Abstract
Iriomoteolide-1a and iriomoteolide-1b are very potent cytotoxic agents, isolated from marine dinoflagellates. We carried out the enantioselective syntheses of the proposed structures of these natural products. However, our analysis of the NMR spectra of the synthetic iriomoteolide-1a and the natural products revealed that the structures of iriomoteolide-1a and iriomoteolide-1b were assigned incorrectly. Based upon our detailed analysis of the spectral data of the synthetic iriomoteolide-1a and the natural products, we rationally designed three diastereomers of the proposed structure of 1 in an effort to assign the correct structures. The key steps of our syntheses of the proposed structures of iriomoteolides involved a highly diastereoselective ene reaction, a carbocupration that utilized a Gilman reagent, a Julia–Kocienski olefination to couple fragments, and Yamaguchi macrolactonization to form the target macrolactone. This synthetic route was then utilized to carry out syntheses of three diastereomers to the proposed structure of 1. These diastereomeric structures show close similarities to natural iriomoteolide-1a; however, there were differences in their spectral data. While natural iriomoteolides exhibited potent cytotoxicies, our preliminary biological evaluation of synthetic iriomoteolide-1a, iriomoteolide-1b, and all three synthetic derivatives did not show any appreciable cytotoxic properties.
1. Introduction
Marine natural products are a great source of structurally intriguing bioactive molecules with novel modes of action [1,2]. The field of marine natural products is immensely important in modern drug discovery. Already, many new approved drugs with interesting biological mechanisms are in pharmacies [3,4]. The field has great potential in modern medicine; however, it is vastly unexplored. The synthesis of these bioactive molecules and exploration of structure activity relationship studies are playing an important role in drug discovery today [5,6]. Iriomoteolide-1a (1) (Figure 1) is a 20-memembered cytotoxic macrolide, which was isolated by Tsuda and co-workers from a benthic HYA024 strain of dinoflagellate Amphidinium sp. collected off Iriomote Island, Japan in 2007 [7,8]. It displayed very potent cytotoxicity against human B lymphocyte DG-75 cells, with an IC50 value of 2 ng/mL. Furthermore, it exhibited cytotoxicity against Epstein–Barr virus (EBV)-infected human lymphocyte Raji cells (IC50 = 3 ng/mL) [7,8]. The initial structure of iriomoteolide-1a (1) was determined based on extensive 2D-NMR studies and mass spectroscopic analyses. The relative and absolute configurations were assigned based on the NMR studies, conformational analyses of derivatives of 1 with Mosher’s reagent. Later, Tsuda et al. reported the isolation of iriomoteolide-1b (2) [8], which was isolated from the same HY A024 strain of dinoflagellate Amphidinium sp. Iriomoteolide-1b (2) is structurally related to iriomoteolide-1a (1). Instead of a 6-membered hemiketal ring at the C9–C13 position and an exo-methylene group at C11 in iriomoteolide-1a (1), iriomoteolide-1b (2) possesses a ketone at C13 conjugated with a Z-double bond at C11–C12 and a hydroxyl group at C9. Treatment of iromoteolide-1a (1) with triethylamine in dichloromethane for 168 h furnished a polar product. The 1H NMR analyses reveal that the product is identical to iriomoteolide-1b (2). However, the IC50 value of 2 against DG-75 cells is found to be less potent than that of 1 (IC50 900 ng/mL) [7,8]. Iriomoteolides targets have attracted considerable synthetic interest, leading to the syntheses of various segments of iriomoteolides [9,10,11,12,13,14,15,16,17]. In addition, synthesis of the proposed structures of iriomoteolides, as well as syntheses of structural variants of iriomoteolides, have been reported [18,19,20,21,22,23]. Thus far, neither the biological mechanism of action nor the correct structures of iriomotelides have been reported. Herein, we report our revised syntheses of the proposed structures of iriomoteolide-1a and -1b. Our convergent and highly stereoselective synthetic route was utilized for the syntheses of three rationally designed structural variants for structural elucidation and biological studies.
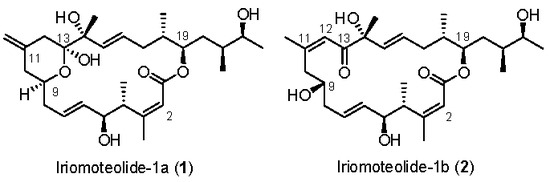
Figure 1.
Proposed structures of iriomoteolide-1a (1) and -1b (2).
