1. Introduction
Metabolites of polyketide pathways are widely recognized as an important class of fungal natural products due to their significant structural and functional diversities, as well as their wide taxonomical distribution [
1,
2]. Attributed from the potent bioactivity, several fungal polyketides have been used as lead compounds in the pharmaceutical industry, which culminates to occupy 20% of the major commercial drugs by these compounds, such as lovastatin and griseofulvin [
3,
4,
5]. Tetraketide-derived diphenyl ethers are structurally distinctive from other subclasses of fungal polyketides in terms of their formation by the condensation of two polyketide derivatives [
6]. Amidst their wide natural distribution, diphenyl ethers are frequently obtained from marine microorganisms, especially from filamentous fungi of the genera
Aspergillus and
Penicillium [
7,
8]. These compounds possess diverse bioactivities, including antibacterial, cytotoxic, enzyme inhibitory and antiviral activities [
9]. Accordingly, these compounds are increasingly applied in cosmetics, pharmaceutical intermediates and chemical products, which in turn have led to extensive studies on their biosynthesis [
9]. Recently, a detailed biosynthetic pathway with the gene cluster and key enzyme system was unveiled for diorcinol, which is a representative of fungal diphenyl ethers, in a marine-derived
A. nidulans strain [
6,
9].
In the process of searching for bioactive compounds from marine-derived fungi, we recently reported the structures of two new sortase A-inhibitory depsihexapeptides from an
Aspergillus ochraceopetaliformis fungus, collected from underwater sediment off the coast of Jeju-do, Korea [
10]. However, LC–UV and LC–ESI–MS analyses, aided by an in-house spectral library of fungal extracts, revealed the presence of a natural product with distinct structural motifs in a liquid culture broth of this strain. More interestingly, the same analyses of this strain from a rice-based semi-solid culture broth revealed the presence of a structurally related but fragmented compound, prompting an extensive chemical investigation.
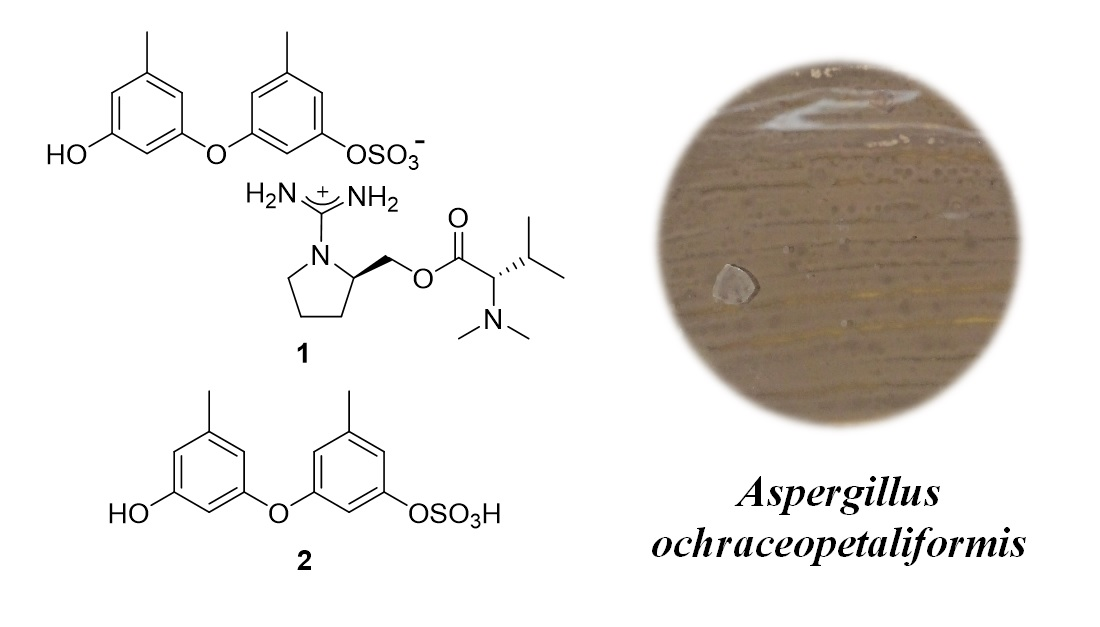
Herein, we report the structures of ochraceopetalin (
1), a novel sulfonated diphenylether-aminol-amino acid guanidinium salt and its aromatic component (
2) (
Figure 1). In the process of isolation and structure determination, ochraceopetaguanidine (
3), a guanidine-bearing aminol amino acid, was structurally elucidated as the other component of
1. Organic salts coupled with organic counterions are occasionally found in marine organisms: psammaplins, bromotyrosines and dihydroxystyrenes, from an association of
Poecillastra sp. and
Jaspis sp. sponges [
11,
12,
13]; suvanines, from a
Coscinoderma mathewsi sponge [
14]; sesterterpene sulfates, from a
Ircinia sp. sponge [
15]; iodotyramine derivative, from the
Didemnum rubem ascidian [
16,
17]. To the best of our knowledge,
1 would be the first of this kind to be obtained from marine-derived fungi. Furthermore, its mixed-biogenetic salt formation, that is derived from polyketide and amino acid pathways, would be remarkably unprecedented. Compound
1 exhibited noticeably stronger cytotoxicity toward the human cancer cell lines K562 and A549 than the other compounds.
