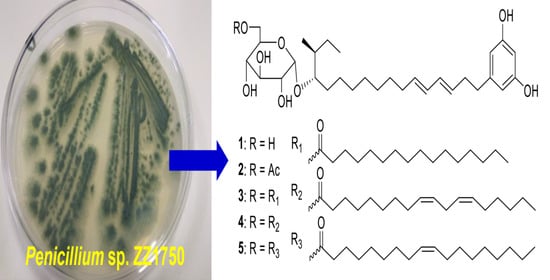
2. Results and Discussion
The marine-sourced strain ZZ1750 (
Figure S1, Supplementary Materials) was identified as
Penicillium sp. ZZ1750 based on its internal transcribed spacer (ITS) rDNA sequence (563 bp,
Figure S2), which was a 100% match to those of six other
Penicillium strains (
Table S1). An EtOAc extract prepared from a large-scale culture of the strain ZZ1750 in rich medium was separated by column chromatography, followed by high performance liquid chromatography (HPLC) purification, to afford compounds
1–
17.
Compound
1 was obtained as a yellowish oil with an optical rotation value of + 76 and a UV absorption at 232 nm. Its molecular formula C
30H
48O
8 was deduced from the HRESIMS (high resolution electrospray ionization mass spectroscopy) ion peak at
m/
z 559.3248 [M + Na]
+ (calcd. for C
30H
48NaO
8+, 559.3247) as well as its
13C-NMR data. Analyses of its
1H,
13C, distortionless enhancement by polarization transfer (DEPT), and heteronuclear multiple quantum correlation (HMQC) spectra showed the presence of ten olefinic carbons, six oxymethines, one oxymethylene, one methine, ten methylenes, and two methyls (
Table 1). A double peak signal at
δH 6.02 (2H, d,
J = 2.0 Hz, H-2 and H-6) showed HMQC correlation with a carbon signal at
δC 106.3 (C-2 and C-6) and also showed HMBC (heteronuclear multiple bond correlation) correlation (
Figure 2) with the same carbon signal (
δC 106.3), indicating the presence of a symmetrical structure unit in
1.
Further analysis of HMBC correlations of H-2/6 (
δH 6.02) with C-3/5 (
δC 158.2), C-4 (
δC 100.1), C-6/2 (
δC 106.3), and C-7 (
δC 35.3) as well as H-4 (
δH 6.00, t, 2.0 Hz) with C-2, C-3, C-5, and C-6 demonstrated that the symmetrical structure unit was a resorcinol derivative with a substituent at the C-1 position. This substituent was made up of two hydroxyalkene and hexose moieties. The hydroxyalkene moiety at C-1 was identified as a octadecene with two pairs of double bonds at C
9-10 and C
11-12, an oxymethine at C-20, and a methyl at C-21 based on the two COSY (correlation spectroscopy) spin systems of H
2-7/H
2-8/H-9/H-10/H-11/H-12/H
2-13/H
2-14/H
2-15 and H
2-18/H
2-19/H-20/H-21(H
3-24)/H
2-22/H
3-23 as well as the key HMBC correlations (
Figure 2) of H-2/6 with C-7, H
2-7 with C-2, C-6, and C-9, H
2-8 with C-1 and C-9, H-10 with C-8 and C-12, H-11 with C-9, H-12 with C-14, H
2-13 with C-11 and C-12, H
2-14 with C-13 and C-16, H
2-15 with C-13 and C-17, H
2-16 with C-18, H
2-18 with C-20, H
2-19 with C-17 and C-20, H
2-22 with C-20, H
3-23 with C-21 and C-22, H
3-24 with C-20, C-21, and C-22.
The large coupling constants of
3JH9-H10 (15.3 Hz) and
3JH11-H12 (14.8 Hz) (
Table 1) indicated 9
E and 11
E geometries. The hexose moiety resonated at
δH 4.70 (1H, d, 3.7 Hz, H-25), 3.15 (1H, dd, 9.5, 3.7, H-26), 3.37 (1H, t, 9.5 Hz, H-27), 3.07 (1H, t, 9.5 Hz, H-28), 3.43 (1H, m, H-29), 3.53 (1H, dd, 11.5, 2.0 Hz, H-30a), 3.46 (1H, dd, 11.5, 4.7 Hz, H-30b), and
δC 98.2 (CH, C-25), 72.1 (CH, C-26), 73.0 (CH, C-27), 70.1 (CH, C-28), 73.0 (CH, C-29), and 60.8 (CH
2, C-30), which were assigned by HMQC, COSY and HMBC correlations. Enzymatic hydrolysis of
1 by
α-glucosidase produced D-glucose and
1a, a new compound. The aldonitrile acetate of the hydrolytic D-glucose was identified by gas chromatography (GC) analysis [
20] (
Figure S3) using the authentic aldonitrile acetates of different sugars (D-glucose, L-glucose, D-galactose, L-galactose) as references. A small coupling constant (3.5 Hz,
3JH25-H26) and a upfield
13C chemical shift (
δC 98.3, C-25, when compared to
δC 103.2 for the anomeric carbon of
β-
d-glucosyl moiety) [
21] indicated an
α anomeric configuration for the glucose, which was further supported by the fact that only
α-glucosidase hydrolyzed this compound. Therefore, the hexose was proved to be an
α-D-glucopyranosyl moiety, which was linked to C-20 as established by the HMBC correlations of H-20 (
δH 3.35) with C-25 and H-25 with C-20 (
δC 81.0). The planar structure of the aglycone
1a was established based on its HRESIMS data as well as
13C,
1H, COSY, and HMQC spectroscopic analyses. The configuration at C-20 of
1a was assigned by the Mosher ester NMR method. Treatment of
1a with (
R)-α-methoxy-α-(trifluoromethyl) phenylacetyl chloride (
R-MTPA-Cl) or
S-MTPA-Cl gave
S-MTPA ester (
1as) or
R-MTPA ester (
1ar). The
1H-NMR chemical shift differences (Δ
δS-R,
Figure 3 and
Table S2) between
1as and
1ar in negative values for H-18 and H-19 and positive values for H-21, H-22, H-23, and H-24 were observed, indicating a 20
S-configuration for
1a.
In order to assign the configuration at C-21 and further support the 20
S-configuration, four possible diastereomers of 20
R,21
S-
1a, 20
S,21
S-
1a, 20
R,21
R-
1a, and 20
S,21
R-
1a were subjected to
13C and
1H-NMR calculations [
22,
23]. The results (
Tables S3–S5) showed that NMR data of
1a was very close to those of the model molecule 20
S,21
S-
1a with DP4
+ probability scores of 99.90% for
13C-NMR data, 68.95% for
1H-NMR data, and 99.98% for all NMR data, determining a 20
S,21
S-configuration for
1a. Based on the combined foregoing evidence, the structure of
1 was elucidated as a new glycosylated alkylresorcinol analogue, which was named peniresorcinoside A. The full assignment of its
13C- and
1H-NMR data (
Table 1) was made by HMQC, COSY, and HMBC correlations (
Figure 2).
