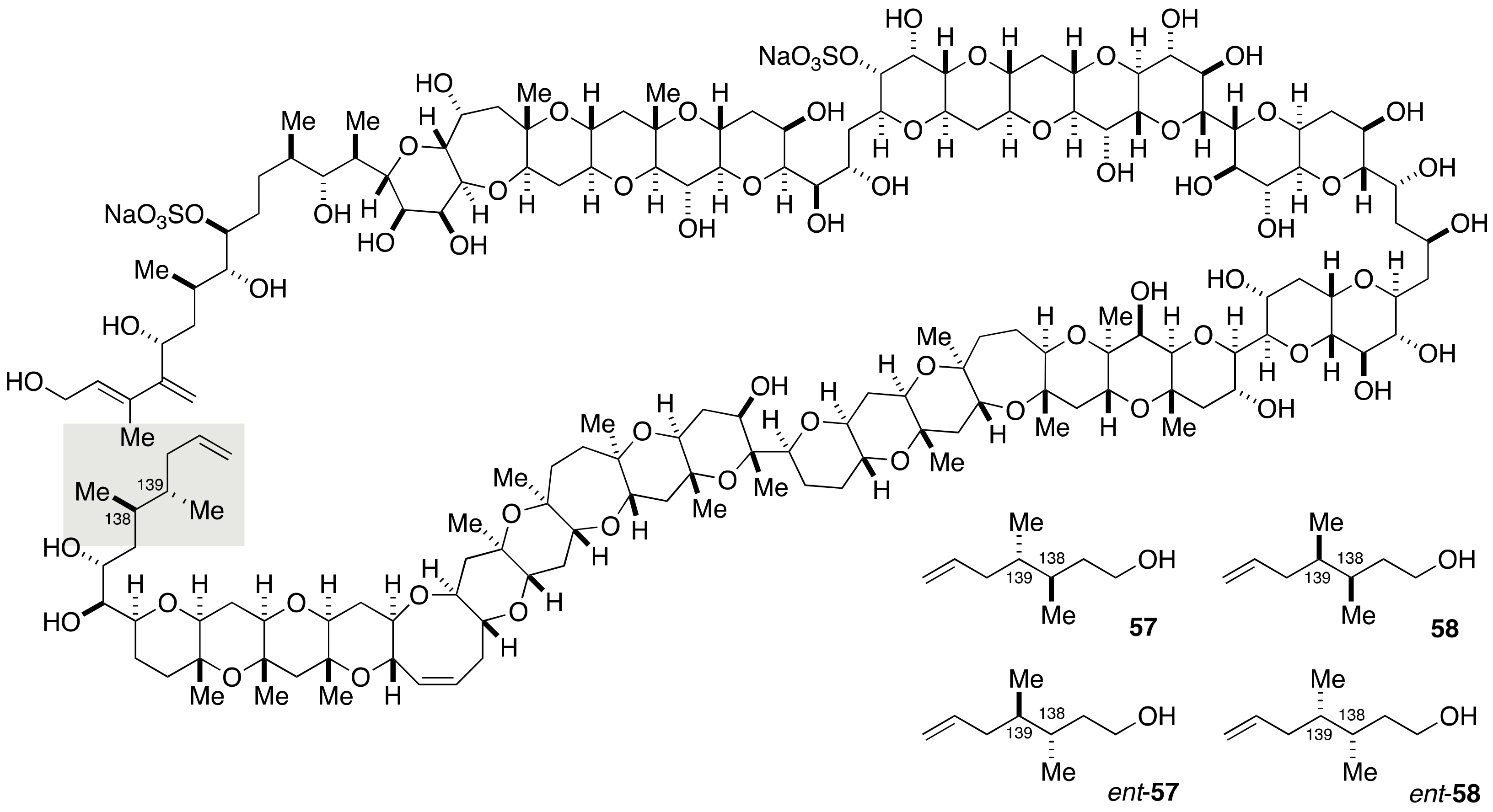
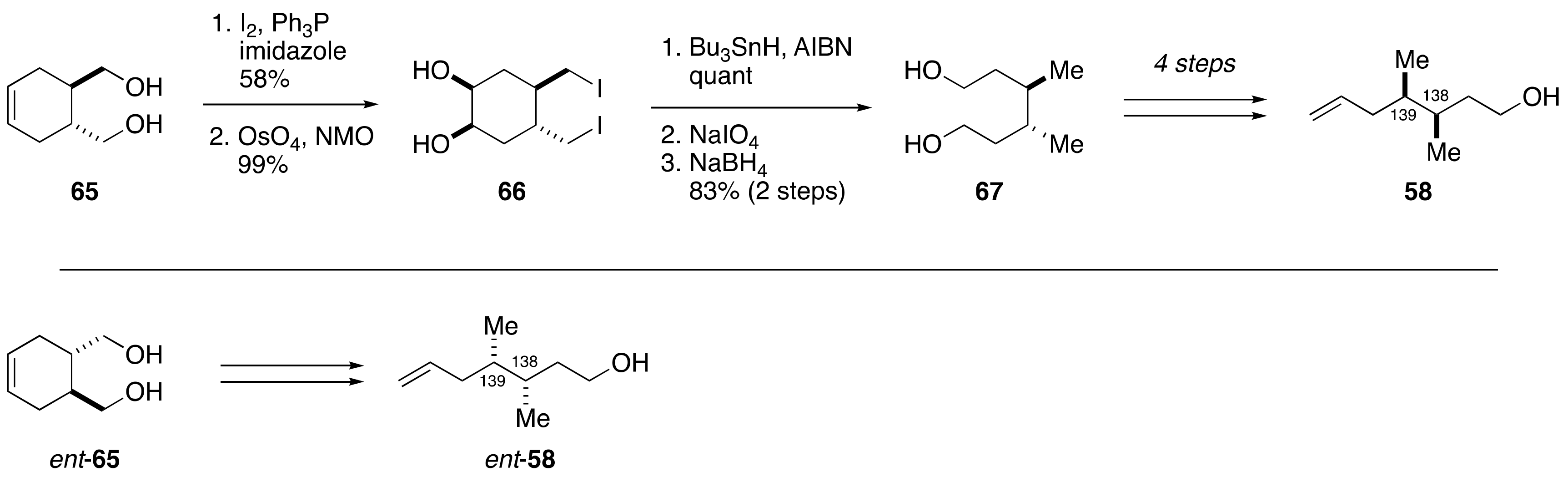
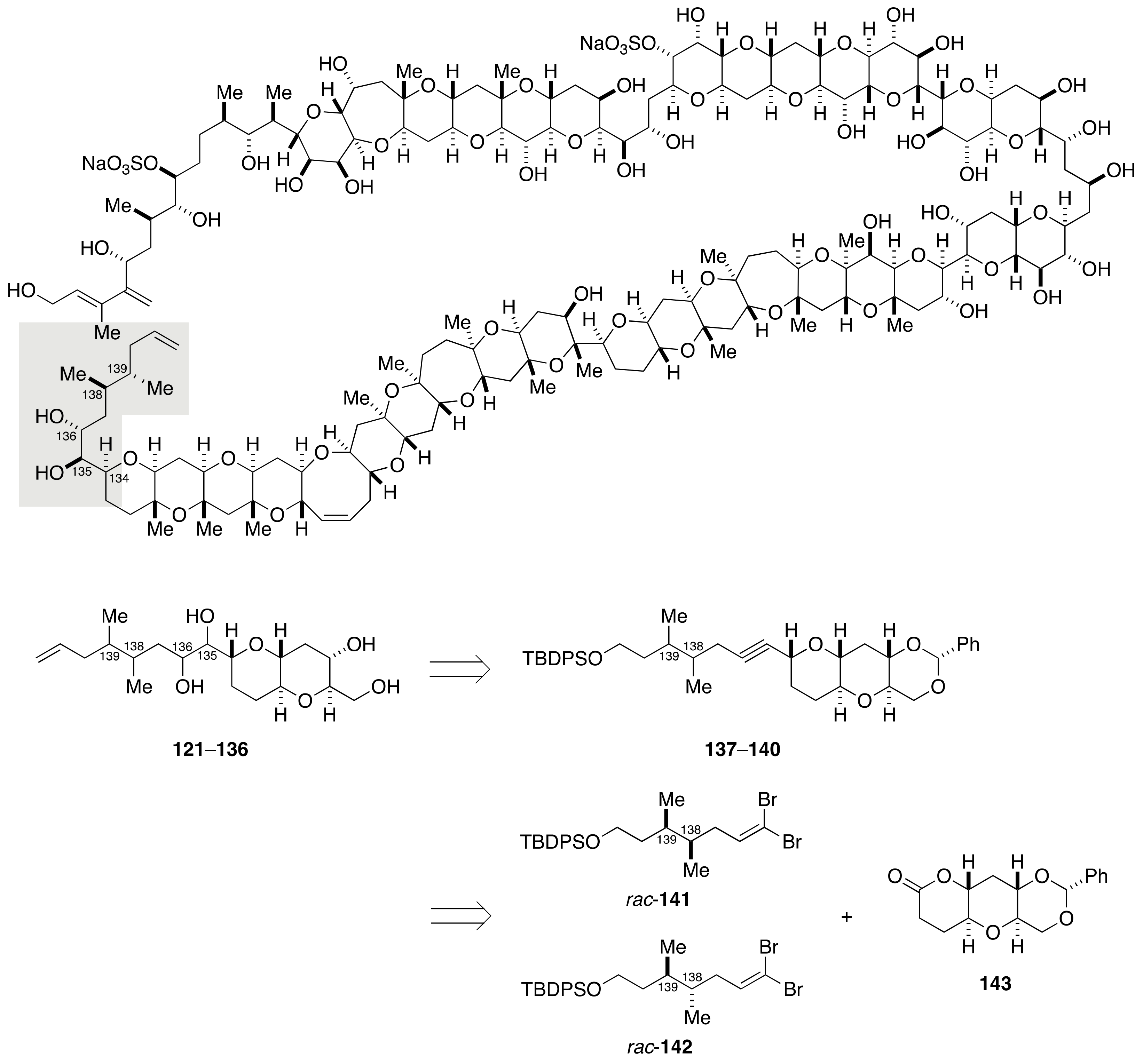
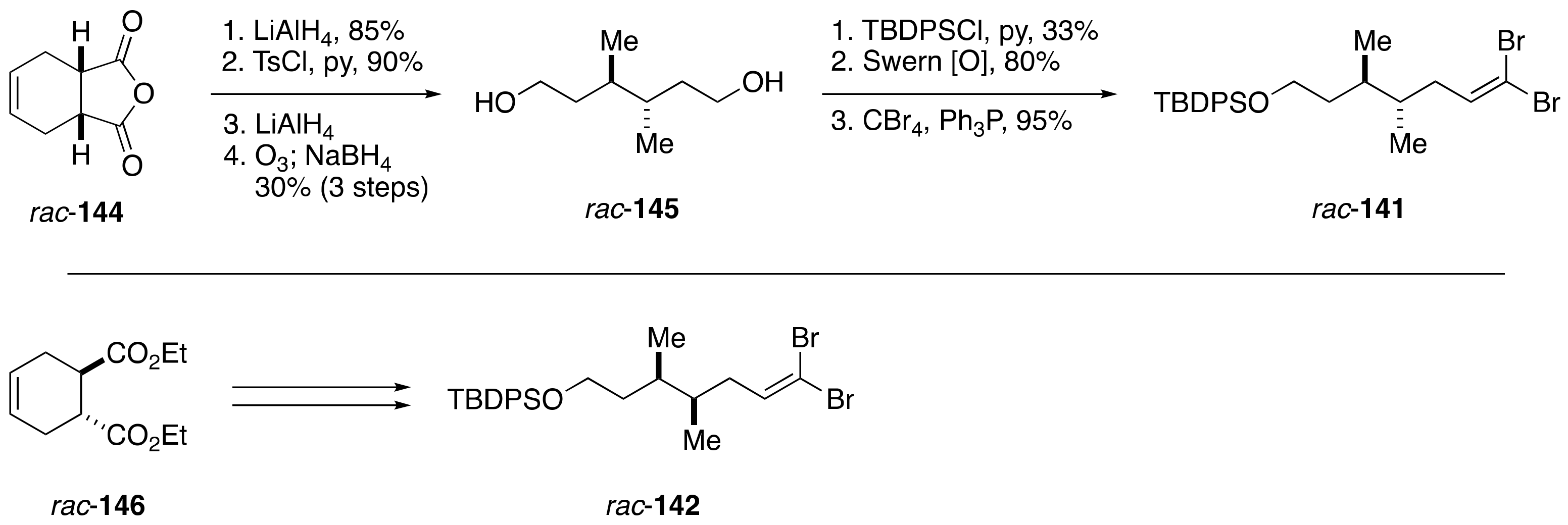
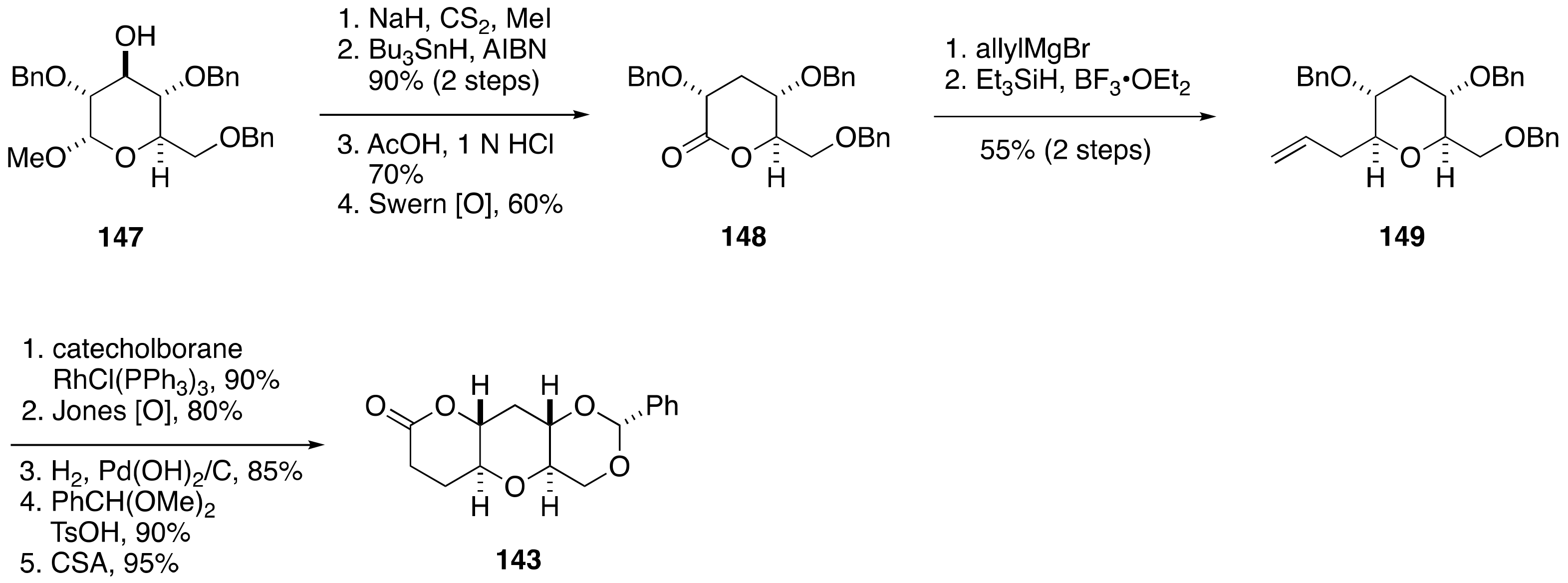
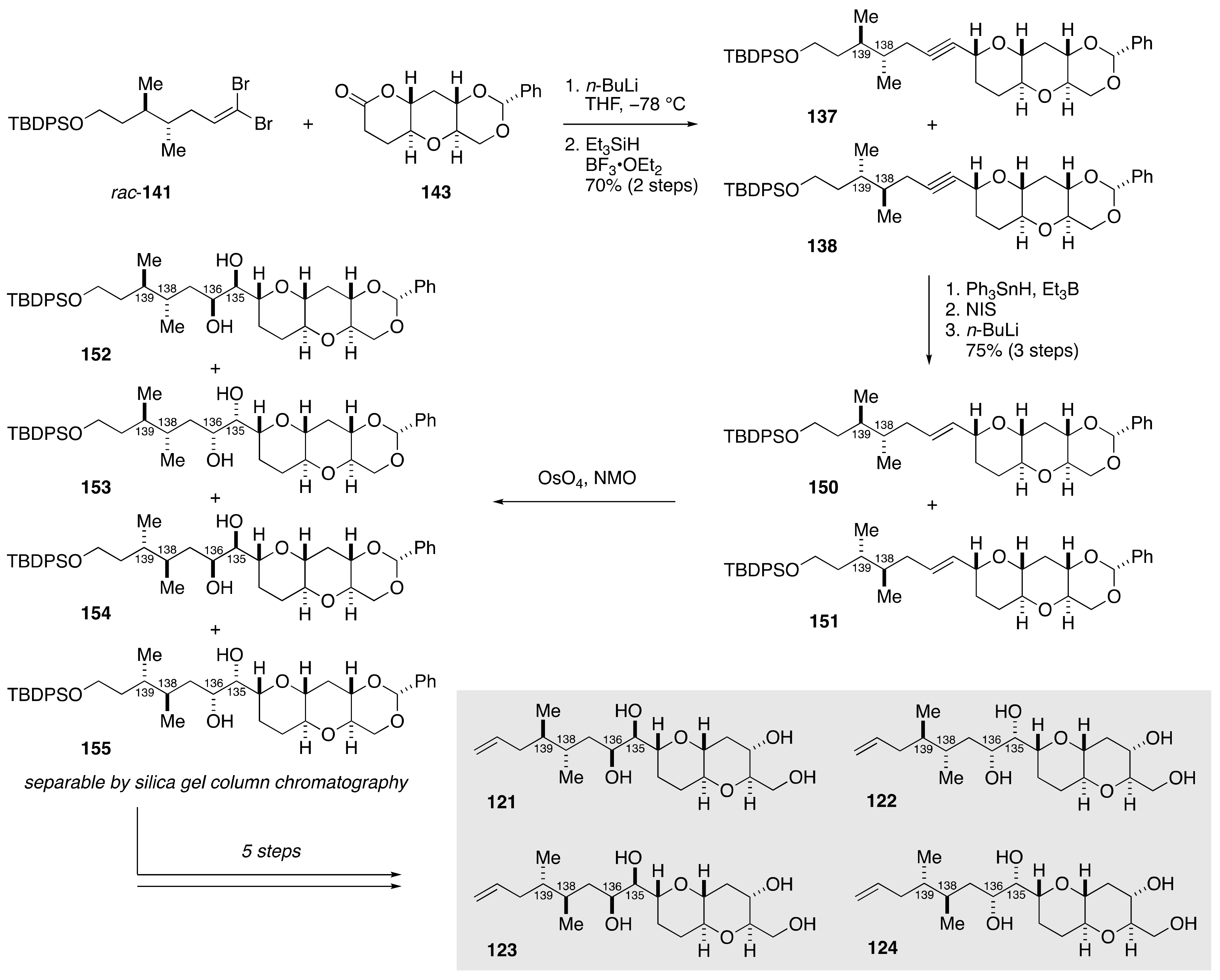
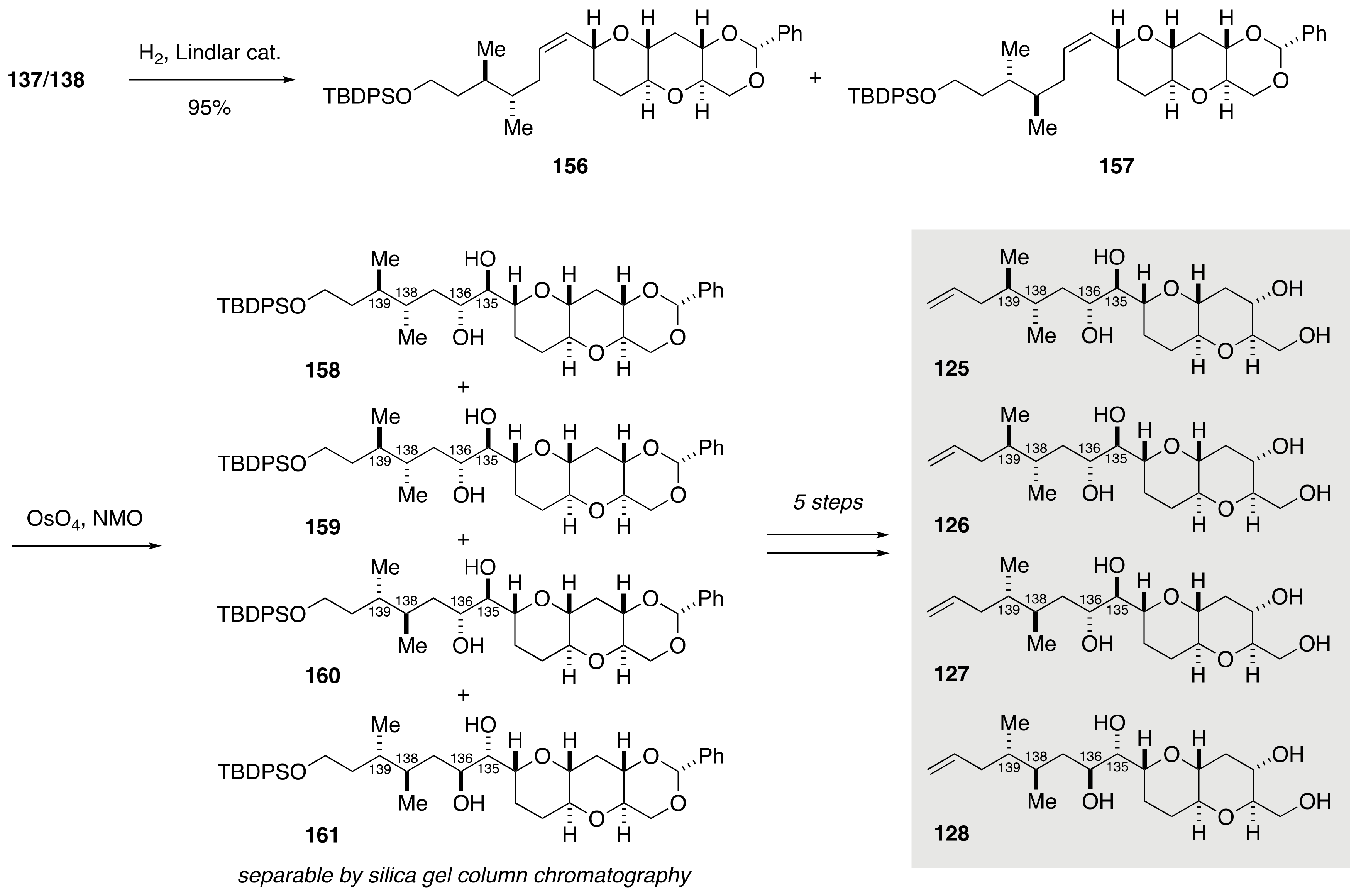
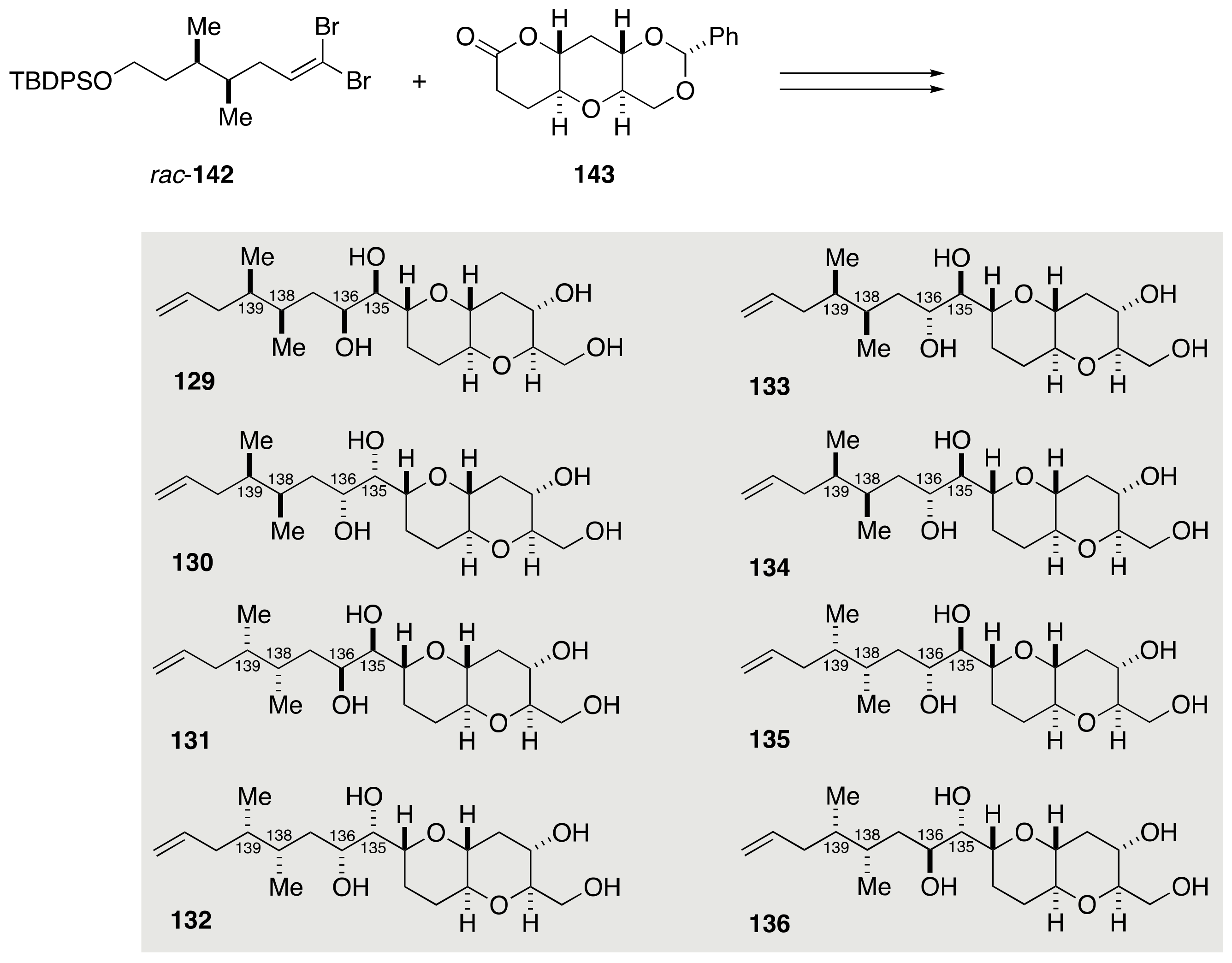
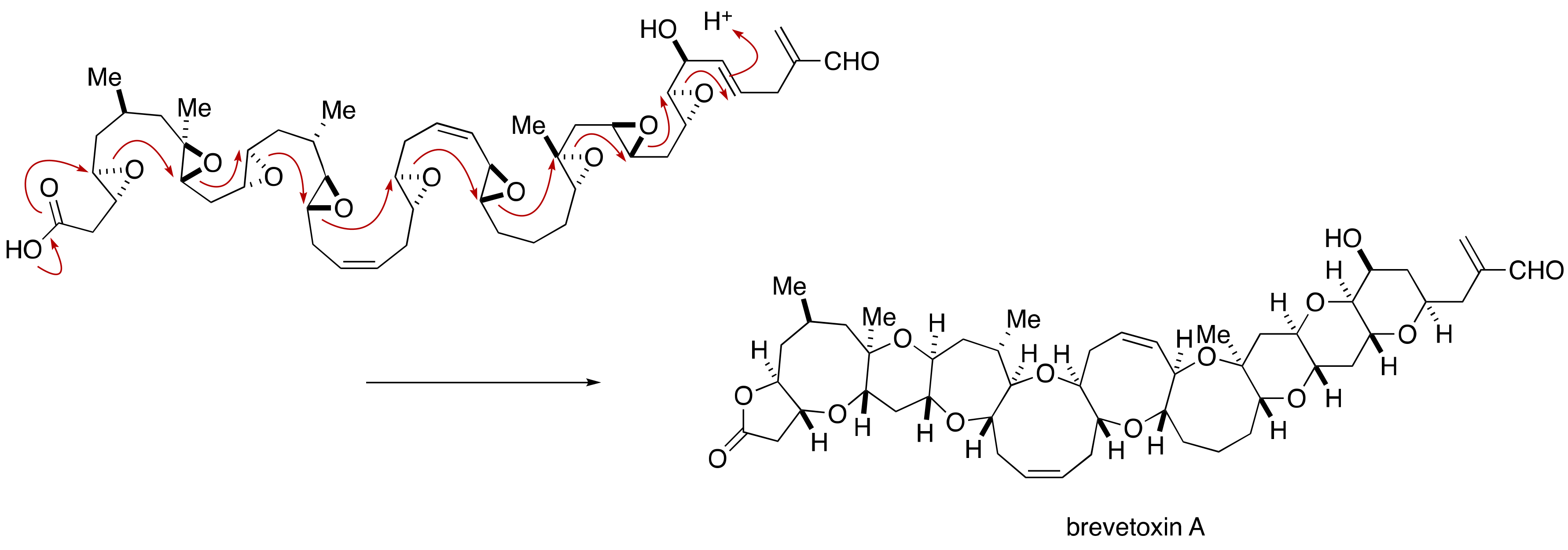
2.1.4. Stereochemical Assignment of the C135–C142 Side Chain and Absolute Configuration of Maitotoxin
2.2. Determination of the Relative Configuration of Maitotoxin by the Kishi Group
2.2.1. Stereochemical Assignment of the C1–C15 Side Chain
2.2.2. Stereochemical Assignment of the C35–C39 Acyclic Portion
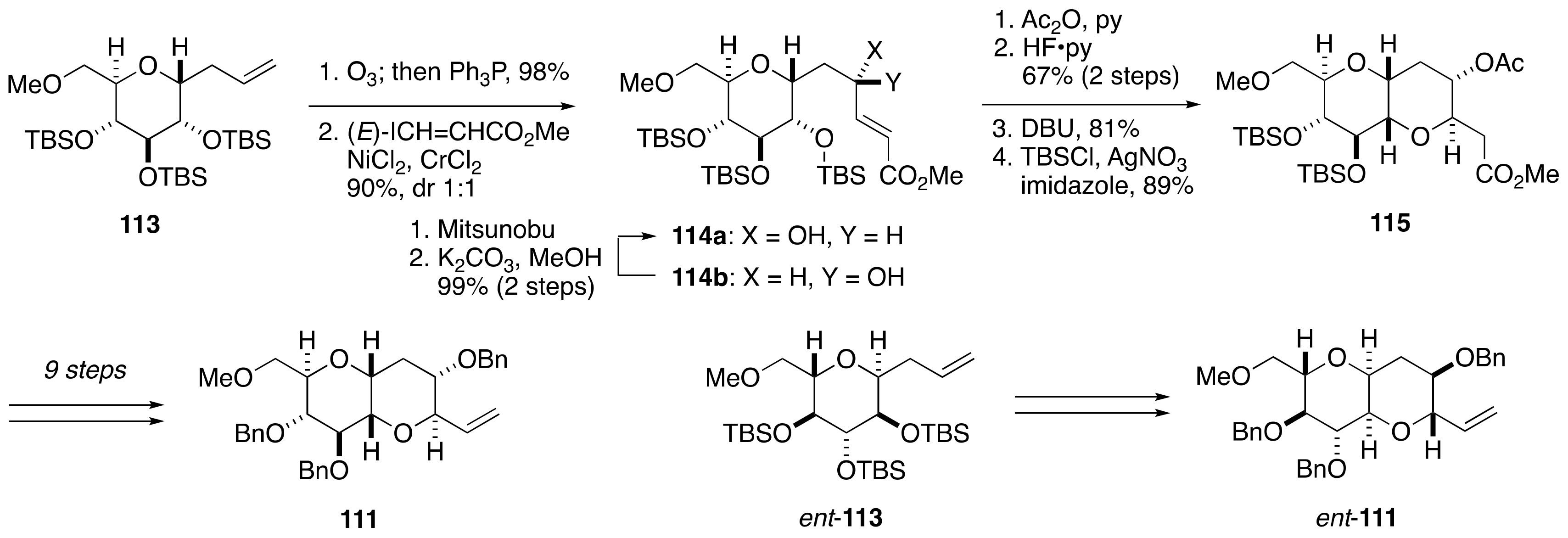
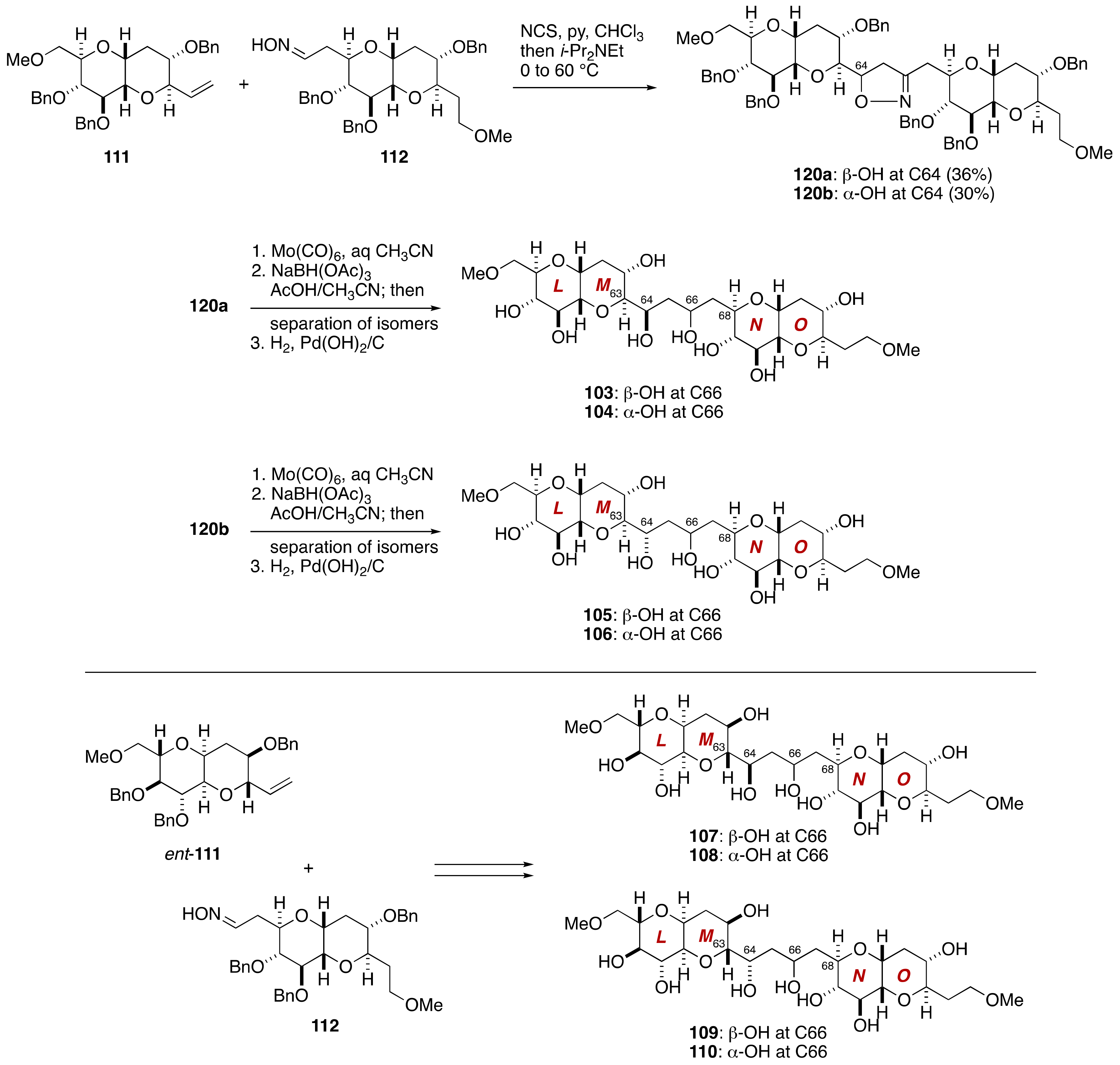
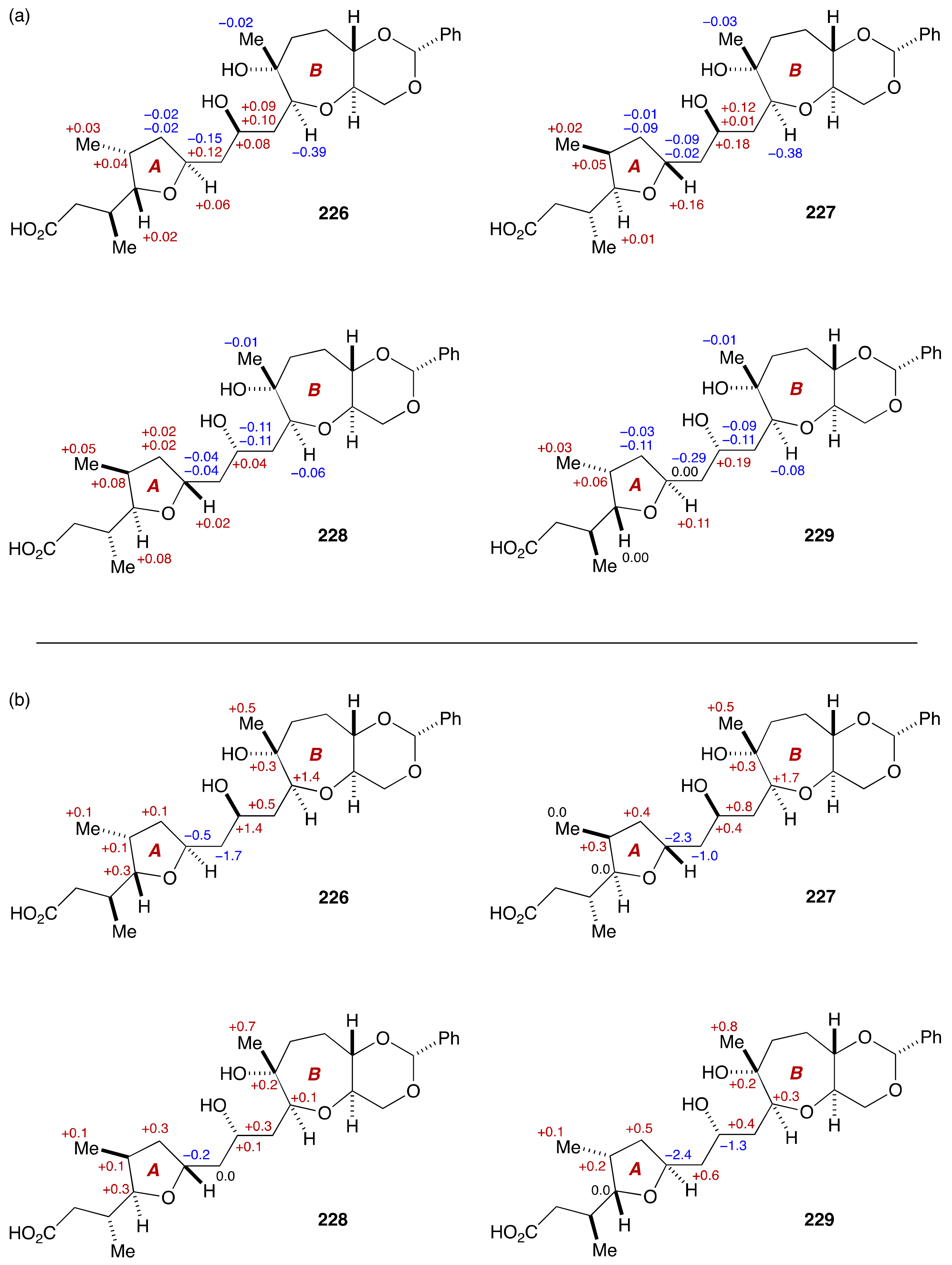
2.2.3. Stereochemical Assignment of the C63–C68 Acyclic Portion
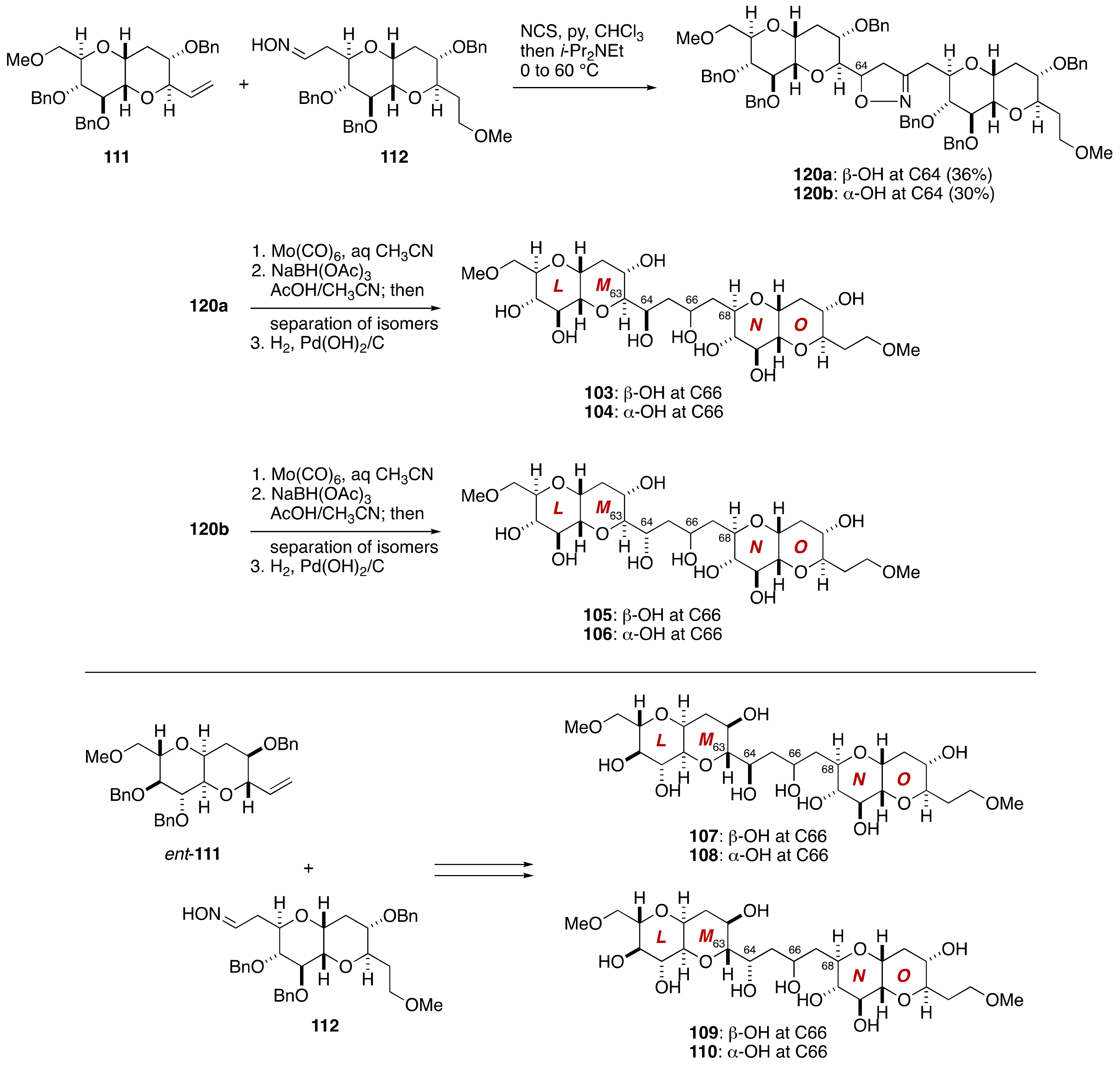
2.2.4. Stereochemical Assignment of the C134–C142 Side Chain
2.3. Proposal of the Alternative Configuration of the JK-Ring Juncture by Gallimore and Spencer, and Its Disproof by Confirmation of the Originally Assigned Configuration of the JK-Ring Juncture by the Nicolaou Group
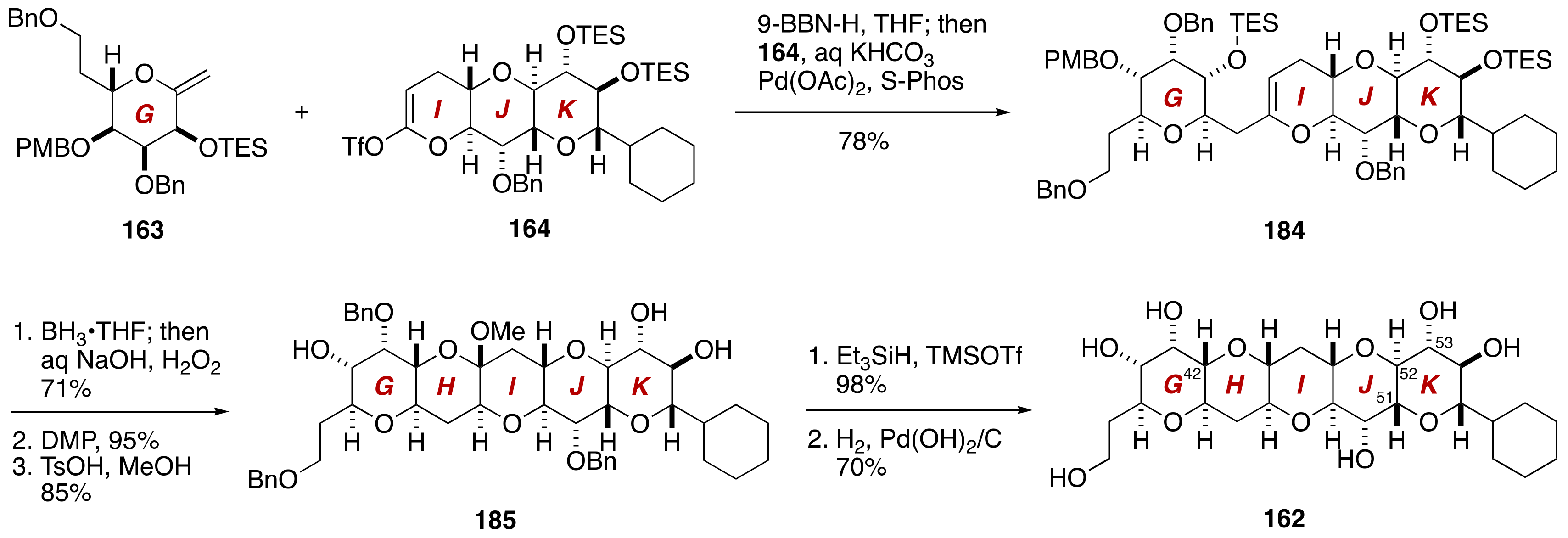
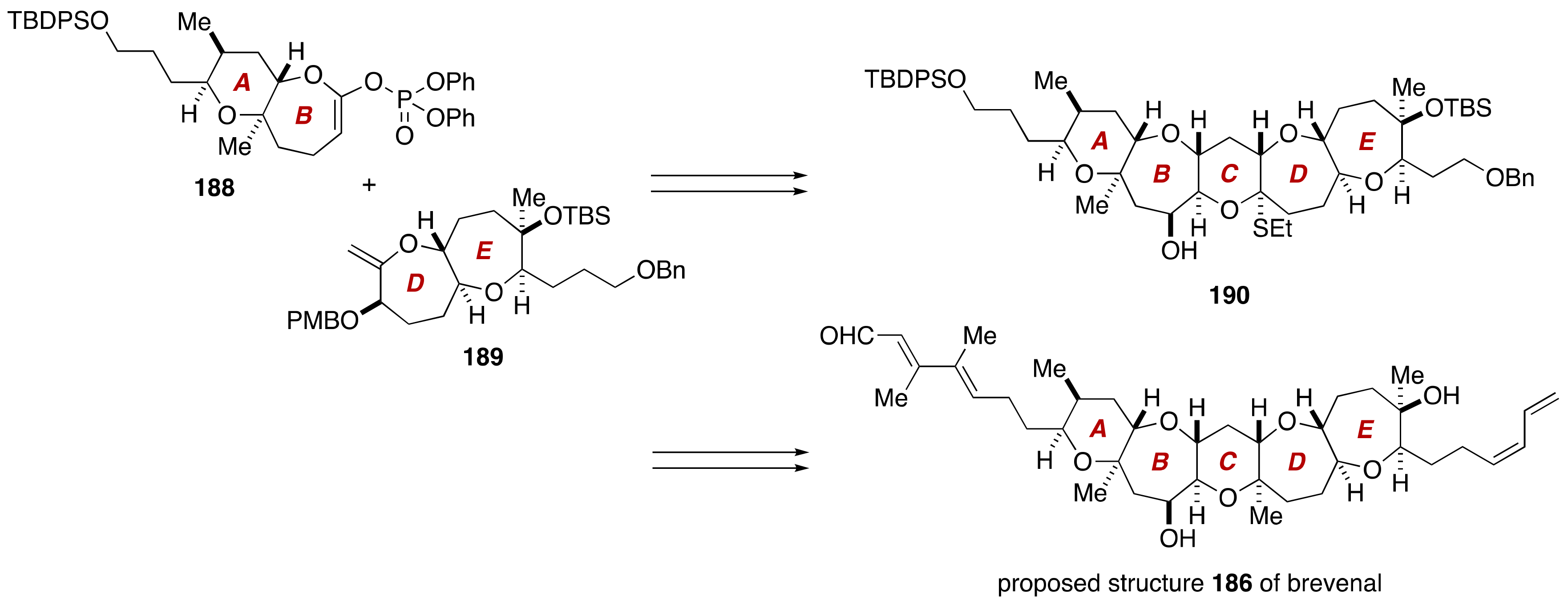
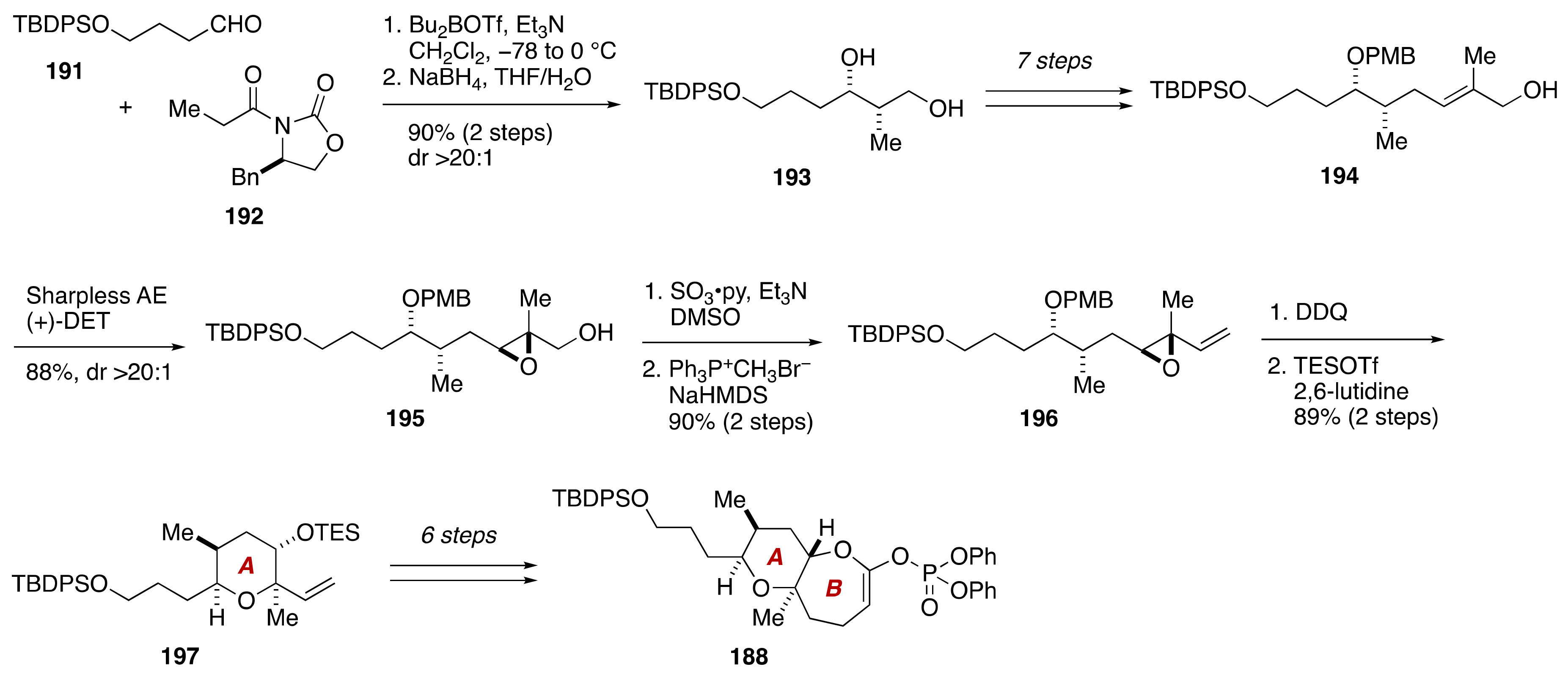
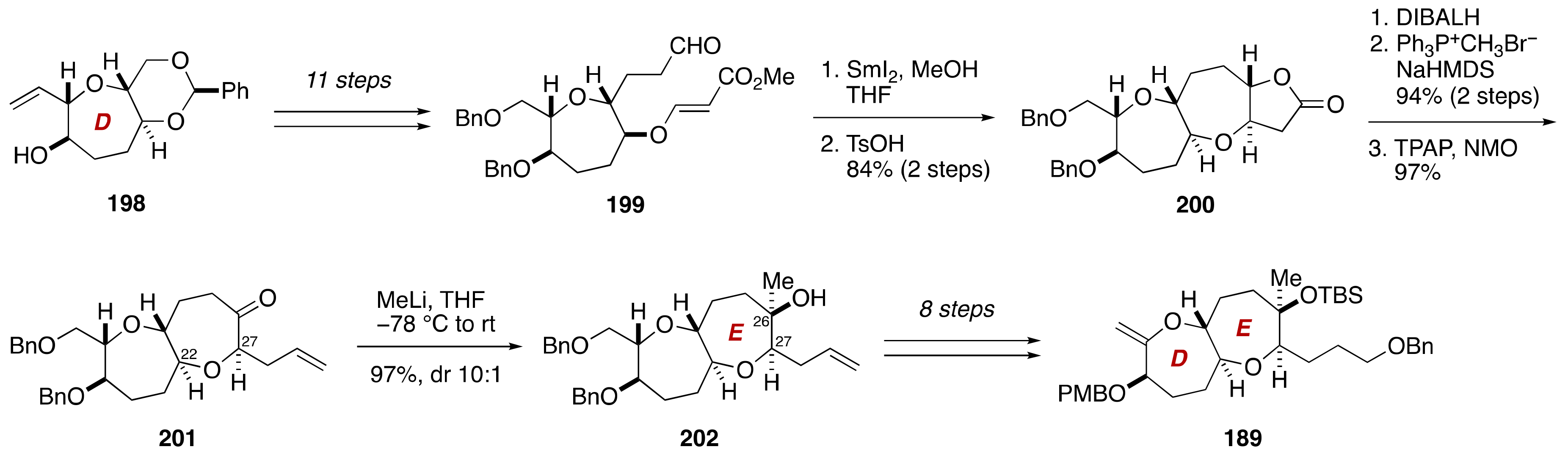
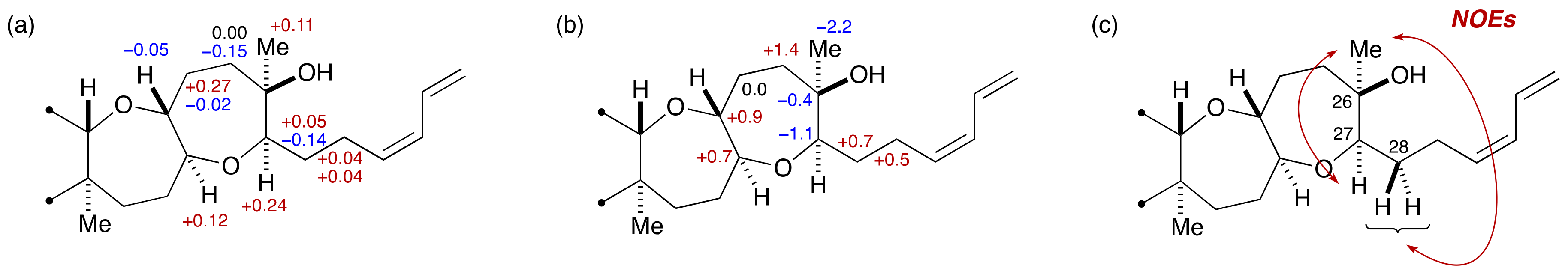
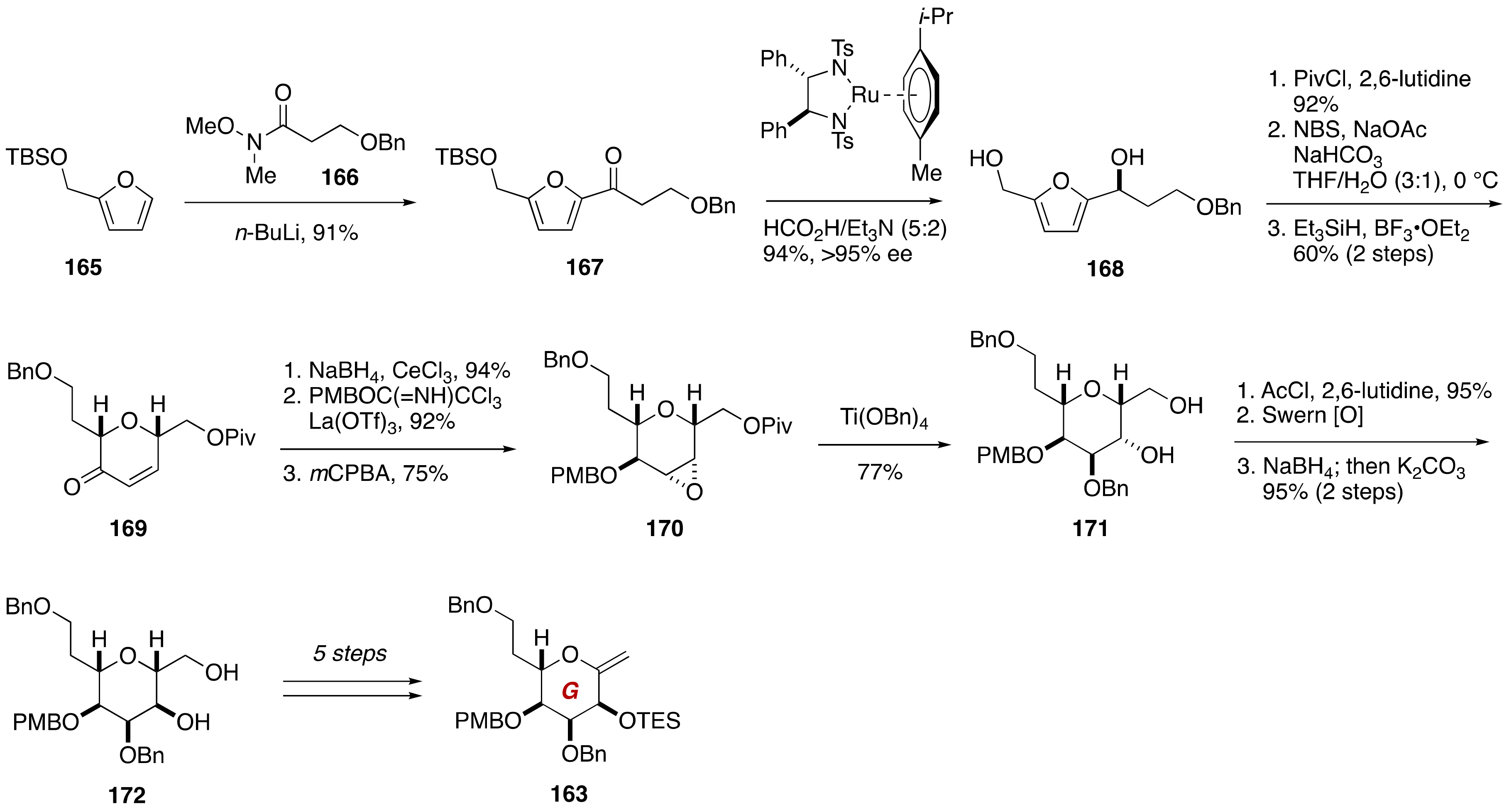
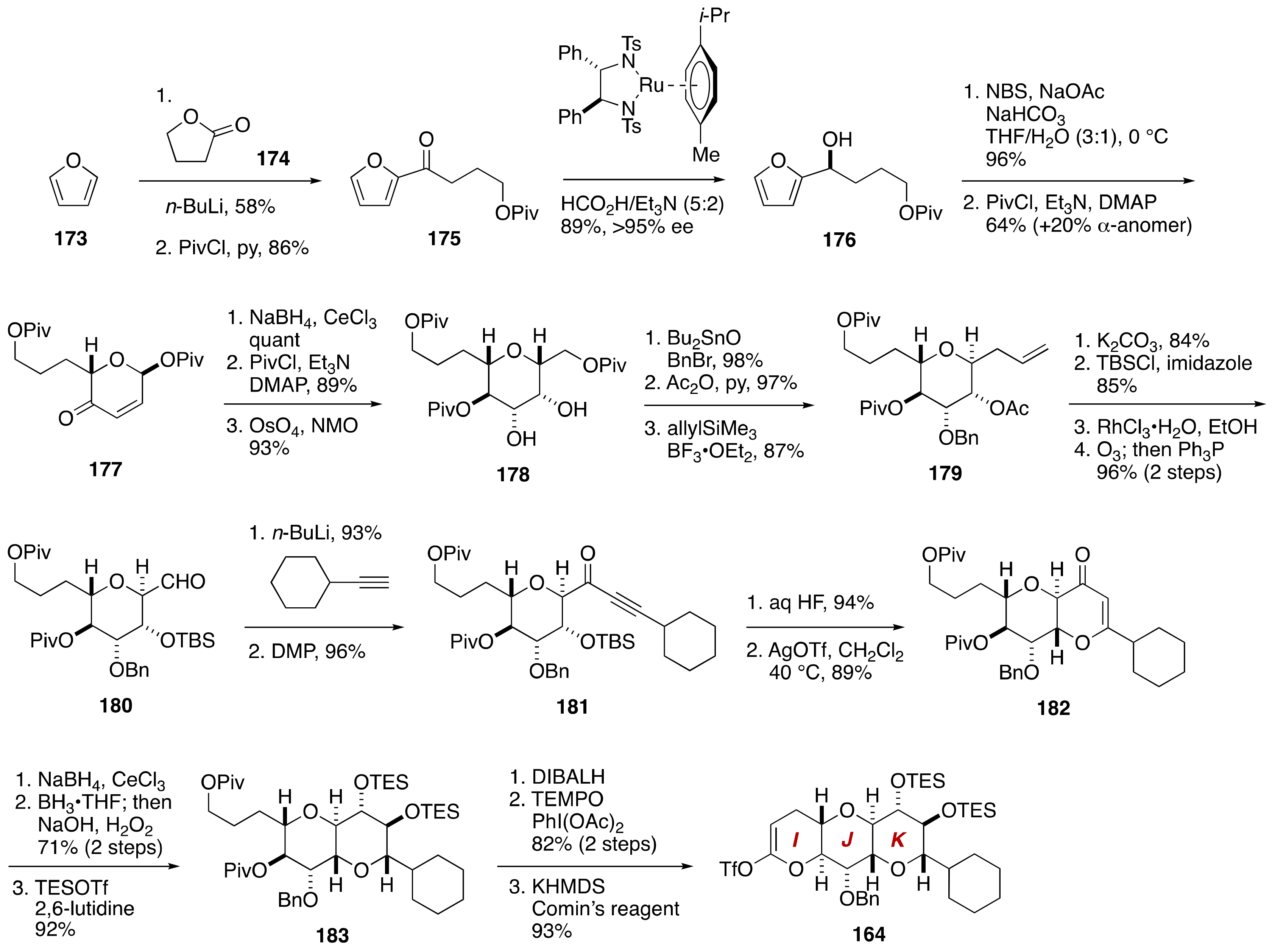
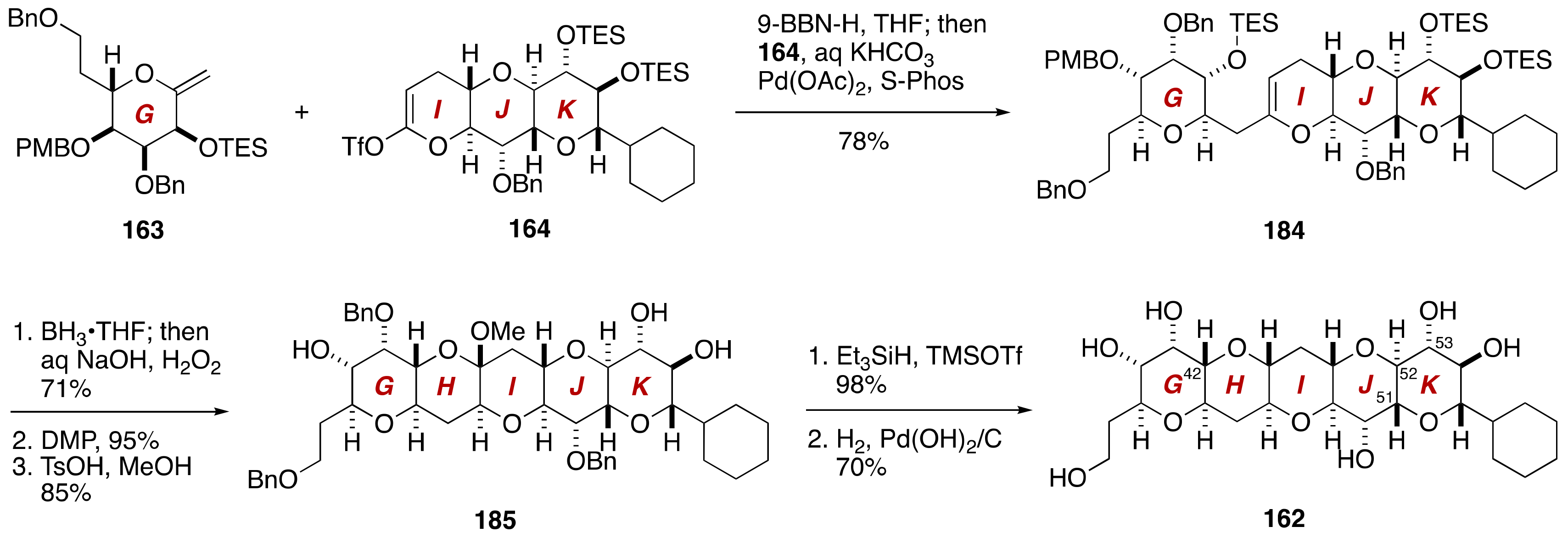
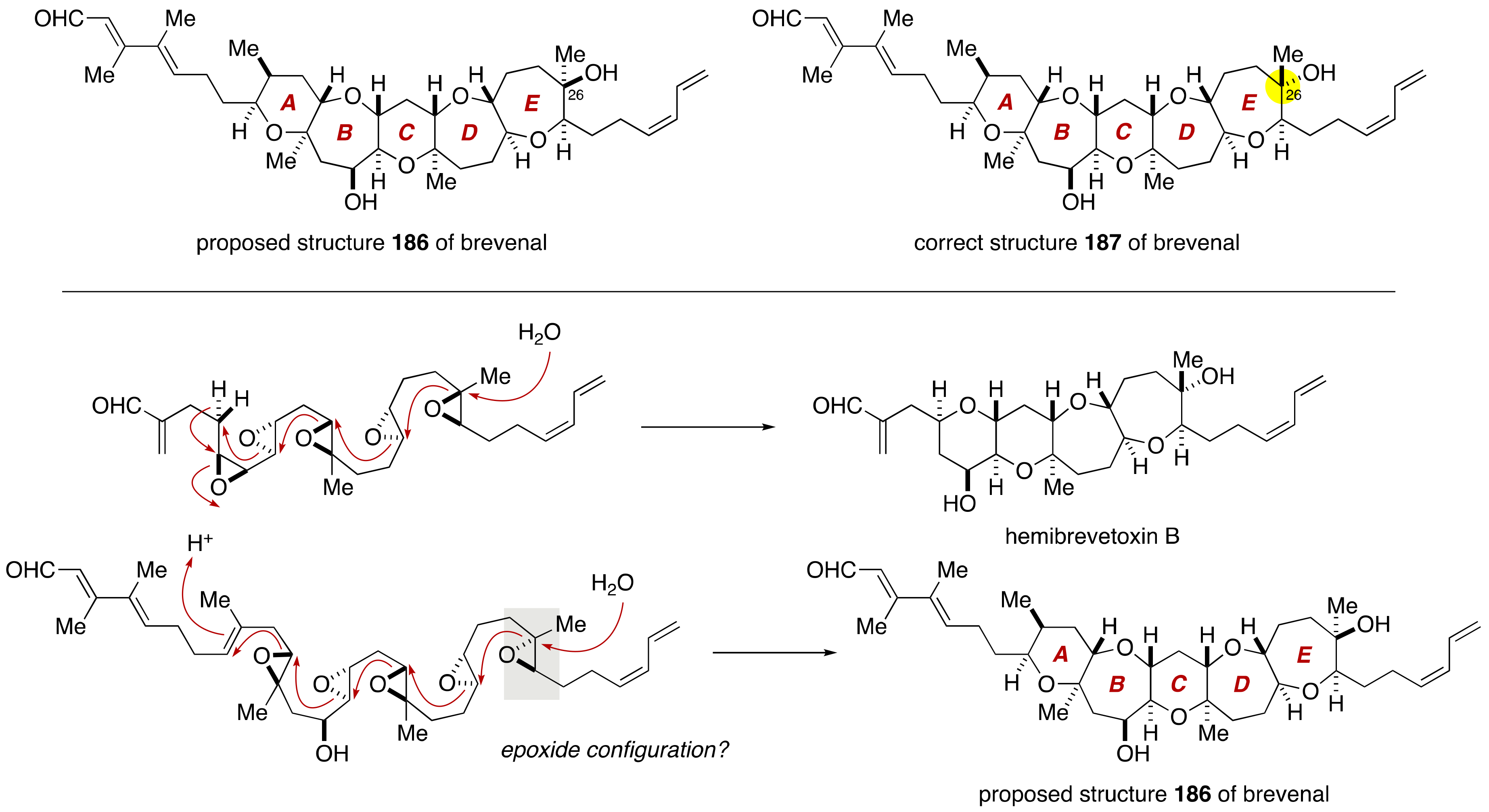
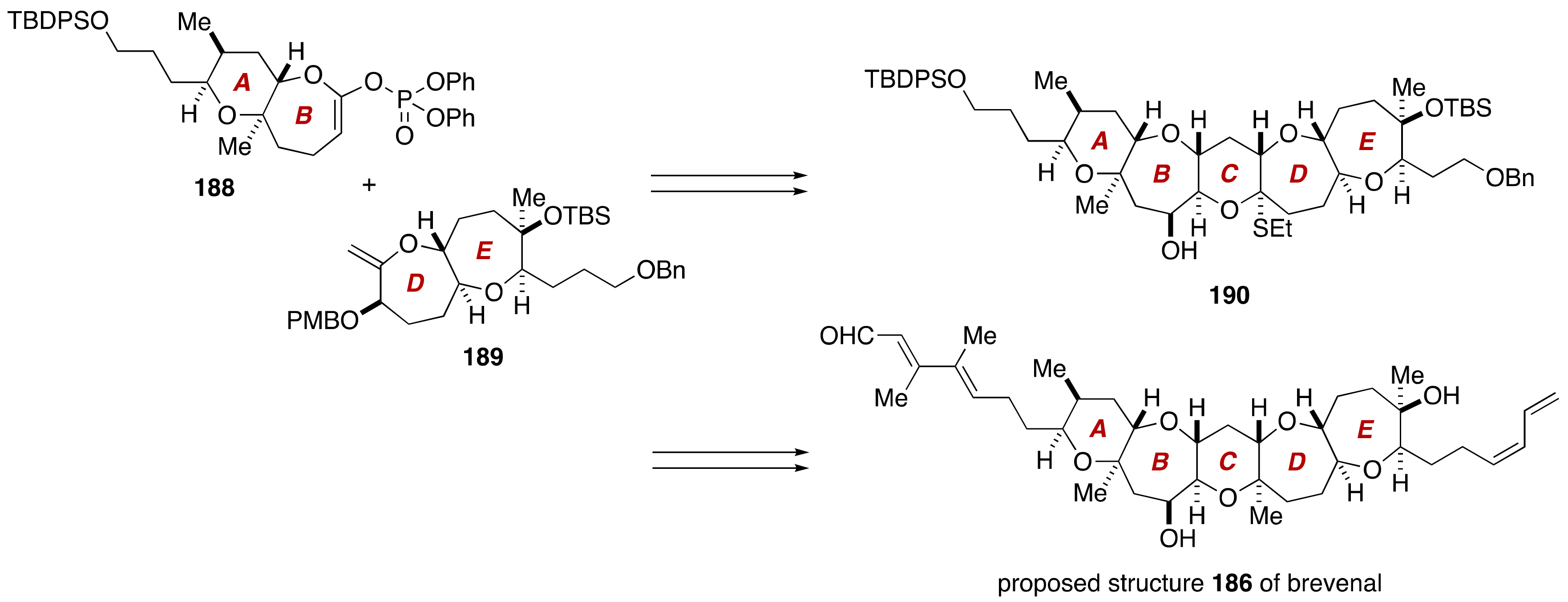
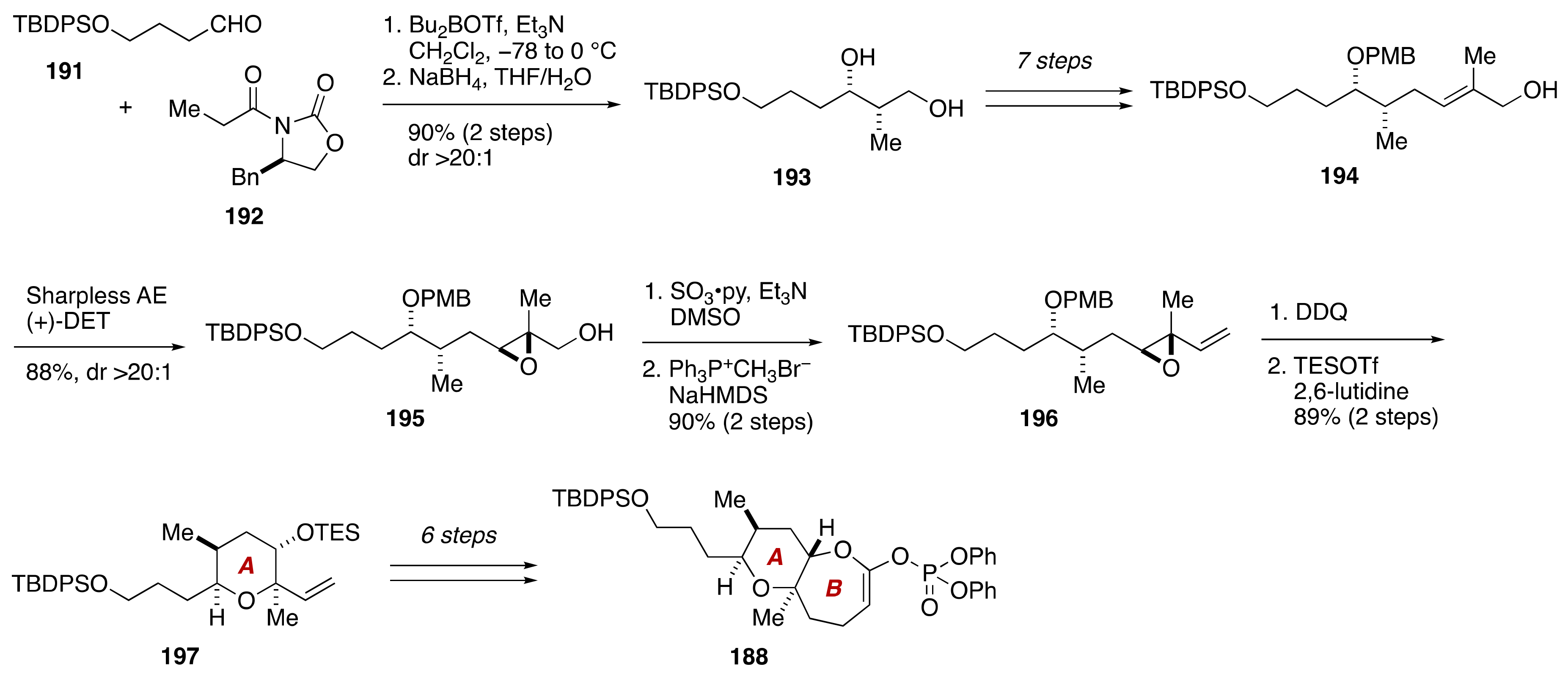
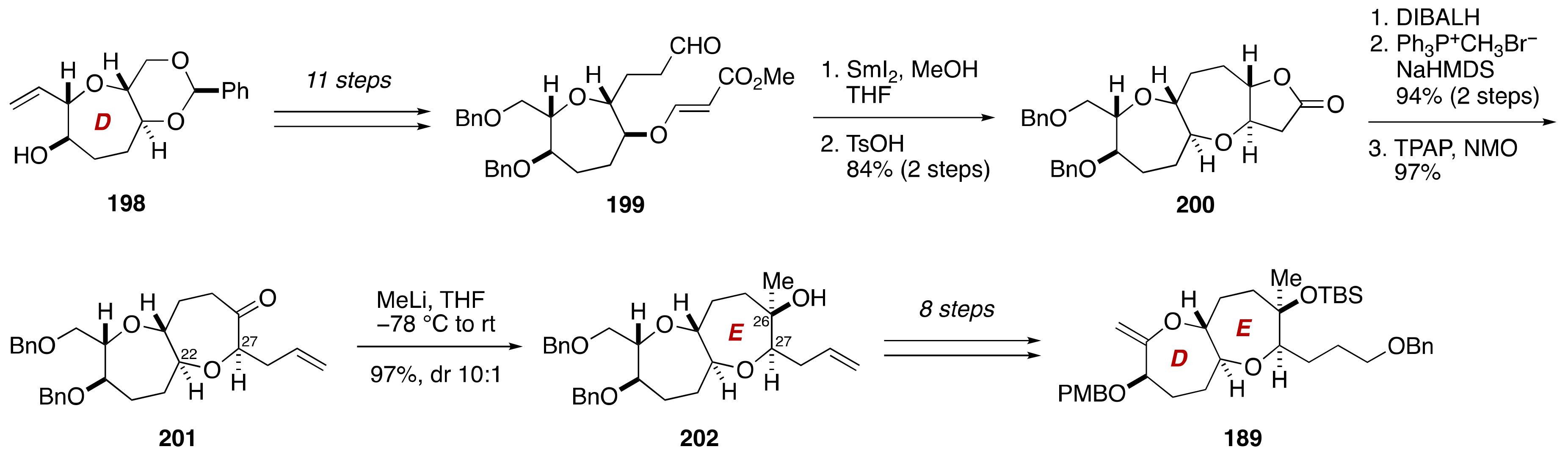
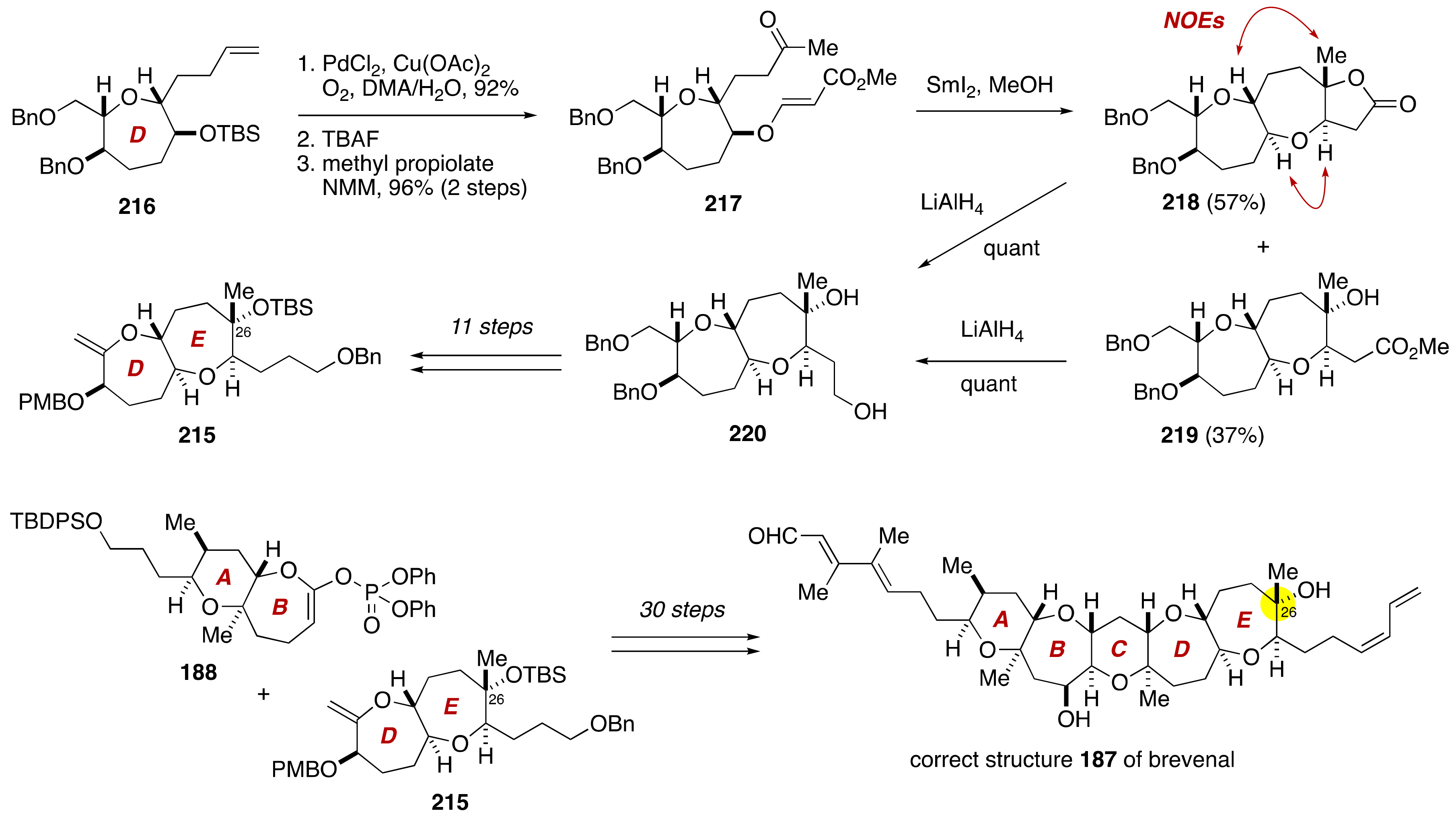
3. Brevenal
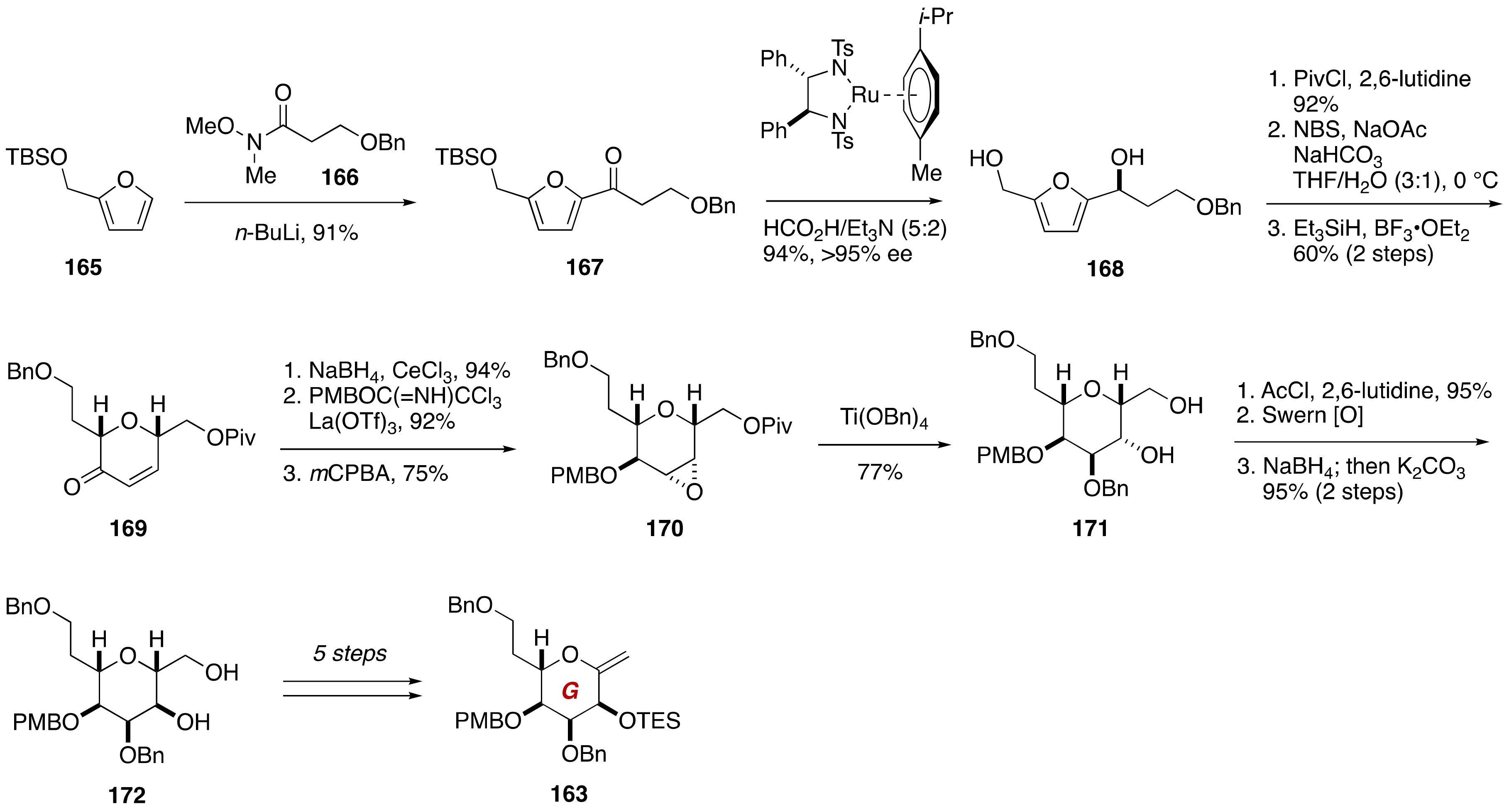
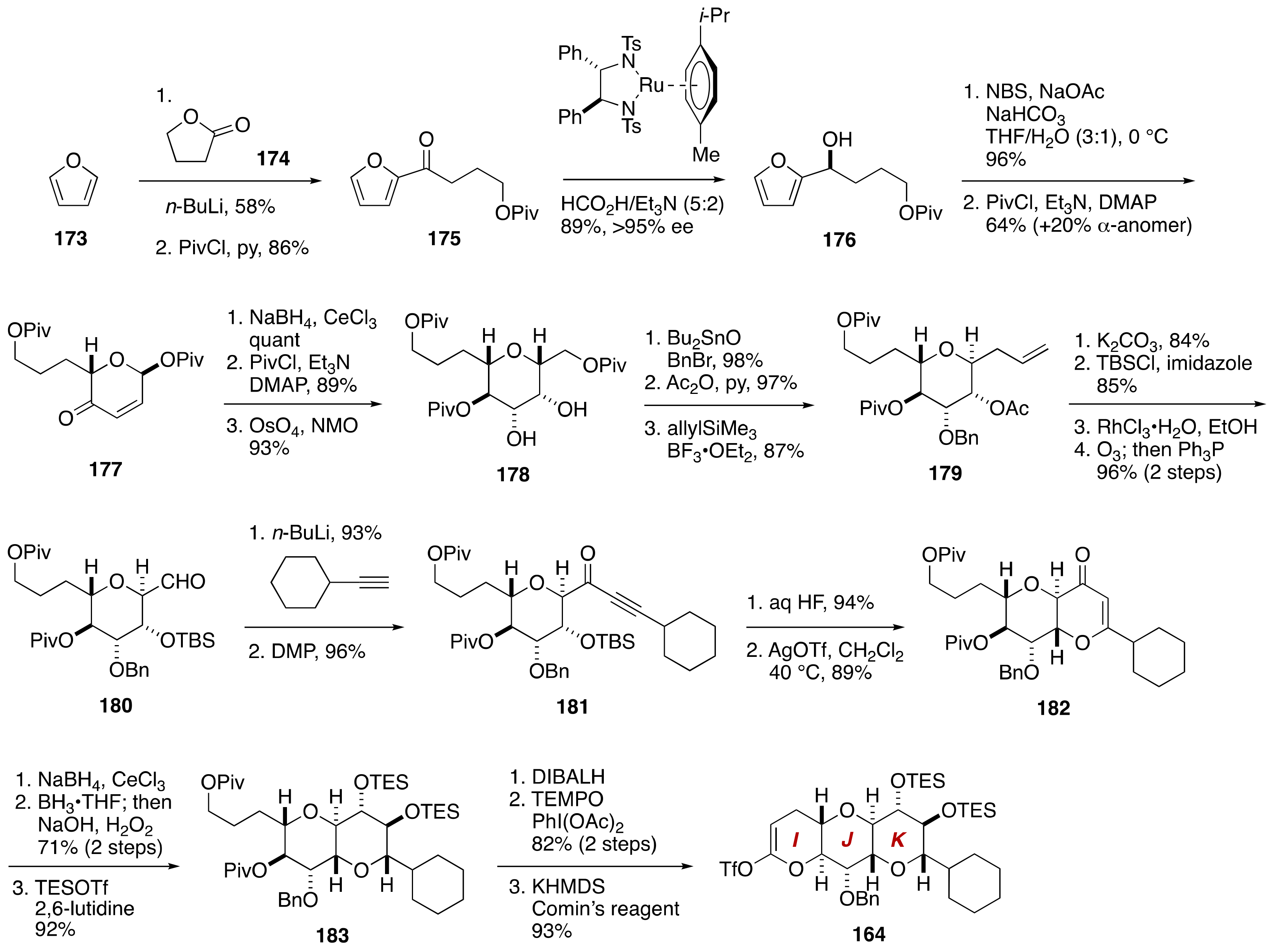
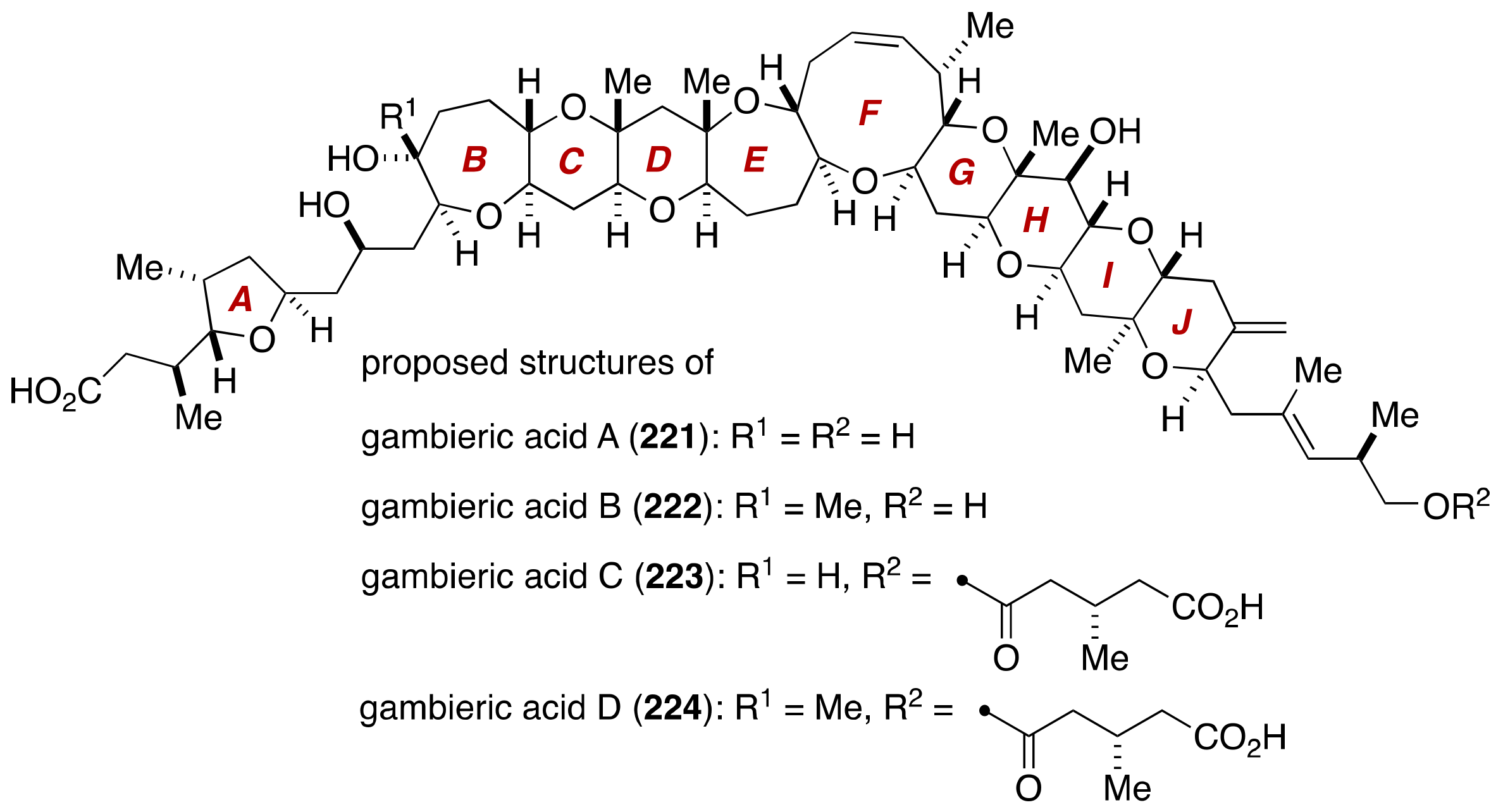
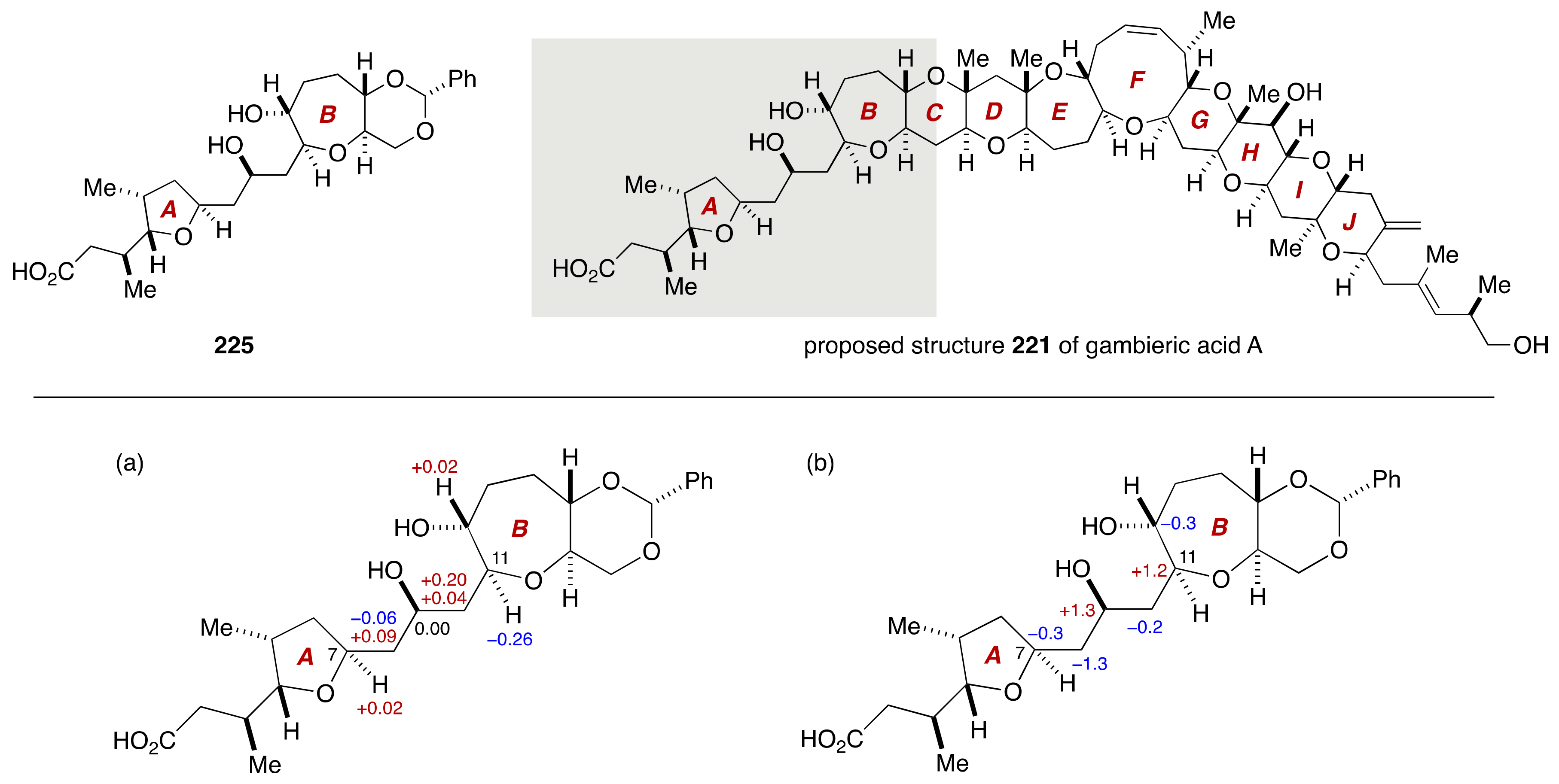
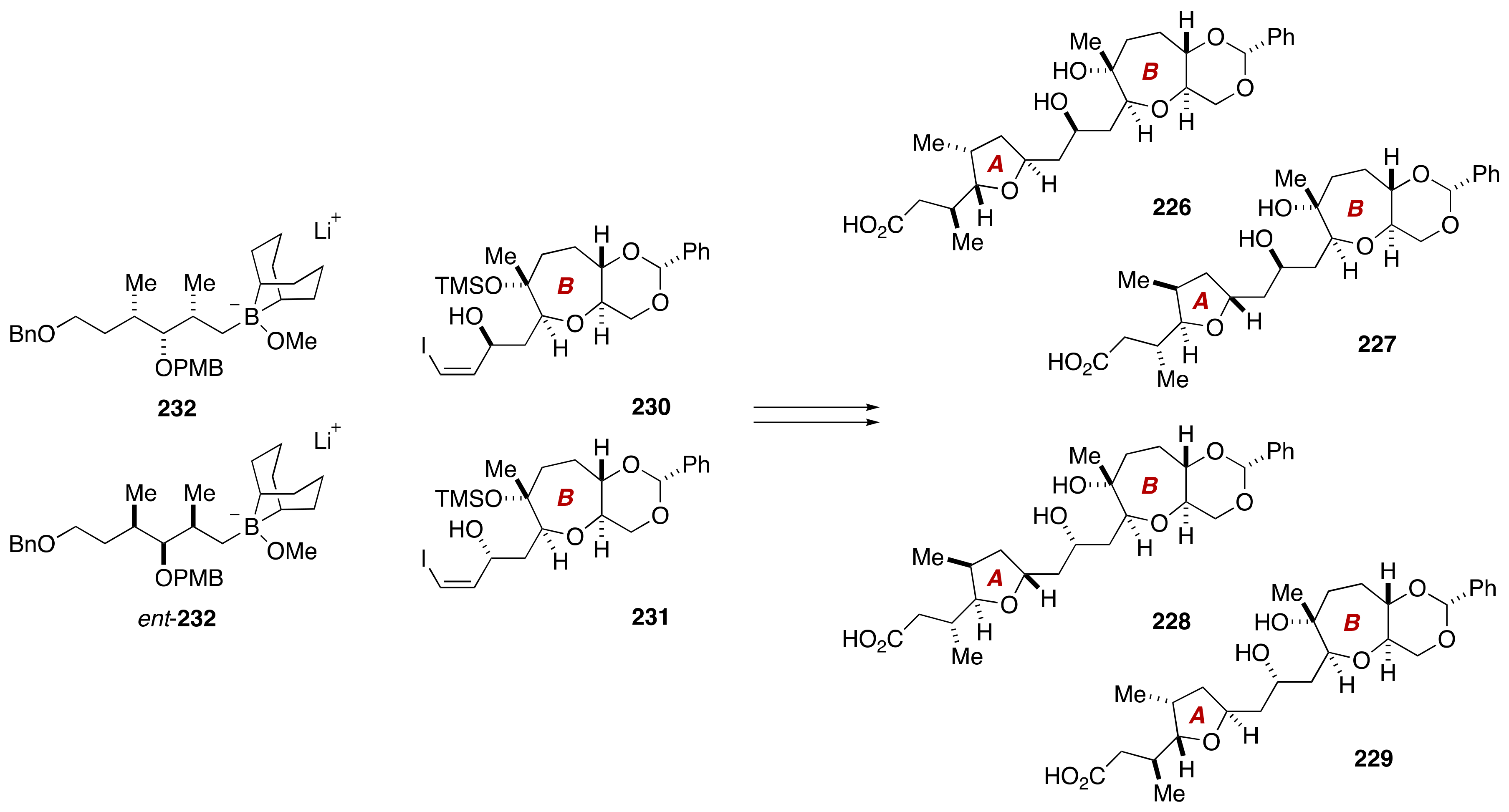
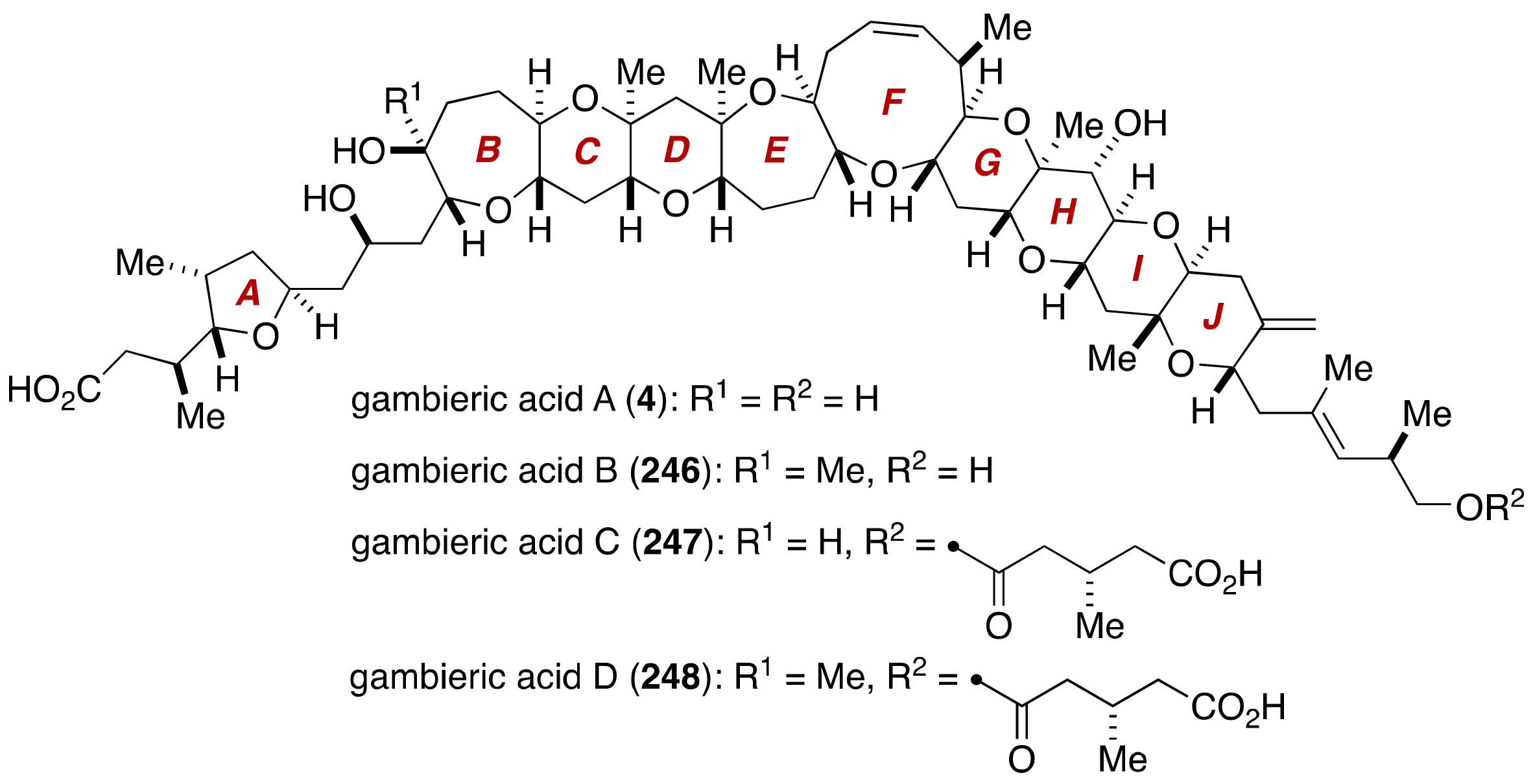
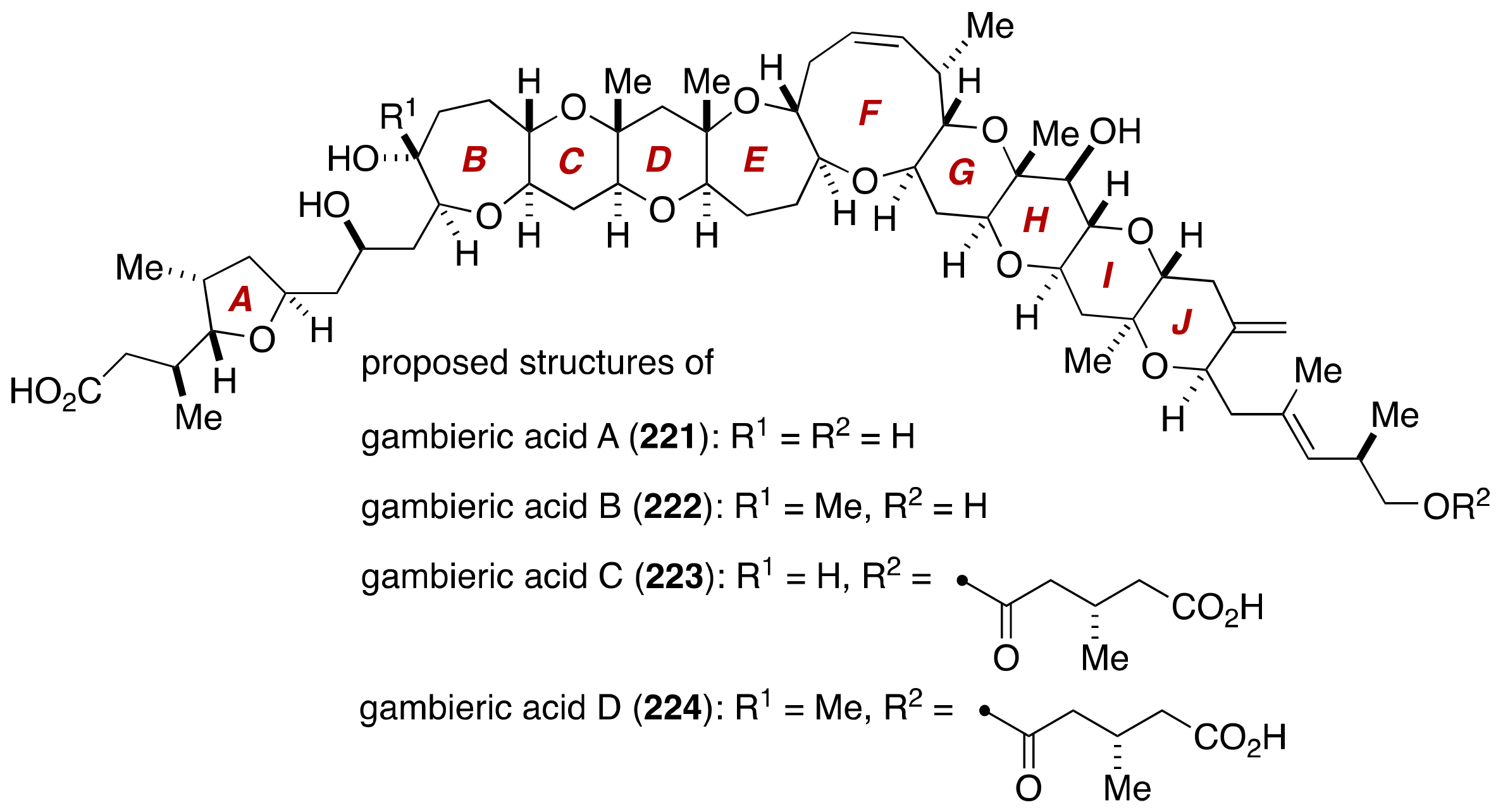
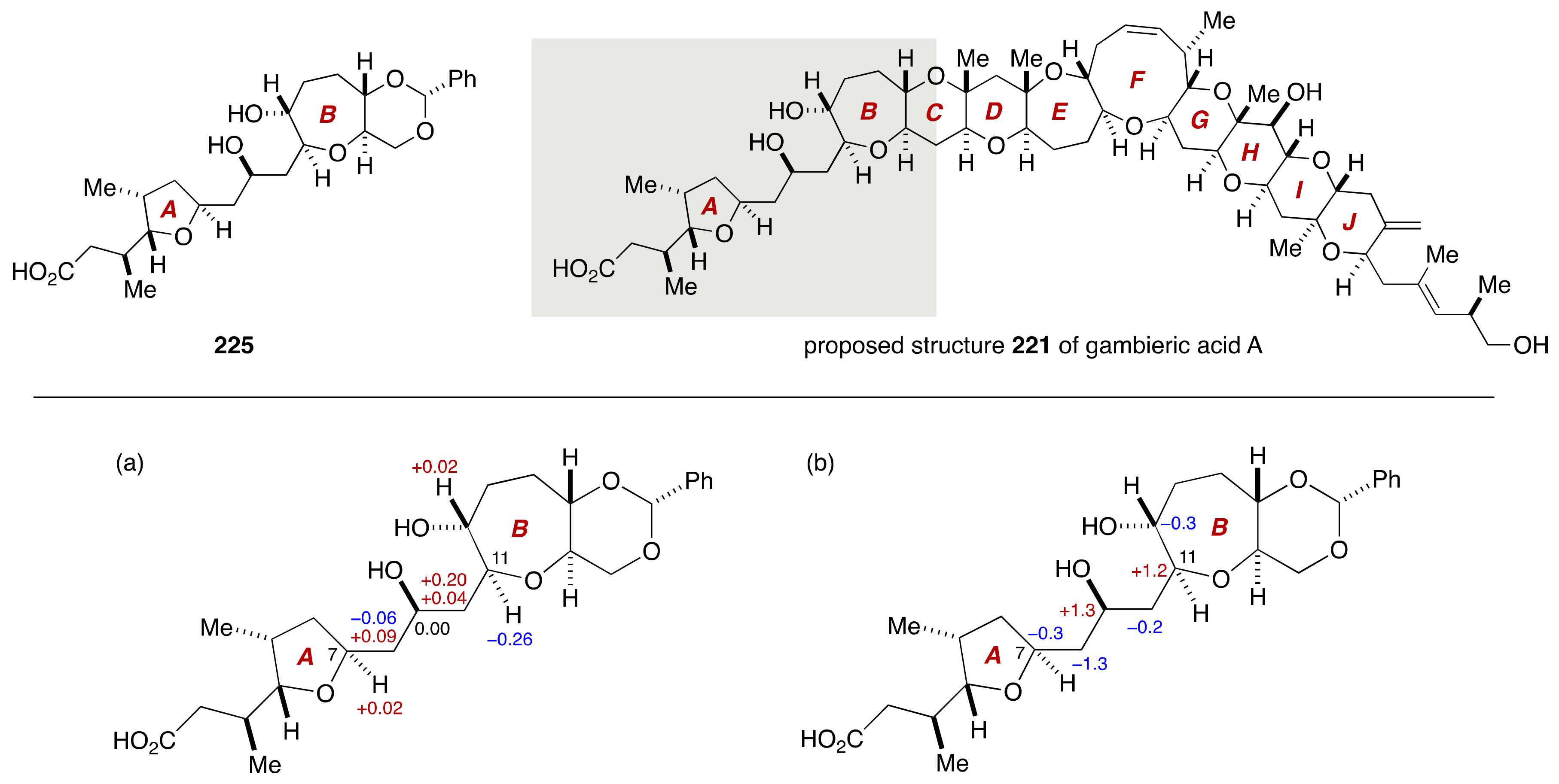
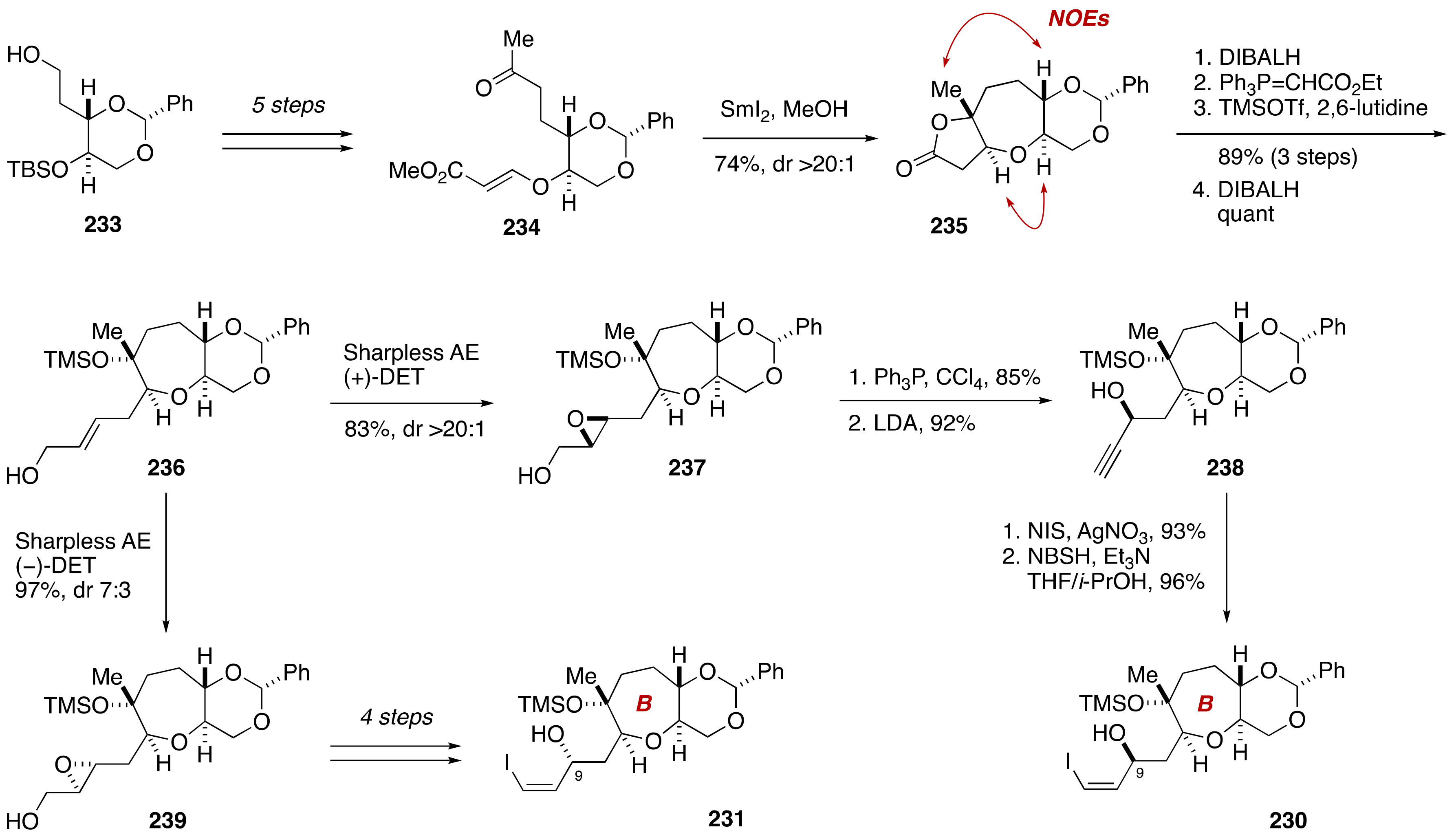
4. Gambieric Acids
5. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yasumoto, T.; Murata, M. Marine Toxins. Chem. Rev. 1993, 93, 1897–1909. [Google Scholar] [CrossRef]
- Murata, M.; Yasumoto, T. The structure elucidation and biological activities of high molecular weight algal toxins: Maitotoxin, prymnesins and zooxanthellatoxins. Nat. Prod. Rep. 2000, 17, 293–314. [Google Scholar] [CrossRef] [PubMed]
- Yasumoto, T. The Chemistry and Biological Function of Natural Marine Toxins. Chem. Rec. 2001, 1, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, G.M.; Lewis, R.J. Ciguatoxins: Cyclic polyether modulators of voltage-gated ion channel function. Mar. Drugs 2006, 4, 82–118. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A.; Cestèle, S.; Yarov-Yarovoy, V.; Yu, F.H.; Konoki, K.; Scheuer, T. Voltage-gated ion channels and gating modifier toxins. Toxicon 2007, 49, 124–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shmukler, Y.B.; Nikishin, D.A. Ladder-shaped ion channel ligands: Current state of knowledge. Mar. Drugs 2017, 15, 232. [Google Scholar] [CrossRef]
- Finol-Urdaneta, R.K.; Belovanovic, A.; Micic-Vicovac, M.; Kinsella, G.K.; McArthur, J.R.; Al-Sabi, A. Marine toxins targeting Kv1 channels: Pharmacological tools and therapeutic scaffolds. Mar. Drugs 2020, 18, 173. [Google Scholar] [CrossRef] [Green Version]
- Van Theemsche, K.M.; Van de Sande, D.V.; Snyders, D.J.; Labro, A.J. Hydrophobic drug/toxin binding sites in voltage-dependent K+ and Na+ channels. Front. Pharmacol. 2020, 11, 735. [Google Scholar] [CrossRef]
- Lin, Y.-Y.; Risk, M.; Ray, S.M.; Van Engen, D.; Clardy, J.; Golik, J.; James, J.C.; Nakanishi, K. Isolation and Structure of Brevetoxin B from the “Red Tide” Dinoflagellate Ptychodiscusbrevis (Gymnodinium breve). J. Am. Chem. Soc. 1981, 103, 6773–6775. [Google Scholar] [CrossRef]
- Nakanishi, K. The chemistry of brevetoxins: A review. Toxicon 1985, 23, 473–479. [Google Scholar] [CrossRef]
- Shimizu, Y. Natural Toxins: Animal, Plant and Microbial; Harris, J.B., Ed.; Clarendon Press: Oxford, UK, 1986; pp. 115–125. [Google Scholar]
- Chou, H.-N.; Shimizu, Y. Biosynthesis of Brevetoxins. Evidence for the Mixed Origin of the Backbone Carbon Chain and Possible Involvement of Dicarboxylic Acids. J. Am. Chem. Soc. 1987, 109, 2184–2185. [Google Scholar] [CrossRef]