2. Results and Discussion
The molecular formula of compound 1 obtained from the YMM liquid culture broth was deduced to be C27H40N4O8S, with 11 degrees of unsaturation, by positive HR–ESI–MS analysis ([M + H]+ m/z 581.2628, calcd 581.2640). This positive mode MS analysis also gave a highly conspicuous cluster of C13H27N4O2 (m/z 271.2123, calcd 271.2129), while a similar analysis in negative mode gave a conspicuous cluster of C14H13O6S (m/z 309.0433, calcd 309.0427), whose sum corresponded well with the molecular formula of 1. This phenomenon of significant discrepancy attributed from the opposite MS ionization modes strongly suggested the presence of ionized partial structures, thus the salt nature of 1. In addition, a strong absorption band was observed in the IR spectrum at 1456 cm−1, which corresponds with a sulfate group, as suggested by the MS-indicated sulfur atom.
The
13C NMR data of compound
1 showed signals of a carbonyl carbon (δ
C 170.5) and thirteen sp
2 carbons (δ
C 160.2–102.7) (
Table 1). Based on the combined
1H NMR and HSQC data, the chemical shifts and splitting patterns (δ
H 6.74–6.16, all s) of corresponding protons were indicative of aromatic moieties. In addition, the odd numbers of sp
2 carbons suggested the presence of an electron-deficient functionality, such as an imine or guanidine. The NMR data of
1 also contained three sp
3 methines (δ
C/δ
H 73.3/2.74, 53.0/3.63 and 26.8/1.94), of which the former two carbons would bear nitrogen, according to their carbon and proton chemical shifts. Among the remaining four aliphatic methylene and six methyl signals in the NMR data, the carbon and proton chemical shifts indicated the presence of an oxymethylene (δ
C/δ
H 64.4/4.19 and 4.12), a nitrogenous methylene (δ
C/δ
H 42.7/3.30 and 3.05) and two nitrogenous methyls (δ
C/δ
H 41.0/2.21 × 2).
The gross structure of
1 was determined by combined
1H-
1H COSY and HMBC experiments. First, all of the aromatic protons (δ
H 6.74–6.16, all s) lacked direct proton–proton couplings, placing them at meta- or para-positions to each other. The HMBC correlations of these and two benzylic methyl protons (δ
H 2.23, H
3-13 and δ
H 2.18, H
3-14) with neighboring carbons readily constructed two 1,3-dioxygenated-5-methyl-benzene moieties (C-1–C-6 and C-13, C-7–C-12 and C-14), accounting for eight degrees of unsaturation (
Figure 2). Despite the identical proton–carbon correlations, the noticeable differences in both the
1H and
13C chemical shifts revealed different oxygenated functionalities between aromatic rings. Aided by the negative mode ESI–MS cluster of C
14H
13O
6S and the IR absorption band of the sulfate group at 1456 cm
−1, the partial structure was interpreted as 9-hydroxy-1-(sulfooxy)-3,7-diphenylether, which is discussed later.
Meanwhile, the COSY data showed the proton spin couplings of an isobutyl group (δC/δH 73.3/2.74, 26.8/1.94, 19.0/0.82 and 19.5/0.92; C-7′–C-10′) which was confirmed by the HMBC correlations among these. Then, additional proton–carbon correlations of the C-7′ methine and C-8′ methine groups placed a carbonyl (δC 170.5, C-6′) and two methyl groups (δC/δH 41.0/2.21 × 2, C-11′ and C-12′) at the adjacent positions. Because the latter methyl groups were determined to be N-CH3 by their chemical shifts, nitrogen must be connected to the methyl groups and C-7′. Thus, an N,N-dimethyl-valine residue was identified. The COSY data also showed a linear array of four methylenes and a methine (δC/δH 42.7/3.30 and 3.05, 24.8/1.78 and 1.57, 29.0/1.78 and 1.57, 53.0/3.63 and 64.4/4.19 and 4.12; C-1′–C-5′). Aided by their 1H and 13C NMR chemical shifts and the crucial HMBC correlation at H2-1′/C-4′, a pyrrolidine moiety was identified for C-1′–C-4′. Similarly, the C-5′ methylene was connected to the C-6′ carbonyl by an ester linkage, according to the HMBC correlation at H2-5′/C-6′. Thus, a prolinol moiety was defined for the C-1′–C-5′ portion.