Compound
2 was also obtained as a yellowish oil with a similar optical rotation value and UV absorption to those of
1. The molecular formula C
32H
50O
9 of
2 was deduced from its HRESIMS ion peak at
m/
z 601.3346 [M + Na]
+, 42 mass units higher than that of
1, corresponding to a -C
2H
2O group. The IR spectrum of
2 showed an absorption at 1724 cm
−1, indicating the presence of a carbonyl group. Detailed comparison of the
13C- and
1H-NMR spectra of
2 and
1 indicated that their chemical shifts bore a very close resemblance, except for two additional
13C-NMR signals (
δC 170.2 and 20.6) and one extra
1H-NMR signal (
δH 1.98, 3H, s) for an acetyl observed in the NMR spectra of
2. The HMBC correlations of H
2-30 (
δH 4.25, dd,
J = 11.7, 1.9 Hz; 3.95, dd,
J = 11.7, 7.3) with C-1′ (
δC 170.2) and H
3-2′ (
δH 1.98) with C-1′ demonstrated the acetyl was at the C-30 position. The difference between the
13C-NMR chemical shifts for C-30 (Δ +3.2 ppm) between
2 and
1 also supported the location of this acetyl group. Compound
2 was thus elucidated as a new glycosylated alkylresorcinol analogue, named peniresorcinoside B. The
13C- and
1H-NMR data (
Table 1) were fully assigned based on the HMQC, COSY, and HMBC correlations (
Figure 2).
Compound
3 was obtained as a yellowish oil. Its HRESIMS spectrum showed ion peaks at
m/
z 775.5719 [M + H]
+ and 797.5543 [M + Na]
+, corresponding to a molecular formula C
46H
78O
9, which was 16 carbons, 30 protons, and one oxygen more than that of
1. Careful analyses of the
13C- and
1H-NMR spectra of
3 and
1 demonstrated that
3 and
1 shared the same glycosylated alkylresorcinol structural backbone. Compared to the NMR signals of
1, compound
3 showed additional NMR signals for a carbonyl (
δC 172.7), a methyl (
δC 13.9;
δH 0.84, 3H, t,
J = 7.0 Hz), and some methylenes, whose NMR signals were high overlapped. Based on these characteristic NMR signals, in consideration of the presence of additional 16 carbons and one oxygen in
3, it was deduced that compound
3 had a structural unit of sixteen-carbon saturated acid (palmitic acid). The HMBC correlations of H
2-30 (
δH 4.26, 1H, d,
J = 10.8; 3.95, 1H, dd,
J = 10.8, 7.3 Hz) with C-1′ (
δC 172.7) established the linkage of this sixteen-carbon unit. Therefore, compound
3 was elucidated as a new glycosylated alkylresorcinol analogue, and named peniresorcinoside C. Its
13C- and
1H- NMR data (
Table 2) were assigned based on the HMQC, COSY, and HMBC correlations (
Figure S4).
The HRESIMS spectrum of
4 gave ion peaks at
m/
z 799.5716 [M + H]
+ and 821.5536 [M + Na]
+, matching a molecular formula of C
48H
78O
9, 18 carbons, 30 protons, and one oxygen more than that of
1. Analyses of the
13C- and
1H-NMR spectra of
4 and
1 led to the conclusion that
4 also had the same glycosylated alkylresorcinol structural unit of
1. Compared to the NMR signals of
1, compound
4 exhibited additional NMR signals for a carbonyl (
δC 172.7), two pairs of double bonds (
δC 129.7, 129.6, 127.8, 127.7;
δH 5.28, 2H, m, 5.31, 2H, m), a methyl (
δC 13.9;
δH 0.84, 3H, t,
J = 7.3 Hz), and some signal-overlapped methylenes. These characteristic NMR signals, together with the fact of additional 18 carbons and one oxygen in
4 mentioned above, suggested that compound
4 had an eighteen-carbon unsaturated fatty acid structural unit. Alkaline hydrolysis of
4 gave peniresorcinoside A (
1) as determined by co-HPLC analysis (
Figure S5) with the authentic sample and 9(
Z),12(
Z)-octadecadienoic acid (linoleic acid,
4a) based on its NMR data (
Table S9) and the comparison with the reference data [
24,
25] as well as co-HPLC analysis (
Figure S6) with standard linoleic acid. The HMBC correlations of H
2-30 (
δH 4.27, 1H, d,
J = 11.2; 3.95, 1H, dd,
J = 11.2, 7.5 Hz) with C-1′ (
δC 172.7) demonstrated this linoleic acid unit at C-30 position. The structure of
4 was thus identified as a new glycosylated alkylresorcinol analogue, named peniresorcinoside D. Its
13C- and
1H-NMR data (
Table 2) were assigned based on the HMQC, COSY, and HMBC correlations (
Figure S4).
The molecular formula C48H80O9 of 5 was determined based on its HRESIMS ions at m/z 823.5694 [M + Na]+, two mass units higher than that of 4. Detailed analyses of the 13C- and 1H-NMR spectra of 5 and 4 indicated that the structures of both compounds had the same glycosylated alkylresorcinol backbone with a difference in the number of double bonds of the eighteen-carbon unsaturated fatty acid unit. The 13C- and 1H-NMR spectra of 5 only exhibited a pair of double bonds (δC 129.7, 129.6; δH 5.29, 2H, m), instead of the two pairs of double bonds in 4.
Alkaline hydrolysis of
5 also gave peniresorcinoside A (
1) as confirmed by co-HPLC analysis (
Figure S5) and octadecenoic acid (oleic acid,
5a), as determined by analyzing its
13C- and
1H-NMR data (
Table S9), comparing with the reference data [
26], and co-HPLC analysis (
Figure S7) with an oleic acid standard. This oleic acid unit connected to C-30 was established through the HMBC correlations of H
2-30 (
δH 4.27, 1H, d,
J = 11.2; 3.95, 1H, dd,
J = 11.2, 7.5 Hz) with C-1′ (
δC 172.7). The structure of
5 was elucidated as a new glycosylated alkylresorcinol analogue, named peniresorcinoside E. The
13C- and
1H-NMR data (
Table 2) of
5 were assigned based on its HMQC, COSY, and HMBC correlations (
Figure S4).