- Lee, M.S.; Qin, G.; Nakanishi, K.; Zagorski, M.G. Biosynthetic Studies of Brevetoxins, Potent Neurotoxins Produced by the Dinoflagellate Gymnodinium breve. J. Am. Chem. Soc. 1989, 111, 6234–6241. [Google Scholar] [CrossRef]
- Vilotijevic, I.; Jamison, T.F. Epoxide-Opening Cascades Promoted by Water. Science 2007, 317, 1189–1192. [Google Scholar] [CrossRef] [Green Version]
- Murata, M.; Legrand, A.M.; Ishibashi, Y.; Yasumoto, T. Structures of Ciguatoxin and Its Congener. J. Am. Chem. Soc. 1989, 111, 8929–8931. [Google Scholar] [CrossRef]
- Murata, M.; Legrand, A.M.; Ishibashi, Y.; Fukui, M.; Yasumoto, T. Structures and Configurations of Ciguatoxin from the Moray Eel Gymnothorax javanicus and Its Likely Precursor from the Dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1990, 112, 4380–4386. [Google Scholar] [CrossRef]
- Satake, M.; Morohashi, A.; Oguri, H.; Oishi, T.; Hirama, M.; Hadara, N.; Yasumoto, T. The Absolute Configuration of Ciguatoxin. J. Am. Chem. Soc. 1997, 119, 11325–11326. [Google Scholar] [CrossRef]
- Nagai, H.; Torigoe, K.; Satake, M.; Murata, M.; Yasumoto, T.; Hirota, H. Gambieric Acids: Unprecedented Potent Antifungal Substances Isolated from Cultures of a Marine Dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1992, 114, 1102–1103. [Google Scholar] [CrossRef]
- Nagai, H.; Murata, M.; Torigoe, K.; Satake, M.; Yasumoto, T. Gambieric Acids, New Potent Antifungal Substances with Unprecedented Polyether Structures from a Marine Dinoflagellate Gambierdiscus toxicus. J. Org. Chem. 1992, 57, 5448–5453. [Google Scholar] [CrossRef]
- Morohashi, A.; Sakate, M.; Nagai, H.; Oshima, Y.; Yasumoto, T. The Absolute Configuration of Gambieric Acids A−D, Potent Antifungal Polyethers, Isolated from the Marine Dinoflagellate Gambierdiscus toxicus. Tetrahedron 2000, 56, 8995–9001. [Google Scholar] [CrossRef]
- Satake, M.; Murata, M.; Yasumoto, T. Gambierol: A New Toxic Polyether Compound Isolated from the Marine Dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1993, 115, 361–362. [Google Scholar] [CrossRef]
- Morohashi, A.; Satake, M.; Yasumoto, T. The absolute configuration of gambierol, a toxic marine polyether from the dinoflagellate, Gambierdiscus toxicus. Tetrahedron Lett. 1998, 39, 97–100. [Google Scholar] [CrossRef]
- Murata, M.; Iwashita, T.; Yokoyama, A.; Sasaki, M.; Yasumoto, T. Partial Structures of Maitotoxin, the Most Potent Marine Toxin from Dinoflagellate Gambierdiscus toxicus. J. Am. Chem. Soc. 1992, 114, 6594–6596. [Google Scholar] [CrossRef]
- Murata, M.; Naoki, H.; Iwashita, T.; Matsunaga, S.; Sasaki, M.; Yokoyama, A.; Yasumoto, T. Structure of Maitotoxin. J. Am. Chem. Soc. 1993, 115, 2060–2062. [Google Scholar] [CrossRef]
- Murata, M.; Naoki, H.; Matsunaga, S.; Satake, M.; Yasumoto, T. Structure and Partial Stereochemical Assignments for Maitotoxin, the Most Toxic and Largest Natural Non-Biopolymer. J. Am. Chem. Soc. 1994, 116, 7098–7107. [Google Scholar] [CrossRef]
- Sasaki, M.; Matsumori, N.; Maruyama, T.; Nonomura, T.; Murata, M.; Tachibana, K.; Yasumoto, T. The Complete Structure of Maitotoxin, Part I: Configuration of the C1–C14 Side Chain. Angew. Chem. Int. Ed. Engl. 1996, 35, 1672–1675. [Google Scholar] [CrossRef]
- Nonomura, T.; Sasaki, M.; Matsumori, N.; Murata, M.; Tachibana, K.; Yasumoto, T. The Complete Structure of Maitotoxin, Part II: Configuration of the C135–C142 Side Chain and Absolute Configuration of the Entire Molecule. Angew. Chem. Int. Ed. Engl. 1996, 35, 1675–1678. [Google Scholar] [CrossRef]
- Murata, M.; Kumagai, M.; Lee, J.S.; Yasumoto, T. Isolation and Structure of Yessotoxin, a Novel Polyether Compound Implicated in Diarrhetic Shellfish Poisoning. Tetrahedron Lett. 1987, 28, 5869–5872. [Google Scholar] [CrossRef]
- Satake, M.; Terasawa, K.; Kadowaki, Y.; Yasumoto, T. Relative Configuration of Yessotoxin and Isolation of Two New Analogs from Toxic Scallops. Tetrahedron Lett. 1996, 37, 5955–5958. [Google Scholar] [CrossRef]
- Takahashi, H.; Kusumi, T.; Kan, Y.; Satake, M.; Yasumoto, T. Determination of the Absolute Configuration of Yessotoxin, a Polyether Compound Implicated in Diarrhetic Shellfish Poisoning, by NMR Spectroscopic Method Using a Chiral Anisotropic Reagent, Methoxy-(2-naphthyl)acetic Acid. Tetrahedron Lett. 1996, 37, 7087–7090. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A. Chasing Molecules That Were Never There: Misassigned Natural Products and the Role of Chemical Synthesis in Modern Structure Eludication. Angew. Chem. Int. Ed. 2005, 44, 1012–1044. [Google Scholar] [CrossRef]
