
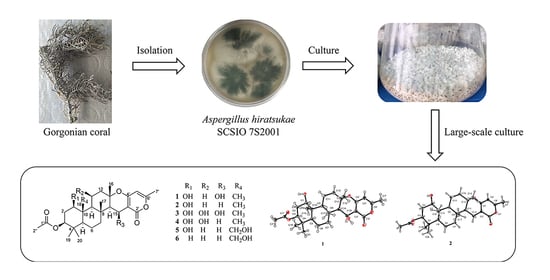
Chevalones H–M: Six New α-Pyrone Meroterpenoids from the Gorgonian Coral-Derived Fungus Aspergillus hiratsukae SCSIO 7S2001

 , , ,
, , ,
Abstract
:
1. Introduction
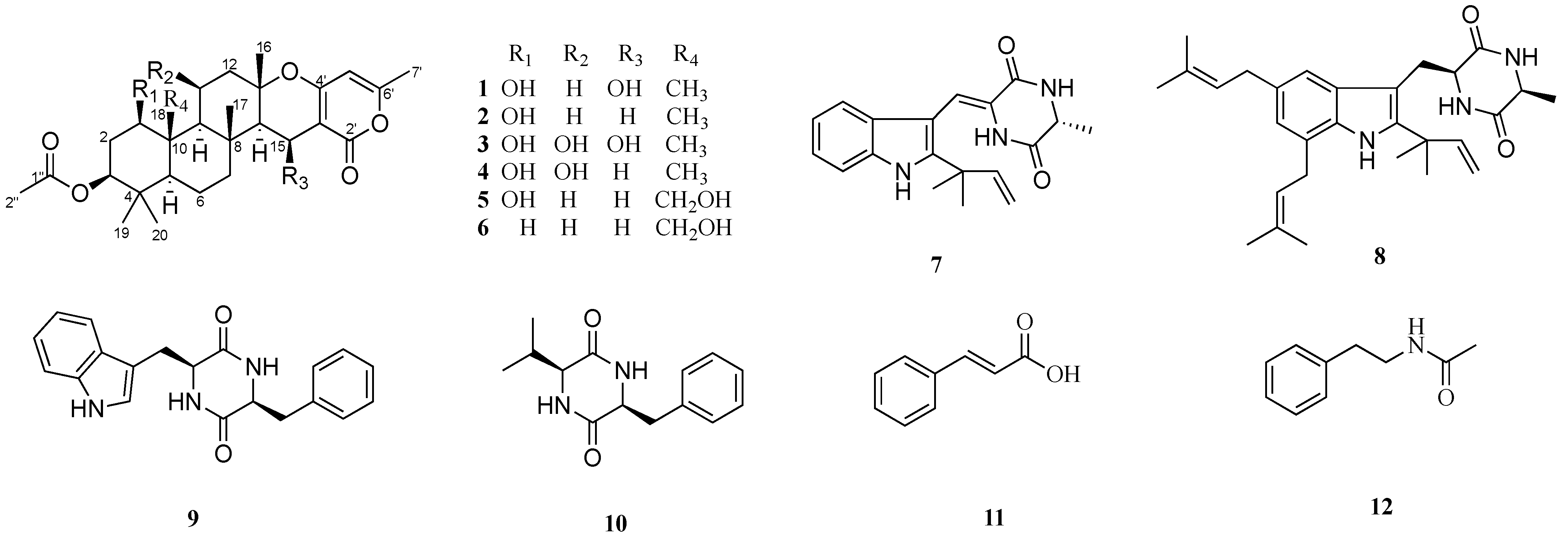
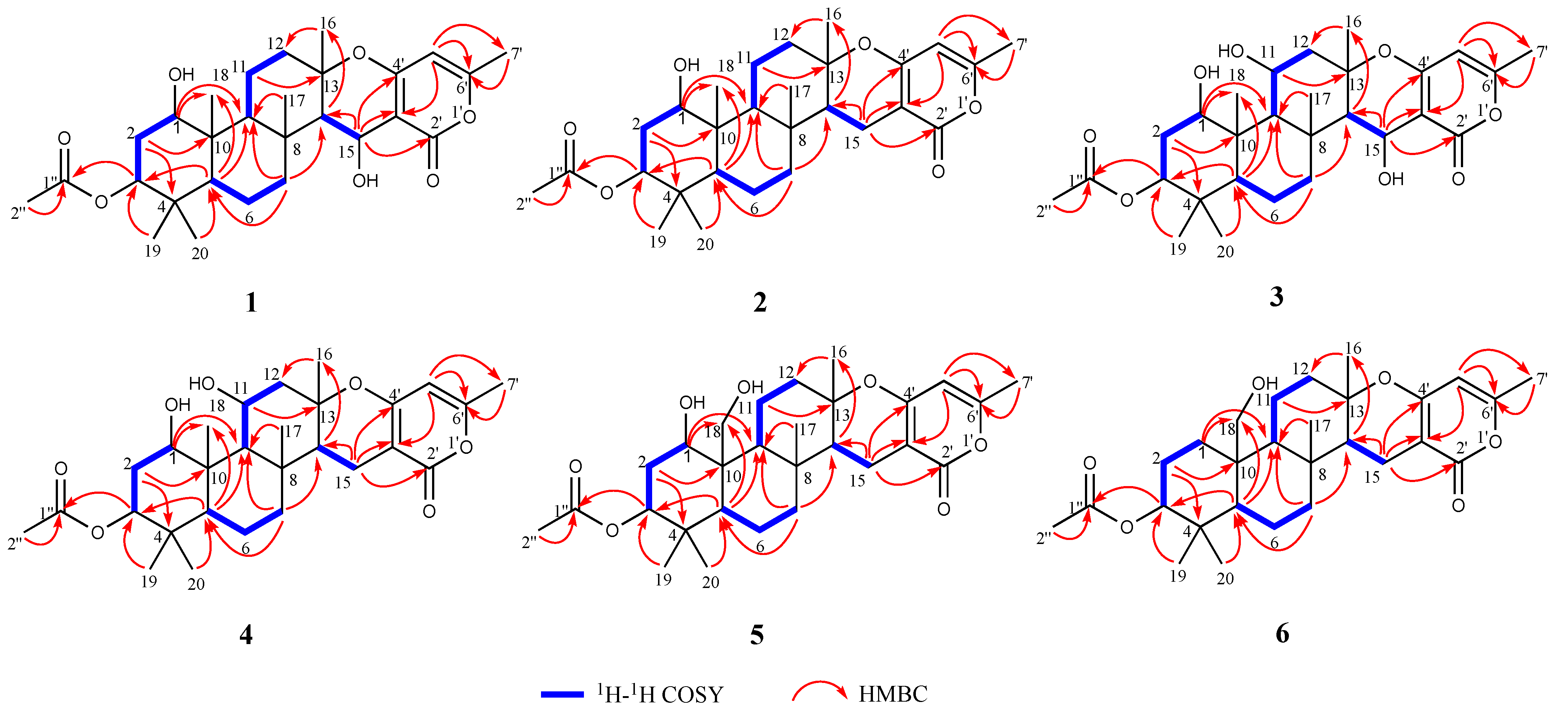
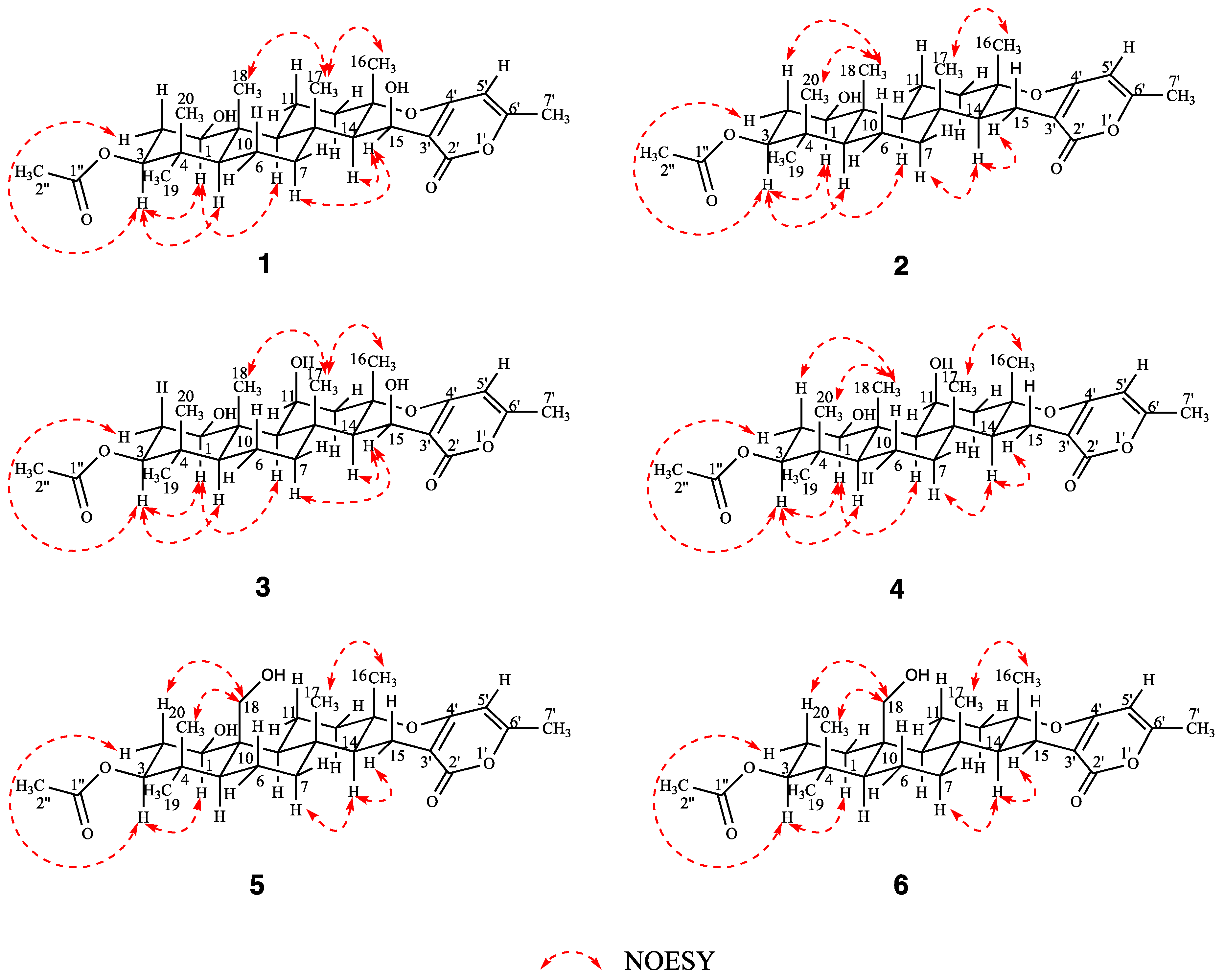
2. Results and Discussion

{kind=link}
{kind=link}
{kind=link}
{kind=link}
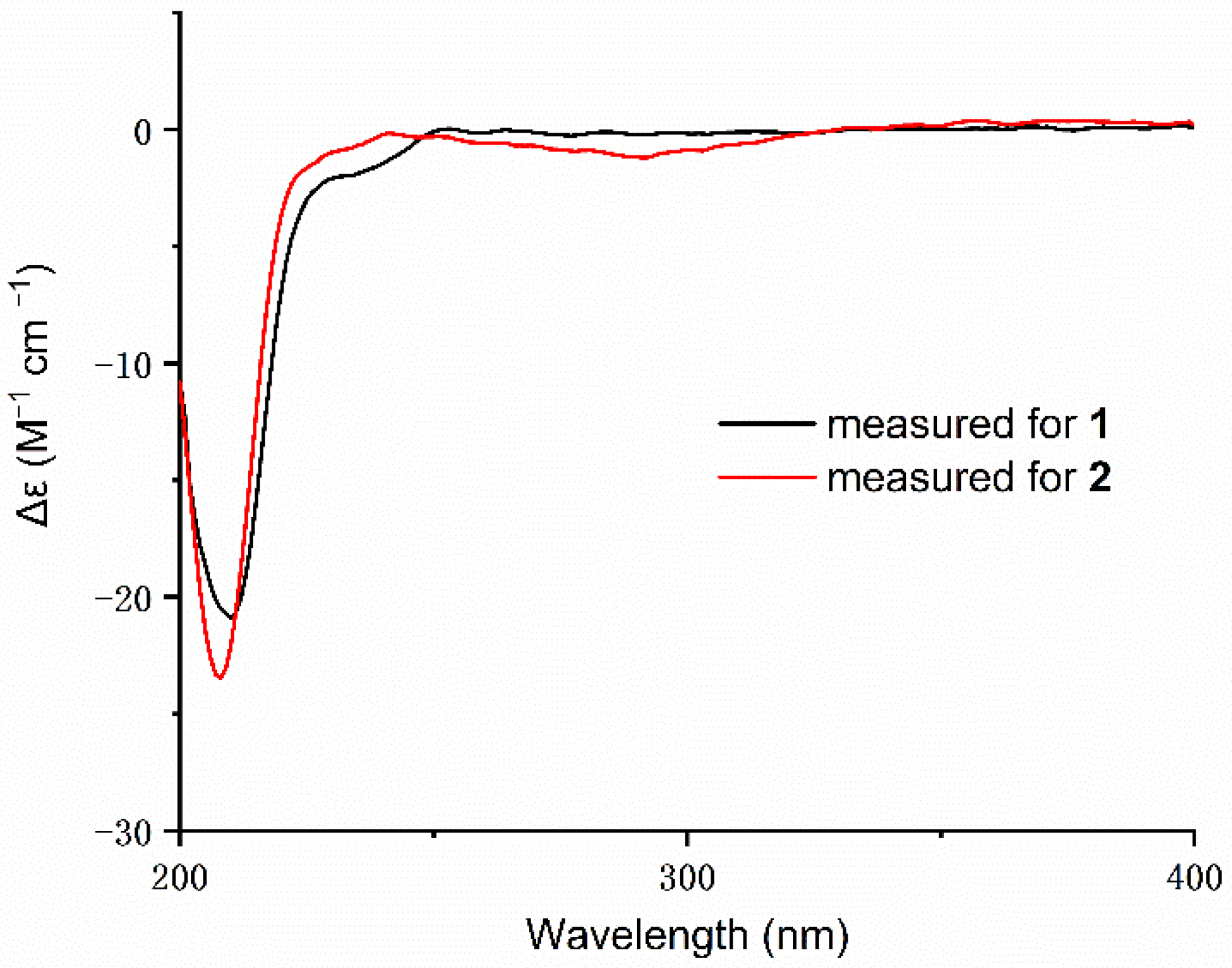{kind=link}
{kind=link}
| NO. | 1 a | 2 a | 3 a | |||
|---|---|---|---|---|---|---|
| δC Type | δH (J in Hz) | δC Type | δH (J in Hz) | δC Type | δH (J in Hz) | |
| 1 | 78.1 CH | 3.51 m | 77.7 CH | 3.54 dd (11.4, 4.6) | 76.4 CH | 3.63 dd (11.2, 4.8) |
| 2 | 34.5 CH2 | 1.69–1.72 m | 34.5 CH2 | 1.67–1.71 m | 33.5 CH2 | 1.93–1.95 m |
| 1.89 dt (12.3, 4.5) | 1.90 dt (12.5, 4.4) | 1.80–1.82 m | ||||
| 3 | 77.3 CH | 4.47 dd (12.2, 4.4) | 77.2 CH | 4.48 dd (12.2, 4.3) | 77.3 CH | 4.45 dd (12.3, 4.2) |
| 4 | 37.9 C | 37.9 C | 37.7 C | |||
| 5 | 53.3 CH | 0.74–0.78 m | 53.3 CH | 0.76 dd (12.1, 2.5) | 54.4 CH | 0.80 dd (12.4, 2.4) |
| 6 | 17.4 CH2 | 1.58–1.60 m | 17.6 CH2 | 1.56–1.58 m | 18.1 CH2 | 1.06–1.08 m |
| 1.66–1.68 m | 1.61–1.63 m | 1.23–1.25 m | ||||
| 7 | 40.5 CH2 | 1.18–1.21 m | 40.8 CH2 | 0.98–1.04 m | 42.0 CH2 | 1.18–1.20 m |
| 2.16–2.18 m | 1.85 dt (12.9, 3.4) | 2.15–2.17 m | ||||
| 8 | 39.6 C | 38.2 C | 39.6 C | |||
| 9 | 61.6 CH | 1.04 dd (11.7, 2.4) | 61.1 CH | 1.10 dd (12.1, 2.4) | 61.5 CH | 0.88 d (3.1) |
| 10 | 43.5 C | 43.5 C | 44.1 C | |||
| 11 | 21.6 CH2 | 1.60–1.62 m | 21.4 CH2 | 1.43–1.45 m | 70.0 CH | 4.79 dt (5.4, 2.7) |
| 2.65–2.69 m | 2.64–2.70 m | |||||