Compound
6 was obtained as orthorhombic crystals from a mixture solvent of EtOAc and CHCl
3 (1:1) and its HRESIMS ion peak at
m/
z 497.3604 [M + Na]
+, corresponding to a molecular formula C
30H
50O
4. However, only 15 carbon signals were observed in the
13C NMR spectrum, suggesting its symmetrical structure. The 15 carbon signals were assigned for six olefin carbons, two oxymethine carbons, three methylene carbons, and four methyl carbons (
Table 3) based on its HMQC spectrum. The half planar structure (C
1-C
15) of
6 was established as a farnesyl derivative by further analyses of its COSY and HMBC correlations (
Figure 4). A HMBC correlation of H
2-12 (
δH 1.92, 2H, m) with C-12′ (
δC 27.9, CH
2), in consideration of its molecular formula, suggested the whole planer structure of
6, which was further confirmed by its crystal structure (
Figure 5) obtained from X-ray diffraction analysis. However, the data from the X-ray diffraction was not good enough for the assignment of the absolute configuration of
6 because of a poor Flack parameter of 0.3 (3). Therefore, a computational method was applied to assign the absolute configuration of
6 by comparing its experimental electronic circular dichroism (ECD) spectrum with the calculated ECD spectra. The X-ray CIF profile of
6 (5
R,8
R,5′
R,8′
R-
6) and its enantiomer (5
S,8
S,5′
S,8′
S-
6) were initially optimized at B3LYP/6-31g (d, p) level in MeOH. The theoretical calculations of ECD were conducted in MeOH using Time-dependent Density functional theory (TD-DFT) at the B3LYP/6-311+g (d, p) level. The results (
Figure 6) showed that the experimental ECD spectrum of
6 was agreement with the calculated ECD curve of the model molecule 5
R,8
R,5′
R,8′
R-
6, indicating a 5
R,8
R,5′
R,8′
R-configuration for
6. Based on the foregoing evidence, the structure of
6 was determined to correspond to a new dimeric farnesene derivative, named penidifarnesylin A. Its
13C and
1H NMR data (
Table 3) were assigned according to the HMQC, COSY, and HMBC correlations (
Figure 4).
The molecular formula of
7 was defined as C
25H
35NO
5 based on its HRESIMS ion peaks at
m/
z 430.2587 [M + H]
+ and 452.2406 [M + Na]. Analyses of its
1H-,
13C- and HMQC NMR spectra demonstrated the presence of two carbonyls, ten olefin carbons, eight olefin protons, three oxymethines, one oxymethylene, one methoxy, one methine, three methylenes, and four methyls (
Table 4). The two carbonyls and ten olefin carbons (five pairs of double bonds) accounted for seven out of the nine degrees of unsaturation required by the molecular formula, suggesting that
7 had a structure with two rings. Two COSY spin systems of H
2-1/H-2(H
3-15)/H-3/H-4/H-5 and H-7/H-8/H-9/H-10/H-11/H
2-12/H
2-13/H
3-14 and the HMBC correlations as described in
Figure 7 determined that the first cycle-related partial structure (
7a,
Figure 7) was a tetrahydropyran derivative. Similarly, the second cycle-related partial structure (
7b) was established as a pyridinone derivative by the COSY correlation of H-23 (
δH 5.93, dd, 8.0, 2.8 Hz) with H-24 (
δH 7.41, d, 8.0 Hz) and the HMBC correlations (
Figure 7). A HMBC correlation of H-4 (
δH 4.84, t, 9.5 Hz) with C-18 (
δC 167.5) demonstrated the connection of the two partial structures of
7a and
7b.
The relative configurations of
7 were assigned based on the coupling constants and nuclear Overhauser effect (NOE) information. A small coupling constant of 5.5 Hz (
3JH2-H3) and the large coupling constants of 9.5 Hz (
3JH3-H4 and
3JH4-H5) (
Table 4) suggested a
β-orientation for H-4 and an
α-orientation for H-2, H-3, and H-5 [
27]. The 6
E geometry was assigned by the NOE correlations of H-7 (
δH 5.97, d, 10.1 Hz) with H-9 (
δH 6.24, dd, 15.0, 10.1 Hz) as well as H-8 (
δH 6.29, dd, 15.0, 10.1 Hz) with H-10 (
δH 6.13, dd, 15.6, 10.1 Hz) and H
3-16 (
δH 1.66, s), whereas the 8
E and 10
E geometries were determined based on their
trans-coupling constants of 15.0 Hz for
3JH8-H9 and 15.6 Hz for
3JH10-H11. These assigned configurations for
7 were the same as those of restrictinol (
7c) [
28,
29]. Based on the forgoing evidences, the structure of
7 was elucidated as a new restrictinol analogue with a unique pyridinone functionality, named penipyridinone A. Its
13C- and
1H-NMR data (
Table 4) were assigned based on the HMQC, COSY, HMBC, and NOESY correlations (
Figure 7).
Based on the NMR data and optical rotation values as well as the comparison with the reported data, compounds
8–
17 were identified as known compounds methyl linoleate (
8) [
30], 12
β-hydroxyverruculogen TR-2 (
9) [
31,
32], 12
β-hydroxy-13
α-methoxyverruculogen TR-2 (
10) [
31,
33], cyclotryprostatin B (
11) [
34], verruculogen (
12) [
34], fumiquinazoline C (
13) [
35], fumiquinazoline J (
14) [
35,
36], brevianamide F (
15) [
37,
38], 2-[(2
R-hydroxypropanoyl)amino] benzamide (
16) [
39], and trypacidin (
17) [
40,
41]. The
13C and
1H NMR data of compounds
8–
17 are reported in
Tables S9–S13.
All isolated compounds
1–1
7 were tested for their antiproliferative activity against human glioma U87MG and U251 cells by using the Sulforhodamine B (SRB) assay [
42]. Doxorubicin (DOX, an anticancer drug) was used as a positive control. The results (
Table 5) showed that peniresorcinosides A (
1) and B (
2) had potent antiglioma activity with IC
50 values of 4.0 and 5.6 µM for U87MG cells and 14.1 and 9.8 µM for U251 cells, respectively. Penidifarnesylin A (
6) also showed antiproliferative activity with IC
50 values of 5.9 µM against U87MG cells and 27.6 µM against U251 cells. However, peniresorcinosides C–E (
3–
5) with a long-chain fatty acid unit only showed antiproliferative activity against U87MG cells with IC
50 values of 53.0, 19.4, and 22.1 µM, respectively. Other tested compounds were inactive at a concentration of 50 µM.