- Maier, M.E. Structural Revisions of Natural Products by Total Synthesis. Nat. Prod. Rep. 2009, 26, 1105–1124. [Google Scholar] [CrossRef] [PubMed]
- Usami, Y. Recent Synthetic Studies Leading to Structural Revisions of Marine Natural Products. Mar. Drugs 2009, 7, 314–330. [Google Scholar] [CrossRef] [PubMed]
- Suyama, T.L.; Gerwick, W.H.; McPhail, K.L. Survey of Marine Natural Product Structure Revisions: A Synergy of Spectroscopy and Chemical Synthesis. Bioorg. Med. Chem. 2011, 19, 6675–6701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, N.Y.S.; Paterson, I. Stereocontrolled Synthesis as an Enabling Tool for the Configurational Assignment of Marine Polyketide Natural Products. Eur. J. Org. Chem. 2020, 2310–2320. [Google Scholar] [CrossRef]
- Yasumoto, T.; Bagnis, R.; Vernoux, J.P. Toxicity of the Surgeonfishes–II. Properties of the Principal Water-Soluble Toxin. Bull. Jpn. Soc. Sci. Fish 1976, 42, 359–365. [Google Scholar] [CrossRef]
- Takahashi, M.; Ohizumi, Y.; Yasumoto, T. Maitotoxin, a Ca2+ Channel Activator Candidate. J. Biol. Chem. 1982, 257, 7287–7289. [Google Scholar] [CrossRef]
- Takahashi, M.; Tatsumi, M.; Ohizumi, Y.; Yasumoto, T. Ca2+ Channel Activating Function of Maitotoxin, the Most Potent Marine Toxin Known, in Clonal Rat Pheochromocytoma Cells. J. Biol. Chem. 1983, 258, 10944–10949. [Google Scholar] [CrossRef]
- Kobayashi, M.; Ohizumi, Y.; Yasumoto, T. The Mechanism of Action of Maitotoxin in Relation to Ca2+ Movements in Guinea-pig and Rat Cardiac Muscles. Br. J. Pharmacol. 1985, 86, 385–391. [Google Scholar] [CrossRef] [Green Version]
- Konoki, K.; Hashimoto, M.; Nonomura, T.; Sasaki, M.; Murata, M.; Tachibana, K. Inhibition of Maitotoxin-Induced Ca2+ Influx in Rat Glioma C6 Cells by Brevetoxins and Synthetic Fragments of Maitotoxin. J. Neurochem. 1998, 70, 409–416. [Google Scholar] [CrossRef] [Green Version]
- Satoh, E.; Ishii, T.; Nishimura, M. The Mechanism of Maitotoxin-Induced Elevation of the Cytosolic Free Calcium Level in Rat Cerebrocortical Synaptosomes. Jpn. J. Pharmacol. 2001, 85, 98–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De la Rosa, L.A.; Alfonso, A.; Vilariño, N.; Vieytes, M.R.; Yasumoto, T.; Botana, L.M. Maitotoxin-Induced Calcium Entry in Human Lymphocytes: Modulation by Yessotoxin, Ca2+ Channel Blockers and Kinases. Cell Signal. 2001, 13, 711–716. [Google Scholar] [CrossRef]
- Sasaki, M.; Nonomura, T.; Murata, M.; Tachibana, K. Synthesis and Stereochemical Confirmation of the cis-Fused L/M and N/O Ring Systems of Maitotoxin. Tetrahedron Lett. 1994, 35, 5023–5026. [Google Scholar] [CrossRef]
- Sasaki, M.; Nonomura, T.; Murata, M.; Tachibana, K. Synthetic Approach toward Complete Structure Determination of Maitotoxin. Stereochemical Assignment of the C63–C68 Acyclic Linkage. Tetrahedron Lett. 1995, 36, 9007–9010. [Google Scholar] [CrossRef]
- Sasaki, M.; Matsumori, N.; Murata, M.; Tachibana, K.; Yasumoto, T. Stereochemical Assignment of the C35–C39 Acyclic Linkage in Maitotoxin: Completion of Stereochemical Determination of C15–C134. Tetrahedron Lett. 1995, 36, 9011–9014. [Google Scholar] [CrossRef]
- Matsumori, N.; Nonomura, T.; Sasaki, M.; Murata, M.; Tachibana, K.; Satake, M.; Yasumoto, T. Long-range Carbon–Proton Coupling Constants for Stereochemical Assignment of Acyclic Structures in Natural Products: Configuration of the C5–C9 Portion of Maitotoxin. Tetrahedron Lett. 1996, 37, 1269–1272. [Google Scholar] [CrossRef]
- Lewis, M.D.; Cha, J.K.; Kishi, Y. Highly Stereoselective Approaches to α- and β-C-glycopyranosides. J. Am. Chem. Soc. 1982, 104, 4976–4978. [Google Scholar] [CrossRef]
- Gao, Y.; Hanson, R.M.; Klunder, J.M.; Ko, S.Y.; Masamune, H.; Sharpless, K.B. Catalytic Asymmetric Epoxidation and Kinetic Resolution: Modified Procedures Including in Situ Derivatization. J. Am. Chem. Soc. 1987, 109, 5765–5780. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Prasad, C.V.C.; Somers, P.K.; Hwang, C.-K. Activation of 6-Endo over 5-Exo Hydroxy Epoxide Openings. Stereoselective and Ring Selective Synthesis of Tetrahydrofuran and Tetrahydropyran Systems. J. Am. Chem. Soc. 1989, 111, 5330–5334. [Google Scholar] [CrossRef]