2. Results and Discussion
2.1. Synthetic Plan
Our retrosynthetic analysis is outlined in Figure 2. Our convergent synthetic strategy involves a Julia–Kocienski olefination [24,25] of aldehyde 3 and sulfone 4. The resulting trans-olefin intermediate was converted to iriomoteolide macrolatone, using Yamaguchi macrolactonization [26] as the key step to build the 20-membered macrolactone. The synthesis of C1–C15 fragment 3 relied upon another Julia–Kocienski olefination from sulfone 5 and aldehyde 6. An ene reaction of aldehyde 7 and olefin 8 was designed to furnish Sulfone 5. A Cu(I)-mediated epoxide ring opening reaction provides the olefin 8 from expoxide 9, which was readily prepared from the known alcohol 10. The synthesis of C16–C23 segment 4 was planned from alkene 11 by hydroboration-oxidation, followed by conversion of the resulting alcohol to sulfone derivative 4. Alkene 11 was be synthesized using an asymmetric crotylboration of the aldehyde derived from 12 as the key step. Optically active alcohol 12 can be conveniently obtained from asymmetric crotylboration of acetaldehyde.
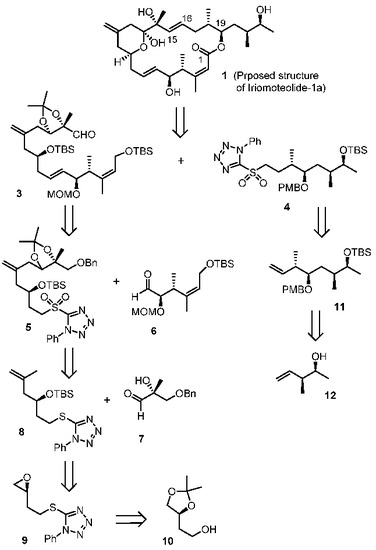
Figure 2.
Retrosynthetic approach to iriomoteolide-1a (1).
2.2. Synthesis of C7–C15 Fragment 5
The synthesis of C7–C15 fragment 5 was planned by using a diastereoselective ene reaction [27,28], as outlined in Scheme 1. Treatment of the known alcohol 10 with 1-phenyl-1H-tetrazole-5-thiol under Mitsunobu’s condition [29] afforded the corresponding sulfide. Deprotection of the acetonide group gave the diol 13. Tosylation of diol 13 in the presence of triethylamine and dibutyltin oxide, followed by treatment of potassium carbonate in a mixture of methanol and dichloromethane, furnished the epoxide 14. Reaction of epoxide 14 with isopropenylmagnesium bromide in the presence of a catalytic amount of copper(I) cyanide resulted in the corresponding alcohol. Interestingly, Curran and co-workers reported that the reaction of the enantiomer of 14 with an alkynyllithium reagent furnished a by-product that resulted from the displacement of 1-phenyltetrazole [30]. The alcohol from 14 was then protected with TBSCl to afford the silyl ether 8. A SnCl4-mediated ene reaction of aldehyde 7 and olefin 8 was carried out to provide the corresponding alcohol in 76% yield as a mixture of diastereomers with good selectivity (8:1 dr). The use of TiCl4 as a Lewis acid led to the desired product but with a lower yield (40%). A chelation-controlled addition between the α–hydroxyl group and the aldehyde resulted in good diastereoselectivity (8:1 dr) for the ene reaction [31,32]. The diol was protected as an acetonide derivative to afford 15. The absolute configuration of the new chiral center at C13 was identified as drawn in 15 (R-configuration) using 1H-NMR NOESY experiments. NOESY between H13 and H15, and NOESY between H12 and H27 were observed, as shown in acetonide 15.
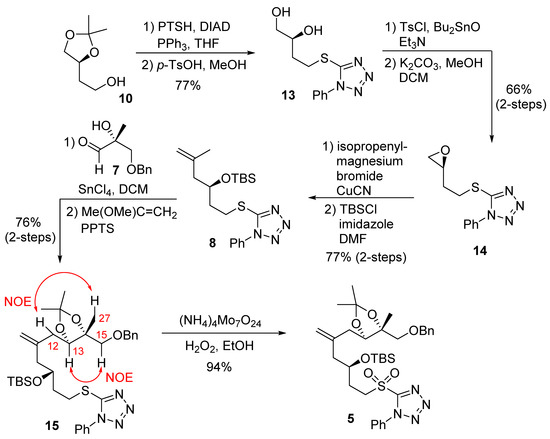
Scheme 1.
Synthesis of C7–15 fragment 5.