Marine-derived Penicillium fungi are still important sources for the discovery of novel bioactive natural products. The current study described the isolation and structural elucidation of 17 metabolites, including new glycosylated alkylresorcinols of peniresorcinosides A–E (1–5), penidifarnesylin A (6), and penipyridinone A (7) as well as some known indole alkaloids with complicated structures, from the marine fungus Penicillium sp. ZZ1750 cultured in rice medium.
Alkylresorcinols are resorcinol units alkylated with a long odd-numbered carbon chain and had high levels (over 500 µg/g) in wheat, rye, and triticale [
43]. It has been reported that some of alkylresorcinols exhibited antimicrobial, antiparasitic, and cytotoxic activities [
44]. Resorcinosides A and B [
45], recently isolated from the marine fungus
Penicillium janthinellum, are the first reported alkylresorcinol derivatives containing a glucose moiety linked to a hydroxy group of the alkyl side chain. Peniresorcinosides A–E (
1–5) are second example of this type of rare glycosylated alkylresorcinols. Peniresorcinosides C–E (
3–
5) have more complicated structures, with a long-chain fatty acid moiety attached to the C-6 position of the glucosyl unit. Evaluation of the antiglioma activity showed that peniresorcinosides A (
1) and B (
2) had potent antiproliferative activity against both glioma U87MG and U251 cells, while peniresorcinosides C–E (
3–
5) only exhibited moderate antiglioma activity against U87MG cells.
The cyclotryprostatin [
34,
46], fumiquinazoline [
35,
46,
47,
48], and verruculogen [
31,
34] indole alkaloids were most frequently isolated from
Aspergillus and
Penicillium fungi. Some of them have reported antibacterial, antifungal, cytotoxic, and antidiabetic activities [
48,
49]. In this study, seven such known indole alkaloids (compounds
9–
15) were obtained from the metabolites produced by the marine-sourced
Penicillium fungus ZZ1750. Fumiquinazoline C (
13) was previously reported to have moderate cytotoxicity against murine lymphocytic leukemia P388 cells [
47]. However, none of these isolated indole alkaloids
9–
15 were active against human glioma U87MG and U251 cells at a concentration of 50 µM.
3. Materials and Methods
3.1. General Experimental Procedures
Melting points (m.p.) were measured with a WRX-4 microscope apparatus (Shanghai Yice Apparatus & Equipment Co. Ltd., Shanghai, China) and was uncorrected. Ultraviolet (UV), optical rotation (OR), electronic circular dichroism (ECD), and infrared radiation (IR) spectra were measured on a METASH UV-8000 (Shanghai METASH Instruments Co. Ltd., Shanghai, China), an Autopol I Automatic polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA), a J-815 spectropolarimeter (JASCO Co. Tokyo, Japan), and a NicoletTM ISTM 10 FT-IR spectrometer (Thermo Fisher Scientific, Waltham, MA, USA), respectively. NMR data were acquired on a JEOL 600 spectrometer (JEOL, Tokyo, Japan) using a standard JEOL pulse sequences for 1D and 2D (HMQC, COSY, HMBC, and NOESY) NMR experiments and chemical shifts were expressed in δ (ppm) relative to DMSO-d6 (δC 39.5, δH 2.50) or MeOH-d4 (δC 49.0, δH 3.31). HRESIMS data were obtained on a 6230 Time of Flight Liquid Chromatography/Mass Spectrometry (TOF LC/MS) spectrometer (Agilent Technologies, Palo Alto, CA, USA). X-ray diffraction analysis was carried out on an Xcalibur Atlas Gemini Ultra diffractometer (Agilent Technologies) with Cu Kα radiation (λ = 1.54184 Å) at 100 K. Silica gel (200–300 mesh, Qingdao Ocean Chemical Plant, Qingdao, China) and octadecyl-functionalized silica gel (ODS, Cosmosil 75C18-Prep, Nacalai Tesque Inc., Kyoto, Japan) were used for column chromatography. HPLC separation was performed on an Agilent 1260 HPLC system using Agilent Zorbax SB-C18 column (250 × 9.2 mm, 5 µm) or a CXTH LC-3000 preparative HPLC system (Beijing Chuangxin Tongheng Science & Technology Co. Ltd., Beijing, China) using a CT-30 column (Fuji-C18, 280 × 30 mm, 10 µm). GC analysis was conducted on a GC-2010 system (Shimadzu, Kyoto, Japan) equipped with an SH-Rtx-5 capillary column (30 m × 0.32 mm, 0.25 µm). Nitrogen gas was used as the carrier gas and flame ionization detection (FID) was used as detector. Both injection port and detector temperatures were fixed at 280 °C. The column temperature was set at 180 to 280 °C in 10 min with an increase of 10 °C/min. Solvents used for this study were purchased from the Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China). Mosher’s reagents (R)-(–)-α-methoxy-α-(trifluoromethyl) phenylacetyl chloride (R-MTPA-Cl) and S-MTPA-Cl were ordered from Aladdin Industrial Corporation (Shanghai, China). Human glioma U87MG (JDS-2568) and U251 (XB-0439) cells were ordered from the Cell Bank of the Chinese Academy of Sciences, Shanghai, China). Linoleic acid (>95.0%) and oleic acid (>99.0%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd, and doxorubicin (DOX, >98.0%) from Sigma-Aldrich (St. Louis, MO, USA). Different culture media were made in the laboratory, including Gauze’s synthetic medium (B, soluble starch 20 g, KNO3 1 g, MgSO4⋅7H2O 0.5 g, NaCl 0.5 g, K2HPO4 0.5 g, FeSO4 0.01 g, agar 20 g, water 1 L), Gauze’s synthetic medium with sea salt (BS, soluble starch 20 g, KNO3 1 g, MgSO4⋅7H2O 0.5 g, NaCl 0.5 g, K2HPO4 0.5 g, FeSO4 0.01 g, agar 20 g, sea salt 35 g, water 1 L), potato dextrose agar medium (PDA, potatoes 200 g, glucose 20 g, agar 20 g, boiled into 1 L of water for 15 min), potato dextrose agar medium with sea salt (PDAS, potatoes 200 g, glucose 20 g, agar 20 g, sea salt 35 g, boiled into 1 L of water for 15 min), William’s E medium (E, yeast 1.0 g, tryptone 5.0 g, FeCl3⋅6H2O 0.17 g, KH2PO4 0.12 g, agar 30 g, water 1 L), William’s E medium with sea salt (ES, yeast 1.0 g, tryptone 5.0 g, FeCl3⋅6H2O 0.17 g, KH2PO4 0.12 g, agar 30 g, sea salt 35 g, water 1 L), international Streptomyces project medium-2 (ISP-2, yeast extract 4 g, malt extract 10 g, dextrose 4.0 g, peptone 5 g, agar 20 g, water 1 L), international Streptomyces project medium-2 with sea salt (ISP-2S, yeast extract 4 g, malt extract 10 g, dextrose 4.0 g, peptone 5 g, agar 20 g, sea salt, 35 g, water 1 L), international Streptomyces project medium-4 (ISP-4, soluble starch 10 g, K2HPO4 1 g, MgSO4⋅7H2O 1 g, NaCl 1 g, (NH4)2SO4 2 g, CaCO3 2 g, FeSO4 1 mg, MnCl2 1 mg, ZnSO4 1 mg, agar 20 g, water 1 L), international Streptomyces project medium-4 with sea salt (ISP-4S, soluble starch 10 g, K2HPO4 1 g, MgSO4⋅7H2O 1 g, NaCl 1 g, (NH4)2SO4 2 g, CaCO3 2 g, FeSO4 1 mg, MnCl2 1 mg, ZnSO4 1 mg, agar 20 g, sea salt, 35 g, water 1 L).