- Evans, D.A.; Chapman, K.T.; Carreira, E.M. Directed Reduction of β-Hydroxy Ketones Employing Tetramethylammonium Triacetoxyborohydride. J. Am. Chem. Soc. 1988, 110, 3560–3578. [Google Scholar] [CrossRef]
- Narasaka, K.; Pai, F.-C. Stereoselective Synthesis of Meso (or Erythro) 1,3-Diols from β-Hydroxyketones. Chem. Lett. 1980, 1415–1418. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, T.; Sato, O.; Hirama, M. Palladium Catalyzed Stereospecific Cyclization of Hydroxy Epoxides. Stereocontrolled Synthesis of cis- and trans-2-Alkenyl-3-hydroxytetrahydropyrans. Tetrahedron Lett. 1990, 31, 4747–4750. [Google Scholar] [CrossRef]
- Cha, J.K.; Christ, W.J.; Kishi, Y. On Stereochemistry of Osmium Tetraoxide Oxidation of Allylic Alcohol Systems. Empirical Rule. Tetrahedron 1984, 40, 2247–2255. [Google Scholar] [CrossRef]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical Determination of Acyclic Structures Based on Carbon–Proton Spin-Coupling Constants. A Method of Configuration Analysis for Natural Products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Saimoto, H.; Tomioka, H.; Oshima, K.; Nozaki, H. Regio- and Stereoselective Ring Opening of Epoxy Alcohols with Organoaluminium Compounds Leading to 1,2-Diols. Tetrahedron Lett. 1982, 23, 3597–3600. [Google Scholar] [CrossRef]
- Roush, W.R.; Adam, M.A.; Peseckis, S.M. Regioselectivity of the Reactions of Trialkylaluminum Reagents with 2,3-Epoxyalcohols: Application to the Synthesis of α-Chiral Aldehydes. Tetrahedron Lett. 1983, 24, 1377–1380. [Google Scholar] [CrossRef]
- Mancuso, A.J.; Swern, D. Activated Dimethyl Sulfoxide: Useful Reagents for Synthesis. Synthesis 1981, 3, 165–185. [Google Scholar] [CrossRef]
- Johnson, M.R.; Nakata, T.; Kishi, Y. Stereo- and Regioselective Methods for the Synthesis of Three Consecutive Asymmetric Units Found in Many Natural Products. Tetrahedron Lett. 1979, 20, 4343–4346. [Google Scholar] [CrossRef]
- Takai, K.; Nitta, K.; Utimoto, K. Simple and Selective Method for RCHO → (E)-RCH=CHX Conversion by Means of a CHX3–CrCl2 System. J. Am. Chem. Soc. 1986, 108, 7408–7410. [Google Scholar] [CrossRef]
- Takai, K.; Kimura, K.; Kuroda, T.; Hiyama, T.; Nozaki, H. Selective Grignard-Type Carbonyl Addition of Alkenly Halides Mediated by Chromium(II) Chloride. Tetrahedron Lett. 1983, 24, 5281–5284. [Google Scholar] [CrossRef]
- Jin, H.; Uenishi, J.; Christ, W.J.; Kishi, Y. Catalytic Effect of Nickel(II) Chloride and Palladium(II) Acetate on Chromium(II)-Mediated Coupling Reaction of Iodo Olefins with Aldehydes. J. Am. Chem. Soc. 1986, 108, 5644–5646. [Google Scholar] [CrossRef]
- Yamashita, M.; Kato, Y.; Suemitsu, R. Selective Reduction of α,β-Unsaturated Carbonyl Compounds by Sodium Hydrotelluride. Chem. Lett. 1980, 847–848. [Google Scholar] [CrossRef]
- Nakata, T.; Tanaka, T.; Oishi, T. Stereoselective Reduction of α-Hydroxy Ketones. Tetrahedron Lett. 1983, 24, 2653–2656. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Hwang, C.-K.; Marron, B.E.; DeFrees, S.A.; Couladouros, E.A.; Abe, Y.; Carroll, P.J.; Snyder, J.P. Bridging of Macrodithionolacotnes to Bicyclic Systems. Synthesis and Modeling of Oxapolycyclic Frameworks. J. Am. Chem. Soc. 1990, 112, 3040–3054. [Google Scholar] [CrossRef] [Green Version]
- Kolb, H.C.; VanNieuwenhze, M.S.; Sharpless, K.B. Catalytic Asymmetric Dihydroxylation. Chem. Rev. 1994, 94, 2483–2547. [Google Scholar] [CrossRef]
- Robins, M.K.; Wilson, J.S.; Hansske, F. Nucleic Acid Related Compounds. 42. A General Procedure for the Efficient Deoxygenation of Secondary Alcohols. Regiospecific and Stereoselective Conversion of Ribonucleosides to 2′-Deoxynucleosides. J. Am. Chem. Soc. 1983, 105, 4059–4065. [Google Scholar] [CrossRef]
- Heathcock, C.H.; Davis, B.R.; Hadley, C.R. Synthesis and Biological Evaluation of a Monocyclic, Fully Functional Analog of Compactin. J. Med. Chem. 1989, 32, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Furuta, K.; Iwanaga, K.; Yamamoto, H. Asymmetric Diels–Alder Reaction. Cooperative Blocking Effect in Organic Synthesis. Tetrahedron Lett. 1986, 27, 4507–4510. [Google Scholar] [CrossRef]
- Zheng, W.; DeMattei, J.A.; Wu, J.-P.; Duan, J.J.-W.; Cook, L.R.; Oinuma, H.; Kishi, Y. Complete Relative Stereochemistry of Maitotoxin. J. Am. Chem. Soc. 1996, 118, 7946–7968. [Google Scholar] [CrossRef]
- Boyle, C.D.; Harmange, J.-C.; Kishi, Y. Novel Structure Eludication of AAL Toxin TA Backbone. J. Am. Chem. Soc. 1994, 116, 4995–4996. [Google Scholar] [CrossRef]