While this new chiral center would be removed in the late stage of the synthesis via oxidation to the corresponding ketone, good diastereoselectivity in the ene reaction simplified the NMR spectra. Sulfide 15 was then oxidized by ammonium molybdate and hydrogen peroxide to afford the sulfone 5 in good yield.
2.2.1. Syntheses of C16–C23 Segment 4 and C1–C6 Fragment 6
The synthesis of C16–C23 segment 4 was carried out, as shown in Scheme 2. Asymmetric crotylboration of acetaldehyde using cis-2-butene and (+)-B-methoxy-diisopinocamphenylborane using the protocol developed by Brown and co-workers [33,34] furnished optically active syn-alcohol 12. As reported previously [19], alcohol functionality was protected as a TBS-ether and hydroboration-oxidation of the alkene under standard condition afforded alcohol 16 in good yield. Swern oxidation of 16 provided the aldehyde, which was subjected to Brown’s asymmetric crotylboration using (−)-B-methoxy-diisopinocamphenylborane and trans-2-butene to afford alcohol 17 in good yield and with excellent diastereoselectivity (10:1). Alcohol 17 was protected as a PMB-ether and hydroboration-oxidation of the olefin furnished alcohol 18. Syntheses of derivatives of 17 and 18 with different protecting groups have been reported [17]. This was converted to sulfone 4 by the Mitsunobu reaction, followed by oxidation of the sulfide to sulfone.
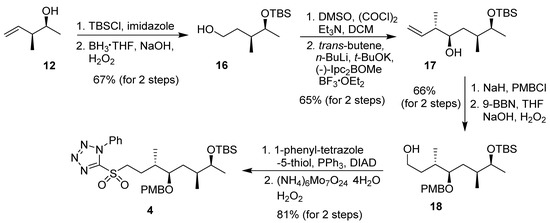
Scheme 2.
Synthesis of C16–C23 fragment 4.
The synthesis of C1–C6 fragment 6 is shown in Scheme 3. The racemic alcohol 19, obtained from the aldol reaction of tert-butylacetate and acrolein, was subjected to immobilized lipase PS-30 catalyzed kinetic resolution in pentane in the presence of excess vinyl acetate, at 30 °C for 19 h, to provide enantio-enriched (R)-19 in 98% ee, along with the corresponding enantiomeric acetate derivative [35]. Treatment of (R)-19 with lithium diisopropylamide, followed by the reaction of the resulting dianion with methyl iodide as described by previously [36], afforded the corresponding anti-alcohol as a single isomer by 1H NMR analysis. The resulting alcohol was protected as a MOM-ether. Reduction of the resulting ester with LAH furnished alcohol 20. Synthesis of derivative of 20 with different protecting groups was reported [17]. Swern oxidation, followed by Corey–Fuchs’ homologation [37] of the aldehyde, provided the corresponding dibromo olefin. Treatment of the dibromide with butyllithium, followed by reaction of the resulting alkynyl anion with methyl chloroformate, furnished alkynyl ester 21 in excellent yield. A carbocupration of alkynyl ester 21 was carried out with freshly prepared Gilman reagent [38] at −40 °C to provide Z-olefin 22 as a single product in excellent isolated yield. The observed NOE between the protons at C2 and Me at C3 is consistent with the assigned Z geometry in ester 22. DIBAL-H reduction of 22, followed by protection of the resulting alcohol with TBSCl, furnished the corresponding silyl ether. Selective oxidative cleavage of the terminal olefin provided the C1–C6 fragment 6.
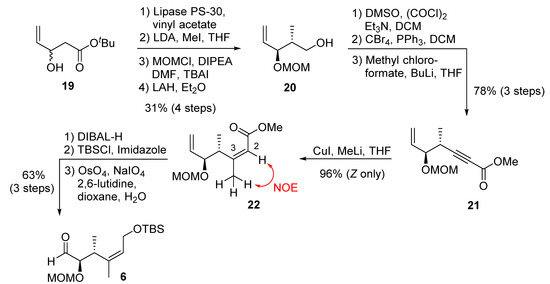
Scheme 3.
Synthesis of C1–C6 fragment 6.