The 13C NMR data of 1 had a remaining nonprotonated carbon (δC 160.2), which was placed at the prolinol nitrogen (C-13′) by the crucial HMBC correlation with H2-1′. The significant deshielding of this carbon must be attributed to the remaining two nitrogen atoms from the MS data. Thus, in conjunction with the C13H27N4O2 cluster observed in the positive ESI–MS analysis, the functionality of C-13′ was assigned to the guanidinium group. Thus, the C-1′–C-13′ portion was defined as 1-(aminoiminomethyl)-prolinol, N,N-dimethylvaline ester and the overall structure of 1, designated as ochraceopetalin, was determined to be a mixed-biogenetic salt.
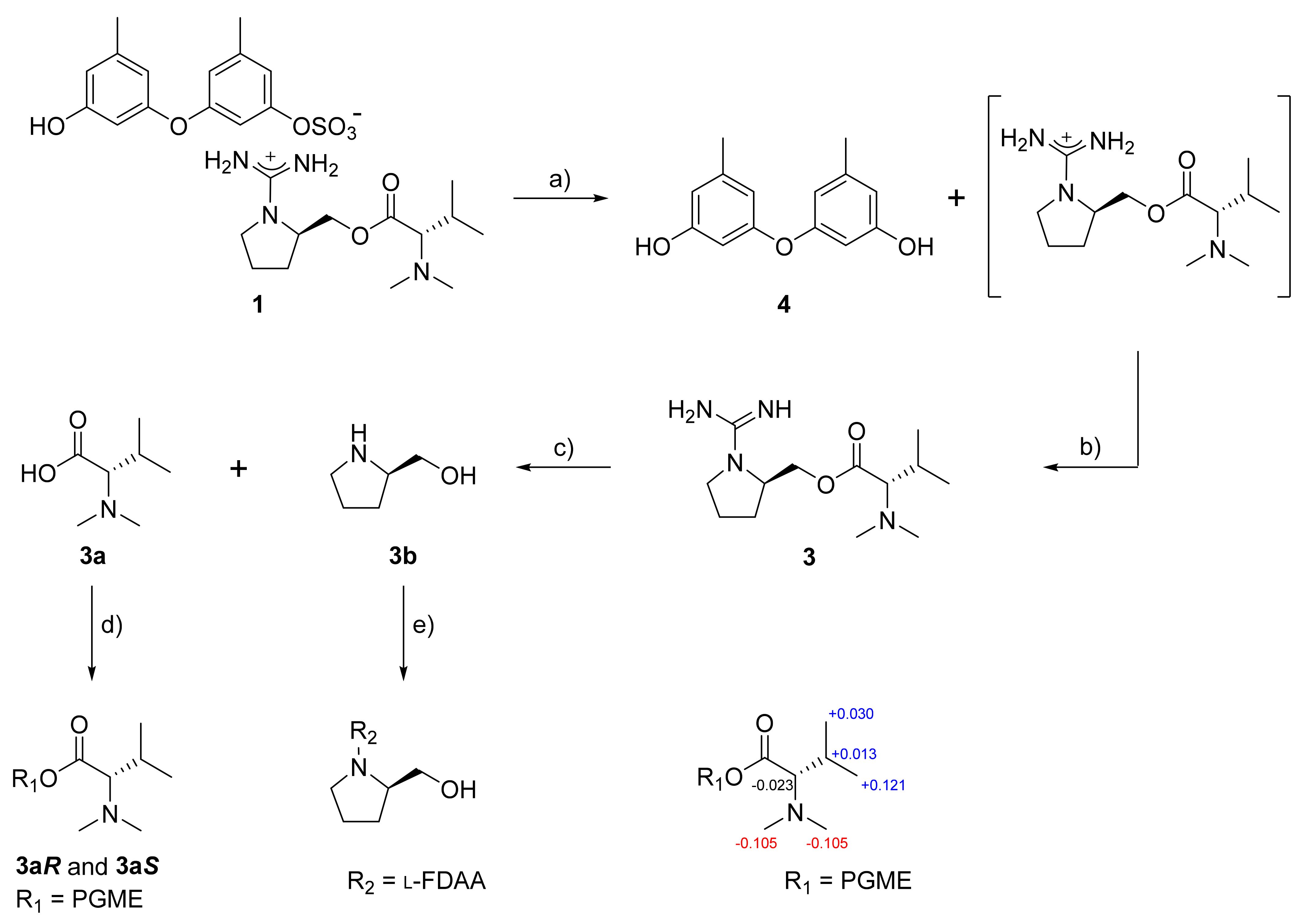
The proposed structure of
1, including the salt nature, was confirmed by a series of pH-dependent degradations (
Figure 3). When
1 was placed in a mildly acidic solution, a compound separated into an organic layer. A combination of HR–ESI–MS and 1D- and 2D-NMR analyses readily identified this compound as diorcinol (
4), a well-known 3,3′-oxybis [5-methylphenol] of diverse fungal origins [
4,
6,
8]. Changing the pH of the remaining aqueous solution to basic conditions brought another compound (
3) into an organic layer. The molecular formula of this compound was deduced to be C
13H
26N
4O
2 by HR–FAB–MS analysis ([M + H]
+ m/z 271.2135, calcd 271.2129) and the positive mode MS analysis of
3 was identical to that of
1. Furthermore, the combined 1D- and 2D-NMR data of
3 were identical to those of the cation portion of
1, which was designated as ochraceopetaguanidine and identified as 1-(aminoiminomethyl)-prolinol,
N,N-dimethylvaline ester. Thus, the structure of
1 was unambiguously defined as a mixed-biogenetic salt derived from polyketide and amino acid pathways. The sulfate [
18,
19] and guanidinium salts of
1 was also supported by the comparison of
13C NMR data between the simple salts bearing these functionalities and corresponding neutral groups (
Figure S27). Compound
1 as a single salt compound was also supported by extensive HPLC analyses, in which
1 was always eluted as single peak under diverse chromatographic conditions (
Figure S28).
The production of
1 was further investigated under different culture conditions. After culturing the same strain in a static semi-solid YMM-rice medium, chromatographic separation of the culture broth yielded compound
2. The molecular formula of
2 was defined to be C
14H
14O
6S by HR–ESI–MS analysis ([M − H]
− m/z 309.0433, calcd 309.0427), which was identical to the data of
1 in the negative mode MS analysis. Furthermore, the 1D- and 2D-NMR data of
2 were identical to the negative ion portion of
1 (
Table 1). In addition,
2 was slowly converted to diorcinol (
4) during prolonged storage, emphasizing their structural relationship. Thus, the structure of
2 was determined to be 1-(sulfooxy)-diorcinol, a sulfate-bearing diphenyl ether of polyketide origin.
Ochraceopetalin (
1) and ochraceopetaguanidine (