- Harmange, J.-C.; Boyle, C.D.; Kishi, Y. Relative and Absolute Stereochemistry of the Fumonisin B2 Backbone. Tetrahedron Lett. 1994, 35, 6819–6822. [Google Scholar] [CrossRef]
- Boyle, C.D.; Kishi, Y. Absolute Configuration at the Tricarballylic Acid Moieties of Fumonisin B1 and AAL Toxin TA. Tetrahedron Lett. 1995, 36, 5695–5698. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tan, C.-H.; Kishi, Y. Stereochemical Assignment of the C21–C38 Portion of the Desertomycin/Oasomycin Class of Natural Products by Using Universal NMR Databases: Prediction. Angew. Chem. Int. Ed. 2000, 39, 4279–4281. [Google Scholar] [CrossRef]
- Tan, C.-H.; Kobayashi, Y.; Kishi, Y. Stereochemical Assignment of the C21–C38 Portion of the Desertomycin/Oasomycin Class of Natural Products by Using Universal NMR Databases: Proof. Angew. Chem. Int. Ed. 2000, 39, 4282–4284. [Google Scholar] [CrossRef]
- Kobayashi, Y.; Tan, C.-H.; Kishi, Y. Toward Creation of a Universal NMR Database for Stereochemical Assignment: Complete Structure of the Desertomycin/Oasomycin Class of Natural Products. J. Am. Chem. Soc. 2001, 123, 2076–2078. [Google Scholar] [CrossRef] [PubMed]
- Roush, W.R.; Ando, K.; Powers, D.B.; Palkowitz, A.D.; Halterman, R.L. Asymmetric Synthesis Using Diisopropyl Tartrate Modified (E)- and (Z)-Crotylboronates: Preparation of the Chiral Crotylboronates and Reactions with Achiral Aldehydes. J. Am. Chem. Soc. 1990, 112, 6339–6348. [Google Scholar] [CrossRef]
- Tadano, K.; Maeda, H.; Hoshino, M.; Iimura, Y.; Suami, T. A novel transformation of four aldoses to some optically pure pseudohexopyranoses and a pseudopentofuranose, carbocyclic analogs of hexopyranoses and pentofuranose. Synthesis of derivatives of (1S,2S,3R,4S,5S)-, (1S,2S,3R,4R,5S)-, (1R,2R,3R,4R,5S)-, (1S,2S,3R,4S,5R)-2,3,4,5-tetrahydroxy-1-(hydroxymethyl)cyclohexanes and (1S,2S,3S,4S)-2,3,4-trihydroxy-1(hydroxymethyl)cyclopentane. J. Org. Chem. 1987, 52, 1946–1956. [Google Scholar]
- Dondoni, A.; Scherrmann, M.-C. Thiazole-Based Synthesis of Formyl C-Glycosides. J. Org. Chem. 1994, 59, 6404–6412. [Google Scholar] [CrossRef]
- Barnes, J.C.; Brimacombe, J.S.; Kabir, A.K.M.S.; Weakley, T.J.R. Higher-Carbon Sugars. Part 12. The Synthesis of New Octitols from D-Glucose and D-Mannose via the Osymlation of Unsaturated Precursors. J. Chem. Soc. Perkin Trans. 1 1988, 12, 3391–3397. [Google Scholar] [CrossRef]
- Mitsunobu, O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis 1981, 1–28. [Google Scholar] [CrossRef]
- Nising, C.F.; Bräse, S. The Oxa-Michael Reaction: From Recent Developments to Applications in Natural Product Synthesis. Chem. Soc. Rev. 2008, 37, 1218–1228. [Google Scholar] [CrossRef] [PubMed]
- Nising, C.F.; Bräse, S. Recent Developments in the Field of Oxa-Michael Reactions. Chem. Soc. Rev. 2012, 41, 988–999. [Google Scholar] [CrossRef]
- Baraldi, P.G.; Barco, A.; Benetti, S.; Manfredini, S.; Simoni, D. Ring Cleavage of 3,5-Disubstituted 2-Isoxazolines by Molybdenum Hexacarbonyl and Water to β-Hydroxy Ketones. Synthesis 1987, 276–278. [Google Scholar] [CrossRef]
- Gallimore, A.R.; Spencer, J.B. Stereochemical Uniformity in Marine Polyether Ladders–Implications for the Biosynthesis and Structure of Maitotoxin. Angew. Chem. Int. Ed. 2006, 45, 4406–4413. [Google Scholar] [CrossRef]
- Satake, M.; Ishida, S.; Yasumoto, T.; Murata, M.; Utsumi, H.; Hinomoto, T. Structural Confirmation of Maitotoxin Based on Complete 13C NMR Assignments and the Three-Dimentional PFG NOESY–HMQC Spectrum. J. Am. Chem. Soc. 1995, 117, 7019–7020. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Frederick, M.O. On the Structure of Maitotoxin. Angew. Chem. Int. Ed. 2007, 46, 5278–5282. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Cole, K.P.; Frederick, M.O.; Aversa, R.J.; Denton, R.M. Chemical Synthesis of the GHIJK Ring System and Further Experimental Support for the Originally Assigned Structure of Maitotoxin. Angew. Chem. Int. Ed. 2007, 46, 8875–8879. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Frederick, M.O.; Burtoloso, A.C.B.; Denton, R.M.; Rivas, F.; Cole, K.P.; Aversa, R.J.; Gibe, R.; Umezawa, T.; Suzuki, T. Chemical Synthesis of the GHIJKLMNO Ring System of Maitotoxin. J. Am. Chem. Soc. 2008, 130, 7466–7476. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A. Cross-Coupling Reactions of Organoboranes: An Easy Way to Construct C–C Bonds. Angew. Chem. Int. Ed. 2011, 50, 6723–6737. [Google Scholar] [CrossRef]