| 12 | 41.8 CH2 | 1.64–1.66 m | 40.6 CH2 | 1.63–1.65 m | 48.7 CH2 | 1.87–1.89 m |
| 2.02 dt (11.9, 3.1) | 2.00–2.04 m | 2.28–2.30 m | ||||
| 13 | 81.9 C | 80.4 C | 81.3 C | |||
| 14 | 56.5 CH | 1.42 d (4.1) | 52.1 CH | 1.46 d (4.5) | 56.6 CH | 1.41 d (4.0) |
| 15 | 60.5 CH | 4.89 d (4.0) | 17.0 CH2 | 2.13 dd (16.7, 13.0) | 60.2 CH | 4.95 d (4.0) |
| 2.42 dd (16.7, 4.7) | ||||||
| 16 | 22.1 CH3 | 1.57 s | 20.6 CH3 | 1.19 s | 23.1 CH3 | 1.77 s |
| 17 | 18.3 CH3 | 1.33 s | 16.4 CH3 | 0.90 s | 19.6 CH3 | 1.69 s |
| 18 | 12.4 CH3 | 0.95 s | 12.4 CH3 | 0.92 s | 14.9 CH3 | 1.29 s |
| 19 | 28.0 CH3 | 0.85 s | 27.9 CH3 | 0.84 s | 27.8 CH3 | 0.84 s |
| 20 | 16.2 CH3 | 0.86 s | 16.1 CH3 | 0.85 s | 16.0 CH3 | 0.88 s |
| 2′ | 165.6 C | 165.6 C | 165.6 C | |||
| 3′ | 101.4 C | 97.9 C | 101.4 C | |||
| 4′ | 163.4 C | 163.5 C | 163.0 C | |||
| 5′ | 101.1 CH | 5.73 s | 100.8 CH | 5.68 s | 101.0 CH | 5.73 s |
| 6′ | 161.6 C | 159.9 C | 161.7 C | |||
| 7′ | 20.0 CH3 | 2.19 s | 19.9 CH3 | 2.17 s | 20.0 CH3 | 2.19 s |
| 1″ | 171.0 C | 171.0 C | 171.0 C | |||
| 2″ | 21.3 CH3 | 2.05 s | 21.3 CH3 | 2.05 s | 21.3 CH3 | 2.06 s |
| NO. | 4 a | 5 b | 6 b | |||
|---|---|---|---|---|---|---|
| δC Type | δH (J in Hz) | δC Type | δH (J in Hz) | δC Type | δH (J in Hz) | |
| 1 | 76.7 CH | 3.59 dd (11.2, 4.9) | 78.9 CH | 3.82 dd (11.1, 5.1) | 33.7 CH2 | 1.73–1.75 m |
| 0.93–0.95 m | ||||||
| 2 | 34.0 CH2 | 1.82–1.84 m | 34.7 CH2 | 1.95–1.98 m | 24.7 CH2 | 1.64–1.69 m |
| 1.89 m | 1.98–2.00 m | |||||
| 3 | 79.0 CH | 4.48 dd (12.3, 4.3) | 77.2 CH | 4.44 dd (12.1, 4.7) | 82.4 CH | 4.49 dd (11.8, 4.7) |
| 4 | 38.7 C | 37.5 C | 38.7 C | |||