3) commonly bear two stereogenic centers in the amino acid portion. For the configurational assignments of these, firstly, a basic hydrolysis of
3 gave
N,N-dimethylvaline and prolinol. The absolute configuration at C-7′ of the valine residue was assigned using the PGME method [
20]. Treatments with (
S)- and (
R)-PGME produced the corresponding PGME amides, respectively. The Δδ
H values between these unambiguously assigned a
d-configuration (
Figure 3). For the C-4′ center of prolinol, the absolute configuration was assigned by using the Marfey reaction [
21]. After the
l-FDAA-prolinol adduct was prepared by condensation with
l-FDAA, its HPLC retention time was compared with those of authentic
l-FDAA-
l-prolinol and
l-FDAA-
d-prolinol, which resulted in assigning the
d-configuration. Thus, both amino acid-derived units of
1 were found to possess
d-configurations. In addition, the guanidinium salt of
1 was supported by the
13C NMR data of
3c, the TFA salt of
3, whose guanidinium bearing C-13′ showed very similar chemical shift with
3 (δ
C 161.47 and 160.39 for
3c and
3, respectively) (
Figure S29). At the same time, the carbon chemical shifts of C-13′ of
1 and
3 also showed similarity with 0.15 ppm and 0.13 ppm differences in DMSO-
d6 (δ
C 160.24 and δ
C 160.39 for
1 and
3, respectively) and MeOH-
d4 solvent (δ
C 161.95 and δ
C 162.08 for
1 and
3, respectively), respectively (
Figures S30–S31, Table S2).
In addition to the structure determination, the structure of
1 and
2 led to an interesting argument. Diorcinol (
4) is widely recognized as a natural product of diverse fungi, such as
Aspergillus versicolor GH-2 [
22],
A. tennesseensis [
23] and
Cordyceps sp. [
24]. The detailed biosynthesis via orsellinic acid, including the gene cluster and enzymatic reactions, was recently determined for a marine-derived
A. nidulans [
6,
9]. However, our results of finding
1 and
2 consecutively (eight times of cultivation) raise the possibility that, in the case of this strain (strain number FJ120) of
A. ochraceopetaliformis, the natural product of the fungus is not diorcinol (
4), but 1-(sulfooxy)-diorcinol (
2), or its salt (
1). Alternatively, it would be possible that naturally produced
4 was sulfonated to
1 or
2 by a biotransformation during the cultivation. It would be also possible to transform
1 by an abiotic process during the cultivation or isolation procedures. To clarify this, time-dependent cultivation and ESI–MS analyses of the broths revealed two interesting phenomena. The first is that
1 and
2 were independently produced after 8 and 6 weeks of cultivation, respectively, under given culturing conditions, while the production of
4 was undetected by LC–ESI–MS analysis (
Figures S24 and S25). The second interesting phenomenon is that distinct products according to the culturing conditions could be attributed to the differences in pH in the culture media,
1, pH 9.0, and
2, pH 5.0. This question of biosynthesis of diorcinol derivatives would be fully answered only by the extensive biosynthetic study on this strain.