3.2. Isolation and Taxonomic Identification of Strain ZZ1750
The strain ZZ1750 was isolated from a sample of marine mud, which was collected from the Arabian Sea near Karachi, Sindh, Pakistan in January 2019. Briefly, the mud sample was air dried at 28 °C for 7 days and the dried sample (1.0 g) was diluted with sterile water to make up the dilutions of 10−2, 10−3, and 10−4 g/mL. Each dilution (200 µL) was transferred on the surface of ten different solid media of B, BS, PDA, PDAS, E, ES, ISP-2, ISP-2S, ISP-4, and ISP-4S on Petri dishes and then incubated at 28 °C for 14 days. The single colony of ZZ1750 was picked from the 10−2 g/mL suspension in PDA medium and then transferred to another PDA medium in dish. After growth for another 7 days at 28 °C, the single pure colony (ZZ1750) that grew well was transferred onto PDA slant medium and stored at 4 °C for further study.
The ITS rDNA sequence analysis of strain ZZ1750 was conducted by Legenomics (Hangzhou Lizhen Biotechnology Co., Ltd., Hangzhou, China). The ITS rDNA sequence was compared to those in the GenBank database using the nucleotide Basic Local Alignment Search Tool (BLAST). The ITS rDNA sequence of strain ZZ1750 has been deposited in GenBank (accession number: MT159428). The strain Penicillium sp. ZZ1750 was preserved at the Laboratory of Institute of Marine Biology and Pharmacology, Ocean College, Zhoushan campus, Zhejiang University, Zhoushan, China.
3.3. Mass Culture of Strain ZZ1750
The colony of strain ZZ1750 from the PDA slant medium was inoculated into a 500 mL Erlenmeyer flask, which contained 250 mL potato dextrose broth (PDB) medium and then incubated for 3 days in a shaker (180 rpm, 28 °C) to produce the seed broth (
Figure S1). The seed broth (5 mL) was transferred into rice medium (40 g rice, 60 mL 3.5% sea salt solution) in 500 mL Erlenmeyer flask and then all these flasks were incubated at 28 °C for 30 days in a static state. For this study, a total of 210 cultured flasks were prepared.
3.4. Extraction and Isolation of Compounds 1–17
The culture of strain ZZ1750 in rice medium in each flask was extracted with EtOAc (250 mL) three times. The combined EtOAc extract was dried in vacuo to give an extract (100 g). This extract was fractionated on a column (160 × 10 cm) of silica gel (1600 g) eluting with a mixture of cyclohexane and EtOAc in different ratios (10:1, 5:1, 2:1, 1:1, 1:2, each 1000 mL) to give four fractions of Frs. A–D based on the results of TLC analyses.
Fr. A was separated by using an Agilent 1260 HPLC system equipped with an Agilent Zorbax SB-C18 column (250 × 9.4 mm, 5 µm; mobile phase: ACN/H2O, 62/38; flow rate: 1.0 mL/min; UV detection: 232 nm) to give 6 (15.0 mg, tR 29.6 min) and 1 (15.2 mg, tR 34.5 min).
Fr. B was first separated on an ODS (200 g) column (530 × 35 mm) eluting with 60%, 70%, 80%, and 90% MeOH (each 1000 mL) to yield four subfractions (SFrs. B1–B4) according to the TLC analytic results. SFr. B1 was further separated on a Fuji-C18 CT-30 column (280 × 30 mm, 10 µm; mobile phase: MeOH/H2O, 75/25; flow rate: 10 mL/min; UV detection: 210 nm) to give 2 (4.0 mg, tR 34.5 min). SFr.B2 was also separated on the same CT-30 column using the same flow rate and same UV detection, but a different mobile phase of MeOH/H2O (70/30), to give 10 (2.5 mg; tR 24.0 min) and 12 (12.2. mg; tR 37.5 min). SFr.B4 was separated on the Zorbax SB-C18 column (mobile phase: MeOH/H2O, 95/5; flow rate: 1.0 mL/min, UV detection: 232 nm) to furnish 4 (2.8 mg, tR 58.8 min), 3 (2.6 mg, tR 72.0 min), and 5 (3.3 mg, tR 76.8 min). Compound 8 (3.2 mg, tR 31.1 min) was obtained from SFr.B5 through purification on the Zorbax SB-C18 column (mobile phase: MeOH/H2O, 98/2; flow rate: 1.0 mL/min, UV detection: 210 nm).
In the same way, SFr. B3 was further separated on the CT-30 column (mobile phase: MeOH/H2O, 67/33; flow rate: 10 mL/min; UV detection: 210 nm) to give parts B3a–B3c. By using the Zorbax SB-C18 column (flow rate: 1.0 mL/min), compounds 9 (20 mg, tR 36.4 min, ACN/H2O, 55/45), 11 (1.5 mg; tR 40.4 min, ACN/H2O, 50/50; UV detection: 210 nm), and 7 (3.8 mg, tR 41 min, MeOH/H2O, 87/13; UV detection of 275 nm) were purified from B3a, B3b, and B3c, respectively.
Similarly, Fr. C was fractionated on the CT-30 column (mobile phase: MeOH/H2O, 60/40; flow rate: 10 mL/min; UV detection: 210 nm) to afford parts C1 and C2. Then, by using the Zorbax SB-C18 column (flow rate: 1.0 mL/min; UV detection: 210 nm), compound 17 (8.0 mg, tR 25.0 min, mobile phase: MeOH/H2O, 65/35) was purified from part C1, 13 (21.0 mg, tR 30.0 min) and 14 (4.7 mg, tR 28.0 min, mobile phase: MeOH/H2O, 63/35) were obtained from part C2.