| 5 | 55.0 CH | 0.91 dd (6.3, 1.8) | 53.8 CH | 0.85–0.87 m | 57.3 CH | 1.07–1.09 m |
| 6 | 19.2 CH2 | 1.67–1.69 m | 17.9 CH2 | 1.53–1.55 m | 18.6 CH2 | 1.56–1.60 m |
| 1.76 dd (12.8, 3.7) | 1.61–1.63 m | 2.16 dd (16.6, 13.0) | ||||
| 7 | 43.5 CH2 | 1.09–1.12 m | 41.8 CH2 | 1.05–1.07 m | 42.7 CH2 | 1.14–1.18 m |
| 1.80–1.82 m | 1.93 dt (13.0, 3.5) | 1.89–1.94 m | ||||
| 8 | 39.0 C | 38.4 C | 38.6 C | |||
| 9 | 61.6 CH | 1.08 d (2.9) | 62.8 CH | 1.18–1.20 m | 62.4 CH | 1.06–1.07 m |
| 10 | 45.2 C | 47.4 C | 43.3 C | |||
| 11 | 70.6 CH | 4.84–4.86 m | 24.4 CH2 | 2.04–2.06 m | 22.8 CH2 | 1.88–1.90 m |
| 2.68–2.70 m | ||||||
| 12 | 47.9 CH2 | 1.90–1.94 m | 41.7 CH2 | 1.51–1.53 m | 42.6 CH2 | 2.01–2.03 m |
| 2.24–2.26 m | 2.00–2.02 m | 1.51–1.53 m | ||||
| 13 | 81.5 C | 80.5 C | 82.3 C | |||
| 14 | 53.2 CH | 1.56 dd (12.8, 4.7) | 52.5 CH | 1.44 dd (12.9, 4.8) | 53.5 CH | 1.50–1.51 m |
| 15 | 17.4 CH2 | 2.40 dd (16.6, 4.8) | 17.2 CH2 | 2.16–2.18m | 17.8 CH2 | 2.39 dd (16.7, 4.7) |
| 2.43 dd (16.7, 4.8) | 1.28–1.32 m | |||||
| 16 | 22.0 CH3 | 1.42 s | 20.1 CH3 | 1.21 s | 20.6 CH3 | 1.23 s |
| 17 | 18.2 CH3 | 1.27 s | 15.5 CH3 | 1.10 s | 16.1 CH3 | 1.10 s |
| 18 | 15.3 CH3 | 1.28 s | 63.5 CH2 | 4.27 d (12.1) | 62.1 CH2 | 3.85 d (12.0) |
| 3.90 d (12.2) | 3.97 d (12.0) | |||||
| 19 | 28.0 CH3 | 0.84 s | 28.2 CH3 | 0.84 s | 29.1 CH3 | 0.89 s |
| 20 | 16.3 CH3 | 0.89 s | 16.2 CH3 | 0.81 s | 17.3 CH3 | 0.87 s |
| 2′ | 167.5 C | 165.5 C | 167.6 C | |||
| 3′ | 98.7 C | 97.8 C | 98.7 C | |||
| 4′ | 165.3 C | 163.5 C | 165.7 C | |||
| 5′ | 102.1 CH | 5.89 s | 100.8 CH | 5.68 s | 102.1 CH | 5.89 s |
| 6′ | 161.8 C | 160.0 C | 161.8 C | |||
| 7′ | 19.5 CH3 | 2.20 s | 19.9 CH3 | 2.19 s | 19.5 CH3 | 2.20 s |