- Sasaki, M.; Fuwa, H.; Inoue, M.; Tachibana, K. New Strategy for Convergent Synthesis of trans-Fused Polyether Frameworks Based on Palladium-Catalyzed Suzuki Cross-Coupling Reaction. Tetrahedron Lett. 1998, 39, 9027–9030. [Google Scholar] [CrossRef]
- Sasaki, M.; Fuwa, H.; Ishikawa, M.; Tachibana, K. A General Method for Convergent Synthesis of Polycyclic Ethers Based on Suzuki Cross-Coupling: Concise Synthesis of the ABCD Ring System of Ciguatoxin. Org. Lett. 1999, 1, 1075–1077. [Google Scholar] [CrossRef]
- Sasaki, M.; Fuwa, H. Convergent Strategies for the Total Synthesis of Polycyclic Ether Marine Metabolites. Nat. Prod. Rep. 2008, 25, 401–426. [Google Scholar] [CrossRef] [PubMed]
- Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Ketones Using a Formic Acid–Triethylamine Mixture. J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar] [CrossRef]
- Achmatowicz, O., Jr.; Bukowsiki, P.; Szechner, B.; Zwierzchowska, Z.; Zamojski, A. Synthesis of Methyl 2,3-Dideoxy-DL-alk-2-enopyranosides from Furan Compounds: A General Approach to the Total Synthesis of Monosaccharides. Tetrahedron 1971, 27, 1973–1996. [Google Scholar] [CrossRef]
- Luche, J.L. Lanthanides in Organic Chemistry. 1. Selective 1,2-Reductions of Conjugated Ketones. J. Am. Chem. Soc. 1978, 100, 2226–2227. [Google Scholar] [CrossRef]
- Caron, M.; Sharpless, K.B. Titanium Isopropoxide-Mediated Nucleophilic Openings of 2,3-Epoxy Alcohols. A Mild Procedure for Regioselective Ring-Opening. J. Org. Chem. 1985, 50, 1557–1560. [Google Scholar] [CrossRef]
- Barder, T.E.; Walker, S.D.; Martinelli, J.R.; Buchwald, S.L. Catalysts for Suzuki–Miyaura Coupling Processes: Scope and Studies of the Effect of Ligand Structure. J. Am. Chem. Soc. 2005, 127, 4685–4696. [Google Scholar] [CrossRef] [PubMed]
- Dess, D.B.; Martin, J.C. A Useful 12-I-5 Triacetoxyperiodinane (the Dess–Martin Periodinane) for the Selective Oxidation of Primary or Secondary Alcohols and a Variety of Related 12-I-5 Species. J. Am. Chem. Soc. 1991, 113, 7277–7287. [Google Scholar] [CrossRef]
- Bourdelais, A.J.; Jacocks, H.M.; Wright, J.L.C.; Bigwarfe, P.M., Jr.; Baden, D.G. A New Polyether Ladder Compound Produced by the Dinoflagellate Karenia brevis. J. Nat. Prod. 2005, 68, 2–6. [Google Scholar] [CrossRef] [Green Version]
- Mattei, C.; Wen, P.J.; Nguyen-Huu, T.D.; Alvarez, M.; Benoit, E.; Bourdelais, A.J.; Lewis, R.J.; Baden, D.G.; Molgó, J.; Meunier, F.A. Brevenal Inhibits Pacific Ciguatoxin-1B-Induced Neurosecretion from Bovine Chromaffin Cells. PLoS ONE 2008, 3, e3448. [Google Scholar] [CrossRef] [Green Version]
- Nguyen-Huu, T.D.; Mattei, C.; Wen, P.J.; Bourdelais, A.J.; Lewis, R.J.; Benoit, E.; Baden, D.G.; Molgó, J.; Meunier, A. Ciguatoxin-Induced Catecholamine Secretion in Bovine Chromaffin Cells: Mechanism of Action and Reversible Inhibition by Brevenal. Toxicon 2010, 56, 792–796. [Google Scholar] [CrossRef]
- Abraham, W.M.; Bourdelais, A.J.; Sabater, J.R.; Ahmed, A.; Lee, T.A.; Serebriakov, I.; Baden, D.G. Airway Responses to Aerosolized Brevetoxins in an Animal Model of Asthma. Am. J. Resp. Crit. Care Med. 2005, 171, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prasad, A.V.K.; Shimizu, Y. The Structure of Hemibrevetoxin-B: A New Type of Toxin in the Gulf of Mexico Red Tide Organism. J. Am. Chem. Soc. 1989, 111, 6476–6477. [Google Scholar] [CrossRef]
- Fuwa, H.; Ebine, M.; Sasaki, M. Total Synthesis of the Proposed Structure of Brevenal. J. Am. Chem. Soc. 2006, 128, 9648–9650. [Google Scholar] [CrossRef]
- Fuwa, H.; Ebine, M.; Bourdelais, A.J.; Baden, D.G.; Sasaki, M. Total Synthesis, Structure Revision, and Absolute Configuration of (−)-Brevenal. J. Am. Chem. Soc. 2006, 128, 16989–16999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ebine, M.; Fuwa, H.; Sasaki, M. Total Synthesis of (−)-Brevenal: A Concise Synthetic Entry to the Pentacyclic Polyether Core. Org. Lett. 2008, 10, 2275–2278. [Google Scholar] [CrossRef]
- Ebine, M.; Fuwa, H.; Sasaki, M. Total Synthesis of (−)-Brevenal: A Streamlined Strategy for Practical Synthesis of Polycyclic Ethers. Chem. Eur. J. 2011, 17, 13754–13761. [Google Scholar] [CrossRef]
- Stille, J.K. The Palladium-Catalyzed Cross-Coupling Reactions of Organotin Reagents with Organic Electrophiles. Angew. Chem. Int. Ed. Engl. 1986, 25, 508–524. [Google Scholar] [CrossRef]