Compounds
1–
4 were tested using a number of bioassays. In cytotoxicity tests, all four compounds were either significantly inhibitory (
1, IC
50 9.5 and 6.8 μM), or less active (
2–
4, IC
50 11–25 μM) against the human cancer cell lines K562 and A549 (
Figure S26 and Table S1). Although its cytotoxicity was not remarkable,
1 was noticeably more potent than its components
2–
4. These microbial compounds failed to inhibit both Gram-positive and Gram-negative bacteria and pathogenic fungi (MIC > 128 μg/mL). They were also inactive against the microbial enzymes sortase A (SrtA), a transpeptidase responsible for the anchoring of surface proteins to the cell wall envelope of Gram-positive bacteria and isocitrate lyase (ICL), a key enzyme in the glyoxylate cycle.
In summary, ochraceopetalin (1), a mixed-biogenetic salt compound and its component 2 were isolated from a marine-derived fungus Aspergillus ochraceopetaliformis. Based on the results of combined spectroscopic and chemical analyses, the structure of 1 was determined to be a sulfonated diphenylether-aminol-amino acid guanidinium of an unprecedented structural class. Compounds 2 and 3, obtained from the different culturing condition and chromatographic processes, respectively, were defined to be the corresponding components of 1, 1-(sulfooxy)-diorcinol and 1-(aminoiminomethyl)-prolinol, N,N-dimethylvaline ester, respectively. Compound 1 exhibited significant cytotoxicity against K562 and A549 cells.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured on a JASCO P1020 polarimeter (Jasco, Tokyo, Japan) using a 1 cm cell. UV spectra were acquired with a Hitachi U-3010 spectrophotometer (Hitachi High-Technologies, Tokyo, Japan). IR spectra were recorded on a JASCO 4200 FT-IR spectrometer (Jasco, Tokyo, Japan) using a ZnSe cell. 1H and 13C NMR spectra were measured in DMSO-d6, CDCl3, or MeOH-d4 solutions on Bruker Avance -400, -500, -600, or -800 instruments (Bruker, Billerica, MA, USA), with solvent peaks at δH 2.50/δC 39.50, δH 7.26/δC 77.16 and δH 3.31/δC 49.00, respectively, as their internal standards. High-resolution ESI mass spectrometric data were obtained at the National Instrumentation Center for Environmental Management (Seoul, Korea) and were acquired using an AB Sciex 5600 QTOF HR-MS instrument (Sciex, Washington, DC, USA). High-resolution FAB mass spectrometric data were obtained at the Korea Basic Science Institute (Daegu, Korea) and were acquired using a JEOL JMS 700 mass spectrometer (Jeol, Tokyo, Japan) with meta-nitrobenzyl alcohol (NBA) as the matrix. Low-resolution ESI–MS data were recorded on an Agilent Technologies 6130 quadrupole mass spectrometer with an Agilent Technologies 1200 series HPLC. Semi-preparative and analytical HPLC separations were performed on a Spectrasystem p2000 equipped with a Spectrasystem RI-150 refractive index detector and a UV-Vis-151 detector (Gilson, Middleton, WI, USA). All the solvents used were of spectroscopic grade or distilled from glass prior to use.
3.2. Fungal Material
The fungal strain
Aspergillus ochraceopetaliformis (strain number FJ120) was isolated from marine sediments collected from Jeju-do, Korea, in July 2007 [
10]. The isolate was identified using standard molecular biology protocols by DNA amplification and sequencing of the ITS region. Genomic DNA extraction was performed using Intron’s i-genomic BYF DNA Extraction Mini Kit, according to the manufacturer’s protocol. The nucleotide sequence was deposited in the GenBank database under accession number KF384187. The 18S rDNA sequence of this strain exhibited 100% identity (588/588) with that of
Aspergillus ochraceopetaliformis strain RKI08-134 (GenBank accession number FJ797698).