| 1″ | 172.6 C | 171.1 C | 172.8 C | |||
| 2″ | 21.0 CH3 | 2.04 s | 21.3 CH3 | 2.06 s | 21.1 CH3 | 2.03 s |
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Fungal Material
3.3. Fermentation and Extraction
3.4. Isolation and Purification
3.5. Crystal Structure Analysis
3.6. Antibacterial Activity
3.7. Cytotoxicity Activity
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kanokmedhakul, K.; Kanokmedhakul, S.; Suwannatrai, R.; Soytong, K.; Prabpai, S.; Kongsaeree, P. Bioactive meroterpenoids and alkaloids from the fungus Eurotium chevalieri. Tetrahedron 2011, 67, 5461–5468. [Google Scholar] [CrossRef]
- Prompanya, C.; Dethoup, T.; Bessa, L.J.; Pinto, M.M.M.; Gales, L.; Costa, P.M.; Silva, A.M.S.; Kijjoa, A. New Isocoumarin Derivatives and Meroterpenoids from the Marine Sponge-Associated Fungus Aspergillus similanensis sp. nov. KUFA 0013. Mar. Drugs 2014, 12, 5160–5173. [Google Scholar] [CrossRef]
- Prata-Sena, M.; Ramos, A.A.; Buttachon, S.; Castro-Carvalho, B.; Marques, P.; Dethoup, T.; Kijjoa, A.; Rocha, E. Cytotoxic activity of Secondary Metabolites from Marine-derived Fungus Neosartorya siamensis in Human Cancer Cells. Phytother Res. 2016, 30, 1862–1871. [Google Scholar] [CrossRef]
- Wang, W.; Du, L.; Sheng, S.; Li, A.; Li, Y.; Cheng, G.; Li, G.; Sun, G.; Hu, Q.; Matsuda, Y. Genome mining for fungal polyketide-diterpenoid hybrids: Discovery of key terpene cyclases and multifunctional P450s for structural diversification. Org. Chem. Front. 2019, 6, 571–578. [Google Scholar] [CrossRef]