- Evans, D.A.; Bartroli, J.; Shih, T.L. Enantioselective Aldol Condensations. 2. Erythro-Selective Chiral Aldol Condensations via Boron Enolates. J. Am. Chem. Soc. 1981, 103, 2127–2129. [Google Scholar] [CrossRef]
- Prashad, M.; Har, D.; Kim, H.-Y.; Repic, O. A New, Economical, Practical and Racemization-Free Method for the Reductive Removal of 2-Oxazolidinones from N-Acyloxazolidinones with Sodium Borohydride. Tetrahedron Lett. 1998, 39, 7067–7070. [Google Scholar] [CrossRef]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A Rapid Esterification by Means of Mixed Anhydride and Its Application to Large-Ring Lactonizaiton. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar] [CrossRef] [Green Version]
- Kadota, I.; Ohno, A.; Matsukawa, Y.; Yamamoto, Y. Synthesis of the H Ring of Gambierol. Tetrahedron Lett. 1998, 39, 6373–6376. [Google Scholar] [CrossRef]
- Hori, N.; Matsukura, H.; Matsuo, G.; Nakata, T. Efficient Strategy for the Iterative Synthesis of trans-Fused Polycyclic Ether via SmI2-Induced Reductive Intramolecular Cyclization. Tetrahedron 2002, 58, 1853–1864. [Google Scholar] [CrossRef]
- Nakata, T. SmI2-Induced Cyclizations and Their Applications in Natural Product Synthesis. Chem. Rec. 2010, 10, 159–172. [Google Scholar] [CrossRef]
- Ley, S.V.; Norman, J.; Griffith, W.P.; Marsden, S.P. Tetrapropylammonium Perruthenate, Pr4N+RuO4−, TPAP: A Catalytic Oxidant for Organic Synthesis. Synthesis 1994, 639–666. [Google Scholar] [CrossRef]
- Fuwa, H.; Sasaki, M.; Tachibana, K. Synthetic Studies on a Marine Polyether Toxin, Gambierol: Stereoselective Synthesis of the FGH Ring System via B-Alkyl Suzuki Coupling. Tetrahedron Lett. 2000, 41, 8371–8375. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Prasad, C.V.C.; Hwang, C.-K.; Duggan, M.E.; Veale, C.A. Cyclizations of Hydroxy Dithioketals. New Synthetic Technology for the Construction of Oxocenes and Related Medium-Ring Systems. J. Am. Chem. Soc. 1989, 111, 5321–5330. [Google Scholar] [CrossRef]
- Fleming, I.; Newton, T.W.; Roessler, F. The Silylcupration of Acetylenes: A Synthesis of Vinylsilanes. J. Chem. Soc. Perkin Trans. 1 1981, 2527–2532. [Google Scholar] [CrossRef]
- Stamos, D.P.; Taylor, A.G.; Kishi, Y. A Mild Preparation of Vinyliodides from Vinylsilanes. Tetrahedron Lett. 1996, 37, 8647–8650. [Google Scholar] [CrossRef]
- Allred, G.D.; Liebeskind, L.S. Copper-Mediated Cross-Coupling of Organostannanes with Organic Iodides at or below Room Temperature. J. Am. Chem. Soc. 1996, 118, 2748–2749. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Reddy, K.R.; Skokotas, G.; Sato, F.; Xiao, X.-Y.; Hwang, C.-K. Total Synthesis of Hemibrevetoxin B and (7aα)-epi-Hemibrevetoxin B. J. Am. Chem. Soc. 1993, 115, 3558–3575. [Google Scholar] [CrossRef]
- Inoue, M.; Hirama, M.; Satake, M.; Sugiyama, K.; Yasumoto, T. Inhibition of brevetoxin binding to the voltage-gated sodium channel by gambierol and gambieric acid-A. Toxicon 2003, 41, 469–474. [Google Scholar] [CrossRef]
- Sakamoto, B.; Nagai, H.; Hokama, Y. Stimulators of Gambierdiscus toxicus (Dinophyceae) Growth: The Possible Role of Gambieric Acid-A as an Endogenous Growth Enhancer. Phycologia 1996, 35, 350–353. [Google Scholar] [CrossRef]
- Fuwa, H.; Suzuki, A.; Sato, K.; Sasaki, M. Stereoselective Synthesis of the AB-Ring Fragment of Gambieric Acid A. Heterocycles 2007, 72, 139–144. [Google Scholar] [CrossRef]
- Fuwa, H.; Goto, T.; Sasaki, M. Stereocontrolled Synthesis of the A/B-Ring Fragment of Gambieric Acid B: Reassignment of the Absolute Configuration of the Polycyclic Ether Region. Org. Lett. 2008, 10, 2211–2214. [Google Scholar] [CrossRef]
- Fuwa, H.; Ishigai, K.; Goto, T.; Suzuki, A.; Sasaki, M. Synthetic Studies on Gambieric Acids, Potent Antifungal Polycyclic Ether Natural Products: Reassignment of the Absolute Configuration of the Nonacyclic Polyether Core by NMR Analysis of Model Compounds. J. Org. Chem. 2009, 74, 4024–4040. [Google Scholar] [CrossRef] [PubMed]
- Fuwa, H.; Kainuma, N.; Tachibana, K.; Sasaki, M. Total Synthesis of (−)-Gambierol. J. Am. Chem. Soc. 2002, 124, 14983–14992. [Google Scholar] [CrossRef]
- Takano, S.; Samizu, K.; Sugihara, T.; Ogasawara, K. Asymmetric Construction of Optically Active 3-Hydroxyalkyne Functionalies. J. Chem. Soc. Chem. Commun. 1989, 1344–1345. [Google Scholar] [CrossRef]
- Myers, A.G.; Zheng, B.; Movassaghi, M. Preparation of the Reagent o-Nitrobenzenesulfonylhydrazide. J. Org. Chem. 1997, 62, 7507. [Google Scholar] [CrossRef]