3.3. Fermentation
The fungal strain was cultured on solid YPG media (5 g of yeast extract, 5 g of peptone, 10 g of glucose, 24.8 g of sea salt and 16 g of agar in 1 L of distilled water) for 7 days. An agar plug (1 cm × 1 cm) was inoculated in 250 mL flask containing 100 mL of YPG media. After 7 days of growth, 10 mL of each culture was transferred to 2.8 L Fernbach flasks containing YMM media (5 g of yeast extract, 5 g of malt extract, 10 g of mannitol and 24.8 g of sea salt in 1000 mL of distilled water). In total, 30 L of YMM media was prepared and cultivated under static conditions for 8 weeks at 30 °C.
The large-scale cultivation of YPG-based seed culture was also performed on semi-solid YMM-rice media (1 g of yeast extract, 1 g of malt extract, 2 g of mannitol and 200 g of rice in 200 mL of artificial seawater) in 2.8 L Fernbach flasks at 30 °C in the static condition for 6 weeks.
3.4. Extraction and Isolation
The entire culture was filtered and extracted with EtOAc (20 L × 3). The solvent was evaporated in vacuo to afford a brown organic gum (4.3 g). The extract was separated by C18 reversed-phase vacuum flash chromatography using sequential mixtures of H2O and MeOH (five fractions of H2O-MeOH, gradient from 80:20 to 0:100), acetone and, finally, EtOAc as the eluents. Based on the results of 1H NMR and LR–ESI–MS analyses, the fraction eluted with H2O-MeOH 40:60 (850 mg) was subjected to semi-preparative reversed-phase HPLC (YMC-ODS-A column, 250 × 10 mm; H2O-MeOH, 50:50, 1.7 mL/min), affording compound 1 (tR = 29.1 min). Compound 1 was further purified by analytical HPLC (YMC-ODS-A column, 250 × 4.6 mm; H2O-MeCN, 75:25, 0.7 mL/min; tR = 22.0 min; 11.1 mg).
The broth (4.2 kg) from semi-solid YMM-rice media was extracted with methanol (21 L × 3) and dichloromethane (21 L × 3). The combined extracts (111.1 g) were dried in vacuo and partitioned between H2O (84.6 g) and n-BuOH (26.4 g). The organic layer was repartitioned between H2O-MeOH (15:85, 14.2 g) and n-hexane (12.2 g). The H2O-MeOH layer was evaporated to obtain an organic extract (14.2 g). The extract was fractionated by C18 reversed-phase vacuum flash chromatography using a sequential mixture of MeCN and H2O as eluents (H2O-MeCN, from 80:20 to 50:50), MeOH, acetone and, finally, EtOAc. On the basis of the result of 1H NMR and LC-MS profile, the fraction (1240 mg) eluted with H2O-MeCN (70:30) was purified by semi-preparative reversed-phase HPLC (YMC-ODS-A column, 250 × 10 mm; H2O-MeOH, 55:45, 1.7 mL/min) to yield compound 2 (tR = 34.5 min). Compound 2 was further purified by analytical HPLC (YMC-ODS-A column, 250 × 4.6 mm; H2O-MeCN, 80:20, 0.7 mL/min; tR = 15.6 min; 13.7 mg).
Ochraceopetalin (
1): white amorphous solid; [α]
25D -5 (
c 0.4, MeOH); UV (MeOH) λ
max (log ε) 209 (3.17), 275 (2.01) nm; IR (ZnSe) ν
max 3347 (br), 2946, 1456, 1031 cm
−1;
1H and
13C NMR data,
Table 1; HR–ESI–MS
m/z 581.2628 [M + H]
+ (calcd for C
27H
41N
4O
8S, 581.2640).
1-(Sulfooxy)-diorcinol (
2): yellow amorphous solid; UV (MeOH) λ
max (log ε) 210 (3.16), 275 (2.19) nm; IR (ZnSe) ν
max 3359 (br), 2833, 1455, 1033 cm
−1;
1H and
13C NMR data,
Table 1; HR–ESI–MS
m/z 309.0437 [M − H]
− (calcd for C
14H
13O
6S, 309.0438).
3.5. pH-Dependent Hydrolysis of Compound 1
Firstly, compound 1 (9.7 mg) was dissolved in water (0.05% TFA, 5 mL). The solution was stood at room temperature for 3 h. The aqueous solution was partitioned with n-BuOH (5 mL) through a separate funnel. Removal of n-BuOH in vacuo yielded pure compound 4 (3.5 mg). To the aqueous fraction, 0.05% of NH4OH (5 mL) was added. After removing water, purification by analytical HPLC (YMC-ODS-A column, 250 × 4.6 mm; 0.7 mL/min; H2O-MeCN, 70:30) afforded compound 3 (tR = 21.1 min; 4.2 mg).