- Ramos, A.A.; Castro-Carvalho, B.; Prata-Sena, M.; Malhão, F.; Buttachon, S.; Dethoup, T.; Kijjoa, A.; Rocha, E. Can marine-derived fungus Neosartorya siamensis KUFA 0017 extract and its secondary metabolites enhance antitumor activity of doxorubicin? An in vitro survey unveils interactions against lung cancer cells. Environ. Toxicol. 2020, 35, 507–517. [Google Scholar] [CrossRef]
- Rajachan, O.; Kanokmedhakul, K.; Sanmanoch, W.; Boonlue, S.; Hannongbua, S.; Saparpakorn, P.; Kanokmedhakul, S. Chevalone C analogues and globoscinic acid derivatives from the fungus Neosartorya spinosa KKU-1NK1. Phytochemistry 2016, 132, 68–75. [Google Scholar] [CrossRef]
- Paluka, J.; Kanokmedhakul, K.; Soytong, M.; Soytong, K.; Kanokmedhakul, S. Meroditerpene pyrone, tryptoquivaline and brasiliamide derivatives from the fungus Neosartorya pseudofischeri. Fitoterapia 2019, 137, 104257. [Google Scholar] [CrossRef]
- Paluka, J.; Kanokmedhakul, K.; Soytong, M.; Soytong, K.; Yahuafai, J.; Siripong, P.; Kanokmedhakul, S. Meroterpenoid pyrones, alkaloid and bicyclic brasiliamide from the fungus Neosartorya Hiratsukae. Fitoterapia 2020, 142, 104485. [Google Scholar] [CrossRef]
- Kuramochi, K.; Ohnishi, K.; Fujieda, S.; Nakajima, M.; Saitoh, Y.; Watanabe, N.; Takeuchi, T.; Nakazaki, A.; Sugawara, F.; Arai, T.; et al. Synthesis and Biological Activities of Neoechinulin A Derivatives: New Aspects of Structure–Activity Relationships for Neoechinulin A. Chem. Pharm. Bull. 2008, 56, 1738–1743. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Li, X.; Li, T.; Dang, H.; Wang, B. Dioxopiperazine Alkaloids Produced by the Marine Mangrove Derived Endophytic Fungus Eurotium Rubrum. Helv. Chim. Acta 2008, 91, 1888–1893. [Google Scholar] [CrossRef]