- Ohtani, I.; Kusumi, T.; Kashman, Y.; Kakisawa, H. High-Field FT NMR Application of Mosher’s Method. The Absolute Configurations of Marine Terpenoids. J. Am. Chem. Soc. 1991, 113, 4092–4096. [Google Scholar] [CrossRef]
- Araki, K.; Saito, K.; Arimoto, H.; Uemura, D. Enantioselective Synthesis of the Spirotetracyclic Carbon Core of Mangicols by Using a Stereoselective Transannular Diels–Alder Strategy. Angew. Chem. Int. Ed. 2004, 43, 81–84. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J.A.; Johns, B.A. Total Synthesis of (+)-Discodermolide. J. Org. Chem. 1998, 63, 7885–7892. [Google Scholar] [CrossRef]
- Fuwa, H.; Ishigai, K.; Hashizume, K.; Sasaki, M. Total Synthesis and Complete Stereostructure of Gambieric Acid A. J. Am. Chem. Soc. 2012, 134, 11984–11987. [Google Scholar] [CrossRef] [PubMed]
- Ishigai, K.; Fuwa, H.; Hashizume, K.; Fukazawa, R.; Cho, Y.; Yotsu-Yamashita, M.; Sasaki, M. Total Synthesis and Biological Evaluation of (+)-Gambieric Acid A and Its Analogues. Chem. Eur. J. 2013, 19, 5276–5288. [Google Scholar] [CrossRef]
- Sun, A.W.; Lackner, S.; Stolz, B.M. Modularity: Adding New Dimensions to Total Synthesis. Trends Chem. 2019, 1, 630–643. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Hakamata, A.; Tsuda, M.; Fuwa, H. Total Synthesis and Stereochemical Revision of Iriomoteolide-2a. Angew. Chem. Int. Ed. 2018, 57, 3801–3805. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, K.; Hakamata, A.; Iwasaki, A.; Suenaga, K.; Tsuda, M.; Fuwa, H. Total Synthesis, Stereochemical Revision, and Biological Assessment of Iriomoteolide-2a. Chem. Eur. J. 2019, 25, 8528–8542. [Google Scholar] [CrossRef]
- Kato, S.; Mizukami, D.; Sugai, T.; Tsuda, M.; Fuwa, H. Total Synthesis and Complete Configurational Assignment of Amphirionin-2. Chem. Sci. 2021, 21, 872–879. [Google Scholar] [CrossRef]
- Lodewyk, M.W.; Siebert, M.R.; Tantillo, D.J. Computational Prediction of 1H and 13C Chemical Shifts: A Useful Tool for Natural Product, Mechanistic, and Synthetic Organic Chemistry. Chem. Rev. 2012, 112, 1839–1862. [Google Scholar] [CrossRef]
- Marcarino, M.O.; Zanardi, M.M.; Cicetti, S.; Sarotti, A.M. NMR Calculations with Quantum Methods: Development of New Tools for Structural Elucidation and Beyond. Acc. Chem. Res. 2020, 53, 1922–1932. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.G.; Goodman, J.M. Assigning Stereochemistry to Single Diastereomers by GIAO NMR Calculation: The DP4 Probability. J. Am. Chem. Soc. 2010, 132, 12946–12959. [Google Scholar] [CrossRef]
- Rodríguez, I.; Genta-Jouve, G.; Alfonso, C.; Calabro, K.; Alonso, E.; Sánchez, J.A.; Alfonso, A.; Thomas, O.P.; Botana, L.M. Gambierone, a Ladder-Shaped Polyether from the Dinoflagellate Gambierdiscus belizeanus. Org. Lett. 2015, 17, 2392–2395. [Google Scholar] [CrossRef] [PubMed]
- Hamamoto, Y.; Tachibana, K.; Holland, P.T.; Shi, F.; Beuzenberg, V.; Itoh, Y.; Satake, M. Brevisulcenal-F: A Polycyclic Ether Toxin Associated with Massive Fish-Kills in New Zealand. J. Am. Chem. Soc. 2012, 134, 4963–4968. [Google Scholar] [CrossRef]
- Igarashi, T.; Satake, M.; Yasumoto, T. Prymnesin-2: A Potent Ichthyotoxic and Hemolytic Glycoside Isolated from the Red Tide Alga Prymnesium parvum. J. Am. Chem. Soc. 1996, 118, 479–480. [Google Scholar] [CrossRef]
- Sasaki, M.; Ebine, M.; Takagi, H.; Takakura, H.; Shida, T.; Satake, M.; Oshima, Y.; Igarashi, T.; Yasumoto, T. Synthesis of the CDE/FG Ring Models of Prymnesins: Reassignment of the Relative Configuration of the E/F Ring Juncture. Org. Lett. 2004, 6, 1501–1504. [Google Scholar] [CrossRef]
















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fuwa, H. Synthesis-Driven Stereochemical Assignment of Marine Polycyclic Ether Natural Products. Mar. Drugs 2021, 19, 257. https://doi.org/10.3390/md19050257
Fuwa H. Synthesis-Driven Stereochemical Assignment of Marine Polycyclic Ether Natural Products. Marine Drugs. 2021; 19(5):257. https://doi.org/10.3390/md19050257
Chicago/Turabian StyleFuwa, Haruhiko. 2021. "Synthesis-Driven Stereochemical Assignment of Marine Polycyclic Ether Natural Products" Marine Drugs 19, no. 5: 257. https://doi.org/10.3390/md19050257
APA StyleFuwa, H. (2021). Synthesis-Driven Stereochemical Assignment of Marine Polycyclic Ether Natural Products. Marine Drugs, 19(5), 257. https://doi.org/10.3390/md19050257