Ochraceopetaguanidine (
3): white amorphous solid; [α]
25D -4 (
c 0.4, MeOH); UV (MeOH) λ
max (log ε) 205 (2.39) nm; IR (ZnSe) ν
max 3362 (br), 2360, 1636 cm
−1;
1H and
13C NMR data,
Table 1; HR-FAB-MS
m/z 271.2135 [M + H]
+ (calcd for C
13H
27N
4O
2, 271.2129).
Diorcinol (
4): colorless gum;
1H and
13C NMR spectra,
Figures S17 and S18; HR–ESI–MS
m/z 231.1012 [M + H]
+ (calcd for C14H15O3, 231.1016).
3.6. Basic Hydrolysis of Compound 3
Compound 3 (3.5 mg) was dissolved in 1 N NaOH (1 mL) and the solution was stirred at 100 °C for 3 h. The separation by analytical HPLC (YMC-ODS-A column, 250 × 4.6 mm; 0.7 mL/min; H2O-MeCN, 65:35) afforded compounds 3a (N,N-dimethylvaline, tR = 15.4 min; 2.2 mg) and 3b (prolinol, tR = 23.8 min; 2.8 mg) as pure compounds.
N,N-Dimethylvaline (3a): white amorphous solid; 1H NMR (DMSO-d6, 400 MHz) δH 3.16 (1H, br s, 6′-OH), 2.66 (1H, d, J = 9.1 Hz, H-7′), 2.29 (6H, s, H-11′, H-12′), 1.90 (1H, dhep, J = 9.1, 6.8 Hz, H-8′), 0.92 (3H, d, J = 6.6 Hz, H-10′), 0.83 (3H, d, J = 6.6 Hz, H-9′); LR–ESI–MS m/z 146.2 [M + H]+.
Prolinol (3b): pale yellow oil; 1H NMR (CDCl3, 400 MHz) δH 3.81 (2H, OH, NH), 3.37 (1H, dd, J = 11.0, 4.0 Hz, H-5′), 3.20 (1H, dd, J = 11.0, 7.0 Hz, H-5′), 3.02 (1H, m, H-4′), 2.71 (1H, m, H-1′), 2.68 (1H, m, H-1′), 1.63–1.52 (3H, m, H-2′,3′), 1.22 (1H, m, H-2′); LR–ESI–MS m/z 102.1 [M + H]+.
3.7. Preparation of (S)- and (R)-PGME Amides of 3a
To a dry DMF solution (500 μL) of compound 3a (0.5 mg, 6.9 mM) and (S)-PGME (1.5 mg, 33 mM), PyBOP (8.5 mg, 33 mM), HOBT (2.2 mg, 33 mM) and N-methylmorpholine (100 μL) were added. After stirring the mixture for 3 h at room temp, a 5% HCl solution and EtOAc were added to the reaction mixture. The EtOAc layer was subsequently washed with saturated NaHCO3 solution and brine. The organic layer was dried over anhydrous Na2SO4. After removing the solvent under vacuum, the residue was purified by reversed-phase HPLC (YMC-ODS column, 250 × 4.6 mm; H2O-MeCN, 25:75) to give (S)-PGME amide 3aS (0.2 mg). Compound 3aR (0.2 mg), the (R)-PGME amide of 3a, was prepared from (R)-PGME in a similar fashion. The molecular formulae of 3aS and 3aR were confirmed as C16H24N2O3 based on LR–ESI–MS data.
(S)-PGME amide of 3a (3aS): white amorphous solid; 1H NMR (CD3OD, 800 MHz) δH 7.393–7.338 (5H, m, PGME-Ar), 5.495 (1H, s, PGME-H-1), 3.697 (3H, s, PGME-OMe), 2.716 (1H, d, J = 9.0 Hz, H-7′), 2.240 (6H, s, H-11′, H-12′), 2.050 (1H, dhep, J = 9.3, 6.7 Hz, H-8′), 0.975 (3H, d, J = 6.7 Hz, H-10′), 0.931 (3H, d, J = 6.7 Hz, H-9′); LR–ESI–MS m/z 293.4 [M + H]+.
(R)-PGME amide of 3a (3aR): white amorphous solid; 1H NMR (CD3OD, 800 MHz) δH 7.373–7.335 (5H, m, PGME-Ar), 5.481 (1H, s, PGME-H-1), 3.704 (3H, s, PGME-OMe), 2.739 (1H, d, J = 9.2 Hz, H-7′), 2.345 (6H, s, H-11′, H-12′), 2.037 (1H, dhep, J = 9.4, 6.7 Hz, H-8′), 0.945 (3H, d, J = 6.7 Hz, H-10′), 0.810 (3H, d, J = 6.7 Hz, H-9′); LR–ESI–MS m/z 293.4 [M + H]+.