- Lin, A.; Du, L.; Fang, Y.; Wang, F.; Zhu, T.; Gu, Q.; Zhu, W. iso-α-Cyclopiazonic acid, a new natural product isolated from the marine-derived fungus Aspergillus flavus C-F-3. Chem. Nat. Compd. 2009, 45, 677. [Google Scholar] [CrossRef]
- López-Cobeñas, A.; Cledera, P.; Sanchez, J.D.; López-Alvarado, P.; Ramos, M.; Avendaño López, C.; Menendez, J.C. Microwave-Assisted Synthesis of 2,5-Piperazinediones under Solvent-Free Conditions. Synthesis 2005, 2005, 3412–3422. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, M.; Yuan, C.; Han, Y.; Jia, Z. Sesquiterpenes and Other Constituents from Cacalia deltophylla. Pharmazie 2003, 58, 596–598. [Google Scholar] [CrossRef]
- Sakai, N.; Moriya, T.; Konakahara, T. An Efficient One-Pot Synthesis of Unsymmetrical Ethers: A Directly Reductive Deoxygenation of Esters Using an InBr3/Et3SiH Catalytic System. J. Org. Chem. 2007, 72, 5920–5922. [Google Scholar] [CrossRef]
- Appendino, G.; Gibbons, S.; Giana, A.; Pagani, A.; Grassi, G.; Stavri, M.; Smith, E.; Rahman, M.M. Antibacterial Cannabinoids from Cannabis sativa: A Structure−Activity Study. J. Nat. Prod. 2008, 71, 1427–1430. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv 2015, 71 Pt 1, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Zhong, W.; Wang, J.; Shi, X.; Wei, X.; Chen, Y.; Zeng, Q.; Xiang, Y.; Chen, X.; Tian, X.; Xiao, Z.; et al. Eurotiumins A–E, Five New Alkaloids from the Marine-Derived Fungus Eurotium sp. SCSIO F452. Mar. Drugs 2018, 16, 136. [Google Scholar] [CrossRef] [Green Version]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, S.; Sova, M.; Ergün, S. Antimicrobial activity of trans-cinnamic acid and commonly used antibiotics against important fish pathogens and nonpathogenic isolates. J. Appl. Microbiol. 2018, 125, 1714–1727. [Google Scholar] [CrossRef]