Finally, Fr. D was separated by using the Zorbax SB-C18 column (mobile phase: ACN/H2O, 40/60; flow rate: 1.0 mL/min; UV detection: 232 nm) to give 15 (2.8 mg, tR 26.6 min) and 16 (2.1 mg, tR 36 min).
Peniresorcinoside A (
1): Yellowish oil; molecular formula C
30H
48O
8; [
α]
20D +76° (
c 0.10, MeOH); UV (MeOH) λ
max (log ε) 232 (3.45) nm; IR (ATR) ν
max 3354, 2912, 2843, 1593, 1452, 1155, 1020, 982, 835 cm
−1;
13C-NMR (150 MHz) and
1H-NMR (600 MHz) data (in DMSO-
d6), see
Table 1; HRESIMS
m/
z 559.3248 [M + Na]
+ (calcd for C
30H
48NaO
8+, 559.3247).
Peniresorcinoside B (
2): Yellowish oil; molecular formula C
32H
50O
9; [
α]
20D +80° (
c 0.10, MeOH); UV (MeOH) λ
max (log ε) 232 (3.46) nm; IR (ATR) ν
max 3398, 2912, 1724, 1606, 1450, 1146, 1034, 609 cm
−1;
13C-NMR (150 MHz) and
1H-NMR (600 MHz) data (in DMSO-
d6), see
Table 1; HRESIMS
m/
z 601.3346 [M + Na]
+ (calcd for C
32H
50NaO
9+, 601.3353).
Peniresorcinoside C (
3): White amorphous powder; molecular formula C
46H
78O
9; [α]
20D +28° (c 0.1, CH
3OH); UV (MeOH) λ
max (log ε) 233 (3.99); IR (ATR) ν
max 3333, 2922, 2855, 1714, 1600, 1510, 1455, 1076, 1018, 841 cm
-1;
13C-NMR (150 MHz) and
1H-NMR (600 MHz) data (in DMSO-
d6), see
Table 2; HRESIMS
m/
z 775.5719 [M + H]
+ (calcd for C
46H
79O
9+, 775.5724) and 797.5543 [M + Na]
+ (calcd for C
46H
78NaO
9+, 797.5544).
Peniresorcinoside D (
4): White amorphous powder; molecular formula C
48H
78O
9; [α]
20D +42° (c 0.1, CH
3OH); UV (MeOH) λ
max (log ε) 233 (4.34); IR (ATR) ν
max 3355, 2924, 2857, 1722, 1592, 1457, 1145, 1104, 1016, 986, 840 cm
-1;
13C-NMR (150 MHz) and
1H-NMR (600 MHz) data (in DMSO-
d6), see
Table 2; HRESIMS
m/
z 799.5716 [M + H]
+ (calcd for C
48H
79O
9+, 799.5724) and 821.5536 [M + Na]
+ (calcd for C
48H
78NaO
9+, 821.5544).
Peniresorcinoside E (
5): White amorphous powder; molecular formula C
48H
80O
9; [α]
20D +40° (c 0.1, CH
3OH); UV (MeOH) λ
max (log ε) 233 (4.22); IR (ATR) ν
max 3343, 2922, 2855, 1716, 1598, 1459, 1149, 1059, 984, 841 cm
-1;
13C-NMR (150 MHz) and
1H-NMR (600 MHz) data (in DMSO-
d6), see
Table 2; HRESIMS
m/
z 823.5694 [M + Na]
+ (calcd for C
48H
80NaO
9+, 823.5700).
Penidifarnesylin A (
6): Orthorhombic crystals (EtOAc:CHCl
3, 1: 1); molecular formula C
30H
50O
4; m.p. 145–148 °C; [
α]
20D +40° (
c 0.10, MeOH); ECD (200 µg/mL, MeOH) λ
max (Δε) 210 (+7.62) nm; UV (MeOH) λ
max (log ε) 210 (4.24) nm; IR (ATR) ν
max 3347, 2921, 2853, 1440, 1376, 1260, 1020 cm
−1;
13C-NMR (150 MHz) and
1H-NMR (600 MHz) data (in DMSO-
d6), see
Table 3; HRESIMS
m/
z 497.3604 [M + Na]
+ (calcd for C
30H
50NaO
4+, 497.3607). Crystal data of penidifarnesylin A (
6): C
30H
50O
4 (
M = 474.70 g/mol), orthorhombic, space group P2
12
12 (no. 18),
a = 7.4649(8) Å,
b = 59.512(10) Å,
c = 9.9356(16) Å,
V = 4413.9(12) Å
3,
Z = 6,
T = 100.00(12) K, μ(Cu Kα) = 0.536 mm
-1,
Dcalc = 1.072 g/cm
3, 29803 reflections measured (5.94° ≤ 2Θ ≤ 148.752°), 8796 unique (
Rint = 0.1650, R
sigma = 0.1369), which were used in all calculations. The final
R1 was 0.0877 (I > 2σ (I)) and
wR2 was 0.2366 (all data). The crystal data and structure refinement parameters of penidifarnesylin A (
6) were also reported in
Table S6. Crystallographic data of penidifarnesylin A (
6) has been deposited at the Cambridge Crystallographic Data Centre (CCDC Number: 1976942). Copies of the data can be obtained free of charge from Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, U.K. [fax (+44)1223-336-033; or e-mail:
[email protected]].
Penipyridinone A (
7): Light-yellow powder; molecular formula C
25H
35NO
5;
13C-NMR (150 MHz) and
1H-NMR (600 MHz) data (in DMSO-
d6), see
Table 4; HRESIMS
m/
z 430.2587 [M + H]
+ (calcd for C
25H
36NO
5+ 430.2593) and 452.2406 [M + Na]
+ (calcd for C
25H
35NNaO
5+ 452.2413).
Methyl linoleate (
8): White amorphous powder; molecular formula C
19H
34O
2;
13C- NMR (150 MHz) and
1H-NMR (600 MHz) data (in DMSO-
d6), see
Table S9; HRESIMS
m/
z 295.2626 [M + H]
+ (calcd for C
19H
35O
2+, 295.2637) and 317.2451 [M + Na]
+ (calcd for C
19H
34NaO
2+, 317.2457).
12
β-Hydroxyverruculogen TR-2 (
9): White amorphous powder; molecular formula C
22H
27N
3O
6; [
α]
20D +51° (
c 0.10, MeOH);
13C-NMR data (150 MHz, in DMSO-
d6), see
Table S10,
1H-NMR data (600 MHz, in DMSO-
d6), see
Table S11; HRESIMS
m/
z 430.1977 [M + H]
+ (calcd for C
22H
28N
3O
6+, 430.1978) and 452.1794 [M + Na]
+ (calcd for C
22H
27N
3NaO
6+, 452.1798).