3.8. Marfey’s Analysis of 3b
Compound 3b (0.5 mg) was dissolved in 12 N HCl (0.5 mL) and heated at 110 °C for 16 h. The solution and traces of HCl were removed by repeated drying under vacuum with distilled water. To the hydrolysate, 1 N NaHCO3 (100 µL) and 1% l- or d-FDAA (50 µL) in acetone were added. The mixture was stirred at 80 °C for 15 min. After quenching the reaction by the addition of 2 N HCl (50 µL), the residue was analyzed using HPLC with an analytical column (YMC-ODS column, 250 × 4.6 mm; H2O-MeCN, 60:40). As standard compounds, l- and d-prolinol were also prepared using this method. The retention times of the l-FDAA-derivatized l-prolinol was 19.686 min and that of l-FDAA-derivatized d-prolinol was 20.160 min. The retention time of the l-FDAA-derivatized 3b was 20.222 min, leading to assignment of the d-configuration.
3.9. Antibacterial, Antifungal, Enzyme-Inhibitory and Cytotoxic Assays
3.9.1. Antibacterial Activity Assay
To investigate the antibacterial activity of isolated compounds, a series of minimal inhibitory concentration (MIC) tests was performed according to the Clinical and Laboratory Standards Institute (CLSI) guide methods [
25]. Three species of Gram-positive bacteria (
S. aureus strain Newman,
E. faecalis ATCC19433 and
E. faecium ATCC19434) and three species of Gram-negative bacteria (
K. pneumoniae ATCC10031,
S. enterica ATCC14028 and
E. coli ATCC25922) were selected as test strains. The MIC of each test compound against six bacterial strains was determined in liquid culture using Mueller–Hinton broth (inoculum concentration, 5 × 10
4 cfu/mL) with the compound (concentration range of 0.06–128 µg/mL) after 24 h incubation at 37 °C. Ampicillin and tetracycline were used as reference compounds.
3.9.2. Antifungal Activity Assay
The antifungal activity of isolated compounds was estimated against
C. albicans ATCC10231,
A. fumigatus HIC6094,
T. rubrum NBRC9185 and
T. mentagrophytes IFM40996 according to the guidelines in CLSI document M38 [
26]. The growth of test fungi was monitored in RPMI 1640 broth (inoculum concentration: 10
4 cells/mL) with the compound concentration range of 0.06–128 µg/mL. Amphotericin B was used as a positive control.
3.9.3. ICL Inhibition Assay
The purification of recombinant ICL from the genomic DNA of
C. albicans ATCC10231 and the evaluation of the effect of isolated compounds on ICL were carried out by the method described previously [
27]. The formation of glyoxylate phenylhydrazone from isocitrate and phenylhydrazine substrates was monitored spectrophotometrically at 324 nm. 3-Nitropropionic acid was used as a positive control.
3.9.4. SrtA Inhibition Assay
The preparation of recombinant SrtA from
S. aureus ATCC6538p and the evaluation of the effect of isolated compounds on SrtA were performed according to a previously described procedure [
27]. The enzyme reaction was carried out at 37 °C for 1 h with 300 µL of buffer (50 mM Tris–HCl, 150 mM NaCl, 5 mM CaCl
2, pH 7.5), 55 µg of purified SrtA, 0.75 µg of synthetic peptide dabcyl-QALPETGEE-edans and test samples at various concentrations. The increase in the fluorescence intensity was recorded by a fluorescence spectrophotometer (excitation, 350 nm; emission, 495 nm). Curcumin and berberine chloride were used as positive controls.
3.9.5. Cytotoxic Assay
The effects of compounds
1–
4 on cell viability were analyzed by MTT assay [
27]. A549 (lung cancer) and K562 (leukemia) cells were purchased from the Korean Cell Line Bank (KCLB), Seoul, Korea. Both cell lines were cultured in RPMI-1640 medium with L-glutamine, 10% fetal bovine serum and 1% penicillin/streptomycin. All the cells were cultured at 37 °C in a humidified atmosphere with 5% CO
2. For the assay, each cell (5 × 10
4 cells/mL) was seeded in 96-well plates (100 µL/well). After 24 h, they were treated with various concentrations of compounds
1–
4. After 24 h of compound treatment, MTT (250 µg/mL) was added to each well and incubated for 4 h. The formazan product was dissolved with 250 µL of DMSO. Absorbances of each well were detected at the wavelength of 595 nm using an ELISA microplate reader (BioTek, Winooski, VT, USA) to determine the cell viability by quantifying the production of formazan. The IC
50 values were calculated using a nonlinear regression analysis (percent survival versus concentration). Doxorubicin was used as a positive control.








{kind=link}
{kind=link}
{kind=link}
{kind=link}