- Qader, M.M.; Hamed, A.A.; Soldatou, S.; Abdelraof, M.; Elawady, M.E.; Hassane, A.S.I.; Belbahri, L.; Ebel, R.; Rateb, M.E. Antimicrobial and Antibiofilm Activities of the Fungal Metabolites Isolated from the Marine Endophytes Epicoccum nigrum M13 and Alternaria alternata 13A. Mar. Drugs 2021, 19, 232. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.H.; Tian, L.; Chen, G.; Xu, N.; Wang, Y.N.; Sun, S.; Pei, Y.-H. Six compounds from marine fungus Y26-02. J. Asian Nat. Prod. Res. 2009, 11, 748–751. [Google Scholar] [CrossRef]

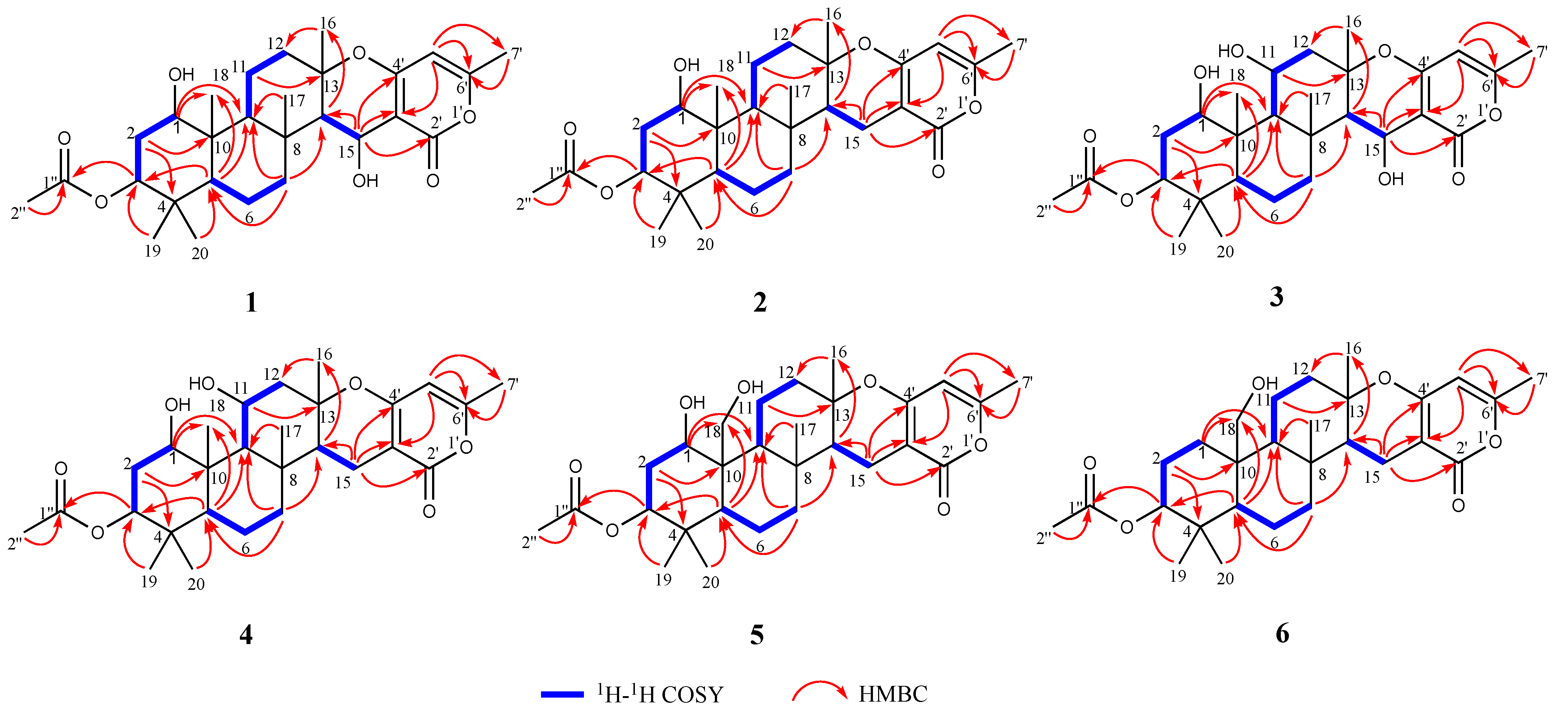



| Compounds | MIC (μg/mL) | |||
|---|---|---|---|---|
| Micrococcus lutea | Klebsiella pneumoniae | Methicillin-Resistant Staphylococcus aureus | Streptococcus faecalis | |
| 1 | 6.25 | 50 | 6.25 | 6.25 |
| 2 | 25 | >100 | 6.25 | 25 |
| 3 | 25 | 25 | 12.5 | >100 |
| 4 | >100 | 6.25 | 25 | 50 |
| 5 | 12.5 | >100 | 12.5 | 12.5 |
| 6 | >100 | >100 | >100 | >100 |
| 7 | >100 | 50 | >100 | 12.5 |
| 8 | >100 | >100 | >100 | >100 |
| 9 | >100 | >100 | >100 | >100 |
| 10 | >100 | >100 | >100 | >100 |
| 11 | >100 | >100 | >100 | >100 |
| 12 | >100 | >100 | >100 | >100 |
| Ciprofloxacin | 0.25 | 0.25 | 0.50 | 0.50 |
| Compounds | IC50 (μM) | |||
|---|---|---|---|---|
| SF-268 | MCF-7 | HepG-2 | A549 | |
| 1 | >128 | >128 | >128 | >128 |
| 2 | 65.64 ± 0.53 | 91.69 ± 6.59 | 107.31 ± 9.83 | 84.54 ± 16.23 |
| 3 | >128 | >128 | >128 | >128 |
| 4 | >128 | >128 | >128 | >128 |
| 5 | 54.78 ± 3.18 | 56.28 ± 2.05 | 58.54 ± 1.52 | 55.33 ± 1.60 |