12
β-Hydroxy-13
α-methoxyverruculogen TR-2 (
10): White powder; molecular formula C
23H
29N
3O
6; [
α]
20D +45° (
c 0.10, MeOH);
13C-NMR data (150 MHz, in DMSO-
d6), see
Table S10,
1H-NMR data (600 MHz, in DMSO-
d6), see
Table S11; HRESIMS
m/
z 444.2123 [M + H]
+ (calcd for C
23H
30N
3O
6+, 444.2135) and 466.1946 [M + Na]
+ (calcd for C
23H
29N
3NaO
6+, 466.1954).
Cyclotryprostatin B (
11): Yellow powder; molecular formula C
23H
27N
3O
5; [
α]
20D +59° (
c 0.10, MeOH);
13C-NMR data (150 MHz, in CDCl
3), see
Table S10,
1H-NMR data (600 MHz, in in CDCl
3), see
Table S11; HRESIMS
m/
z 426.2029 [M + H]
+ (calcd for C
23H
28N
3O
5+, 426.2029) and 448.1837 [M + Na]
+ (calcd for C
23H
27N
3NaO
5+, 448.1848).
Verruculogen (
12): White powder; molecular formula C
27H
33N
3O
7; [
α]
20D −37.8° (
c 0.10, MeOH);
13C-NMR data (150 MHz, in DMSO-
d6), see
Table S10,
1H-NMR data (600 MHz, in DMSO-
d6), see
Table S11; HRESIMS
m/
z 512.2410 [M + H]
+ (calcd for C
27H
34N
3O
7+, 512.2397) and 534.2202 [M + Na]
+ (calcd for C
27H
33N
3NaO
7+, 534.2216).
Fumiquinazoline C (
13): Light-yellow amorphous powder; molecular formula C
24H
21N
5O
4; [
α]
20D −60.8° (
c 0.10, MeOH);
13C-NMR data (150 MHz, in DMSO-
d6), see
Table S12,
1H-NMR data (600 MHz, in DMSO-
d6), see
Table S13; HRESIMS
m/
z 444.1666 [M + H]
+ (calcd for C
24H
22N
5O
4+, 444.1672) and 466.1486 [M + Na]
+ (calcd for C
24H
21N
5NaO
4+, 466.1491).
Fumiquinazoline J (
14): White amorphous powder; molecular formula C
21H
16N
4O
2; [
α]
20D −68° (
c 0.10, MeOH);
13C-NMR data (150 MHz, in DMSO-
d6), see
Table S12,
1H-NMR data (600 MHz, in DMSO-
d6), see
Table S13; HRESIMS
m/
z 357.1343 [M + H]
+ (calcd for C
21H
17N
4O
2+, 357.1352) and 379.1156 [M + Na]
+ (calcd for C
21H
16N
4NaO
2+, 379.1171).
Brevianamide F (
15): White colorless solid; molecular formula C
16H
17N
3O
2; [
α]
20D −53° (
c 0.10, MeOH);
13C-NMR data (150 MHz, in DMSO-
d6), see
Table S12,
1H-NMR data (600 MHz, in DMSO-
d6), see
Table S13; HRESIMS
m/
z 284.1396 [M + H]
+ (calcd for C
16H
18N
3O
2+, 284.1399) and 306.1213 [M + Na]
+ (calcd for C
16H
17N
3NaO
2+, 306.1218).
2-[(2
R-Hydroxypropanoyl) amino] benzamide (
16): White amorphous powder; molecular formula C
10H
12N
2O
3; [
α]
20D +30° (
c 0.10, MeOH);
13C-NMR data (150 MHz, in DMSO-
d6), see
Table S12,
1H-NMR data (600 MHz, in DMSO-
d6), see
Table S13; HRESIMS
m/
z 209.0921 [M + H]
+ (calcd for C
10H
13N
2O
3+, 209.0926) and 231.0744 [M + Na]
+ (calcd for C
10H
12N
2NaO
3+, 231.0746).
Trypacidin (
17): White powder; molecular formula C
18H
16O
7; [
α]
20D −50° (
c 0.10, MeOH);
13C-NMR data (150 MHz, in DMSO-
d6), see
Table S12,
1H-NMR data (600 MHz, in DMSO-
d6), see
Table S13; HRESIMS
m/
z 345.0969 [M + H]
+ (calcd for C
18H
17O
7+, 345.0974) and 367.0787 [M + Na]
+ (calcd for C
18H
16NaO
7+, 367.0794).
3.5. Enzymatic Hydrolysis of Peniresorcinoside A (1)
Peniresorcinoside A (
1, 3.0 mg) was equilibrated at 37 °C for 5 min in a solution of water (2.0 mL) and 0.1 M phosphate buffer (pH = 7.0, 2.0 mL) and then 0.5 mL of α-glucosidase solution (0.2 M potassium phosphate solution containing 1 mM EDTA and 0.05% Tween-20, pH 7.0) was added. The mixture was incubated at 37 °C for 1 h and then 2.0 mL of 0.2 M Na
2CO
3 solution were added to terminate the enzymatic reaction. The enzymatic product was extracted with EtOAc (each 5 mL) three times to give an EtOAc extract and a water solution. The EtOAc extract was separated on an Agilent Zorbax SB-C18 column (250 × 9.4 mm, 5 µm; mobile phase: MeOH/H
2O, 85/15; flow rate: 1.0 mL/min, UV detection: 210 nm) to furnish
1a (1.8 mg, t
R 37.4 min). The aqueous solution was dried under reduced pressure to afford a residue which was first treated with 10 mg hydroxylamine hydrochloride in 2 mL pyridine at 90 °C for 30 min in a water bath and then mixed with 2 mL acetic anhydride at 90 °C for 1 h in a water bath. Finally, the reaction products were dried in vacuo and dissolved in 2 mL chloroform for GC analysis. The aldonitrile acetate of sugar in
1 was identified as the aldonitrile acetate of D-glucose (t
R 7.20 min) (
Figure S3) by GC analysis with aldonitrile acetates of D-glucose (t
R 7.20 min), L-glucose (t
R 7.28 min), D-galactose (t
R 7.40 min), and L-galactose (t
R 7.45 min) as references.
Compound
1a: White amorphous powder; molecular formula C
24H
38O
3;
13C-NMR (150 MHz) and
1H-NMR (600 MHz) data (in MeOH-
d4), see
Table 1; HRESIMS
m/
z: 375.2893 [M + H]
+ (calcd for C
24H
39O
3+, 375.2899) and 397.2715 [M + Na]
+ (calcd for C
24H
38NaO
3+, 397.2719).