| 6 | >128 | >128 | >128 | >128 |
| 7 | >128 | >128 | >128 | >128 |
| 8 | 12.75 ± 1.43 | 9.29 ± 0.80 | >128 | 20.11 ± 2.31 |
| 9 | >128 | >128 | >128 | >128 |
| 10 | >128 | >128 | >128 | >128 |
| 11 | >128 | >128 | >128 | >128 |
| 12 | >128 | >128 | >128 | >128 |
| Adriamycin | 1.94 ± 0.01 | 2.00 ± 0.04 | 2.16 ± 0.05 | 2.16 ± 0.07 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.-Y.; Zeng, Q.; Chen, Y.-C.; Zhong, W.-M.; Xiang, Y.; Wang, J.-F.; Shi, X.-F.; Zhang, W.-M.; Zhang, S.; Wang, F.-Z. Chevalones H–M: Six New α-Pyrone Meroterpenoids from the Gorgonian Coral-Derived Fungus Aspergillus hiratsukae SCSIO 7S2001. Mar. Drugs 2022, 20, 71. https://doi.org/10.3390/md20010071
Chen X-Y, Zeng Q, Chen Y-C, Zhong W-M, Xiang Y, Wang J-F, Shi X-F, Zhang W-M, Zhang S, Wang F-Z. Chevalones H–M: Six New α-Pyrone Meroterpenoids from the Gorgonian Coral-Derived Fungus Aspergillus hiratsukae SCSIO 7S2001. Marine Drugs. 2022; 20(1):71. https://doi.org/10.3390/md20010071
Chicago/Turabian StyleChen, Xia-Yu, Qi Zeng, Yu-Chan Chen, Wei-Mao Zhong, Yao Xiang, Jun-Feng Wang, Xue-Feng Shi, Wei-Min Zhang, Si Zhang, and Fa-Zuo Wang. 2022. "Chevalones H–M: Six New α-Pyrone Meroterpenoids from the Gorgonian Coral-Derived Fungus Aspergillus hiratsukae SCSIO 7S2001" Marine Drugs 20, no. 1: 71. https://doi.org/10.3390/md20010071
APA StyleChen, X.-Y., Zeng, Q., Chen, Y.-C., Zhong, W.-M., Xiang, Y., Wang, J.-F., Shi, X.-F., Zhang, W.-M., Zhang, S., & Wang, F.-Z. (2022). Chevalones H–M: Six New α-Pyrone Meroterpenoids from the Gorgonian Coral-Derived Fungus Aspergillus hiratsukae SCSIO 7S2001. Marine Drugs, 20(1), 71. https://doi.org/10.3390/md20010071