3.6. MTPA Esterification of Compound 1a
Compound 1a (0.8 mg) was dissolved in the anhydrous pyridine (0.5 mL) and then either (R)-α-methoxy-α-(trifluoromethyl)-phenylacetyl chloride (R-MTPA-Cl, 45 µL) or S-MTPA-Cl (45 µL) was added. The mixtures were stirred at 45 °C for 48 h until adding 1 mL MeOH to terminate the reaction. The reaction mixtures were dried under reduced pressure to give a residue. (S)-MTPA ester 1as (0.4 mg, tR 25.1 min, MeOH/H2O, 100/0) or (R)-MTPA ester 1ar (0.4 mg, tR 24.9 min, MeOH/H2O, 100/0) was obtained from the residue by HPLC purification using an Agilent Zorbax SB-C18 column (250 × 9.2 mm, 5 µm) at a flow rate of 1.0 mL/min and UV detection of 210 nm.
Compound
1as: Molecular formula C
54H
59F
9O
9;
1H-NMR data (600 MHz, in MeOH-
d4), see
Table S2; HRESIMS
m/
z: 1045.3910 [M + Na]
+ (calcd for C
54H
59F
9NaO
9+, 1045.3913).
Compound
1ar: Molecular formula C
54H
59F
9O
9;
1H-NMR data (600 MHz, in MeOH-
d4), see
Table S2; HRESIMS
m/
z: 1045.3911 [M + Na]
+ (calcd for C
54H
59F
9NaO
9+, 1045.3913).
3.7. Alkaline Hydrolysis of Peniresorcinosides D (4) and E (5)
Peniresorcinoside D (4, 1.6 mg) was hydrolyzed in 4 mL 3 N NaOH at 40 °C for 2 h in a water bath. The reaction mixture was neutralized with 3 N HCl and then extracted with EtOAc (each 5 mL) three times to give an EtOAc extract. This EtOAc extract was separated on a Zorbax SB-C18 column (250 × 9.4 mm, 5 µm; mobile phase: MeOH/0.1%TFA-H2O, 93/7; flow rate: 1.0 mL/min; UV detection: 210 nm) to give 1 (0.8 mg, tR 12.8 min) and 4a (0.5 mg, tR 35.1 min). In the same way, alkaline hydrolysis of peniresorcinoside E (5, 2.1 mg) produced 1 (1.2 mg, tR 34.5 min) and 5a (0.7 mg, tR 45.2 min).
Compound
4a: Light yellow oil; molecular formula C
18H
32O
2;
13C NMR (150 MHz) and
1H NMR (600 MHz) data (in DMSO-
d6), see
Table S9; HRESIMS
m/
z 279.2316 [M − H]
+ (calcd for C
18H
31O
2−, 279.2324).
Compound
5a: Colorless oil; molecular formula C
18H
34O
2;
13C NMR (150 MHz) and
1H NMR (600 MHz) data (in DMSO-
d6), see
Table S9; HRESIMS
m/
z 283.2645 [M + H]
+ (calcd for C
18H
35O
2+, 283.2637) and 305.2445 [M + Na]
+ (calcd for C
18H
34NaO
2+, 305.2457).
3.8. 13C- and 1H-NMR Calculations
Monte Carlo conformational searches were carried out by means of the Spartan’s 10 software using Merck Molecular Force Field (MMFF). The conformers with Boltzmann-population of over 5% for NMR calculations were initially optimized at B3LYP/6-31g (d, p) level in MeOH. Gauge-independent atomic orbital (GIAO) calculations of
13C and
1H NMR chemical shifts were accomplished by density functional theory (DFT) at the mPWLPW91-SCRF (DMSO)/6-311+g (d, p) level with the PCM solvent continuum model in Gaussian 09 software. The calculated NMR data of the lowest energy conformers for model molecules 20
R,21
S-
1a, 20
S,21
S-
1a, 20
R,21
R-
1a, and 20
S,21
R-
1a were averaged according to the Boltzmann distribution theory and their relative Gibbs free energy. The
13C-NMR and
1H-NMR chemical shifts for TMS were calculated by the same protocol as reported in the reference [
23] and the experimental and calculated data of the isomeric compounds were analyzed by the improved probability DP4
+ method [
23]. A significant higher DP4
+ probability score of the model molecules suggested the correctness of its configuration.
3.9. ECD Calculations
The X-ray CIF profile of 5S,8S,5′S,8′S-6 was initially optimized at B3LYP/6-31g (d, p) level in MeOH. The theoretical calculation of ECD was conducted in MeOH using Time-dependent Density functional theory (TD-DFT) at the B3LYP/6-311+g (d, p) level. Under the same conditions, the enantiomer 5R,8R,5′R,8′R-6 was also calculated. Rotatory strengths for a total of 30 excited states were calculated. ECD spectra were generated using the program SpecDis 1.6 (University of Würzburg, Würzburg, Germany) and GraphPad Prism 5 (University of California San Diego, San Diego, CA, USA) from dipole-length rotational strengths by applying Gaussian band shapes with sigma = 0.2 eV.
3.10. Sulforhodamine B (SRB) Assay
Human glioma U87MG and U251 cells were cultured in Minimum Essential Medium (MEM, Gibco, Thermo Fisher Scientific Inc., Waltham, MA, USA) and Dulbecco’s Modified Eagle Medium (DMEM, Gibco) with 10% FBS, respectively. All cells were incubated at 37°C in a humidified incubator with 5% CO
2 incubator. Cells from the third repeated culture were used for experiments. The SRB assay as describe in previous publication [
42] was used to evaluate the antiproliferative activity of all isolated compounds
1–
17 against human glioma U87MG and C251 cells. Doxorubicin (DOX) was used as a positive control. Briefly, glioma cells in logarithmic growth (4 × 10
3 cells/well) were plated in a 96-well plate, treated with different concentrations of each tested compound after 24 h of cells adhesion, and then incubated for 72 h. After that, the treated cells were fixed with 50 µL of 50% cold TCA (trichloroacetic acid) solution at 4 °C for 1 h, washed with distilled water five times, and then dried at 37 °C in a drying oven. The dried cells were stained with 50 µL of 0.4% SRB for 15 min, rinsed with 1% glacial acetic acid solution five times, then dried at 37 °C. Finnally, the dried dye was dissolved in 100 µL of 10 mM Tris buffer and the optical density (OD) value measured at 515 nm on a microplate reader (BioTech, Winooski, VT, USA). The cell viability (%) was calculated from the formula of T
OD/C
OD × 100% (T
OD: OD value of tested compound; C
OD: OD value of negative control) and IC
50 value was obtained based on the cell viability (%) by logistic calculation using SPSS software.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}