With sulfone 5 and aldehyde 6 in hand, total syntheses of the proposed structures of iriomoteolide-1a and 1b were successfully achieved. The synthesis featured two successive Julia–Kocienski olefinations. As shown in Scheme 4, the first Julia–Kocienski reaction between sulfone 5 and aldehyde 6 afforded trans-olefin 23 in excellent yield. Removal of the benzyl ether, followed by DMP oxidation [39,40] of the resulting alcohol, afforded aldehyde 3. A second Julia–Kocienski reaction of aldehyde 3 and sulfone 4 furnished E-olefin 24 as the only isolated product in good yield. Removal of PMB ether, followed by selective removal of the primary TBS-ether with NH4F, resulted in allylic alcohol 25. Oxidation of 25 with MnO2 followed by NaClO2 afforded the corresponding carboxylic acid [41]. Yamaguchi macrolactonization furnished macrolactone 26 in good yield. Macrolactone 26 was converted to the proposed structures of irimoteolide-1a and irimoteolide-1b, as shown in Scheme 5. Treatment of 26 with HF•Py followed by aqueous AcOH resulted in tetraol derivative 27. Bromocatecholborane promoted the removal of the MOM group and furnished the corresponding pentaol derivative. Treatment of the free alcohols with TESCl and DMAP selectively provided TES- ether derivative 28 in good yield. DMP oxidation [39,40] of the secondary alcohol provided the corresponding ketone and removal of the TES groups with exposure to HF•Py furnished iriomoteolide-1a (1) in 56% yield and iriomoteolide-1b (2) in 17% yield after silica gel chromatography. The 1H-NMR and 13C-NMR of synthetic iriomoteolide-1a and iriomoteolide-1b did not match with the reported data for these natural products [7,8].
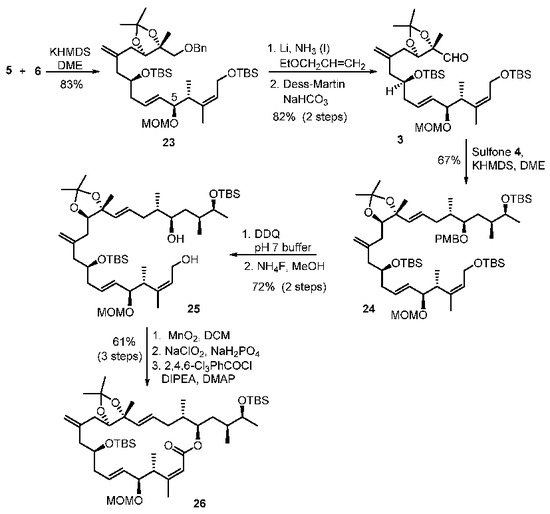
Scheme 4.
Synthesis of macrolactone intermediate 26.
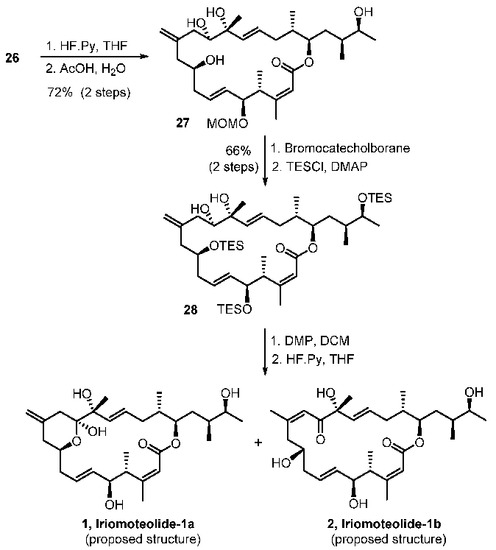
Scheme 5.
Synthesis of proposed structures of iriomoteolide-1a and -1b.
Although the 1H NMR and 13C NMR spectral data of our synthetic iriomoteolide-1a (1) are comparable to those of independent work reported from other groups [18,20], neither data of 1 nor data of 2 matched those of natural iriomoteolide-1a and -1b (Please see Supplementary Materials for NMR comparison). This suggested that the structures of both natural iriomoteolide-1a and iriomoteolide-1b have been assigned incorrectly. While there are many minor differences, the major discrepancies involve the 1H and 13C shifts at C4 (3.98 ppm and 40.6 ppm, respectively, for synthetic iriomoteolide-1a compared to 2.46 ppm and 47.9 ppm, respectively, for the natural product). In addition, there is a distinction of chemical shifts at C24 (1.96 and 20.8 ppm, respectively, for synthetic 1 compared to 2.12 and 23.8 ppm, respectively, for natural 1). These discrepancies reveal that the α,β-unsaturated double bond configuration might be E instead of Z. In addition, the structure may be an epimer at the C4 and C5 positions. Based on the NMR analysis, three diastereomers, 29, 30 and 31 (Figure 3), were designed. The syntheses of these structural variants were carried out utilizing our convergent synthetic route.
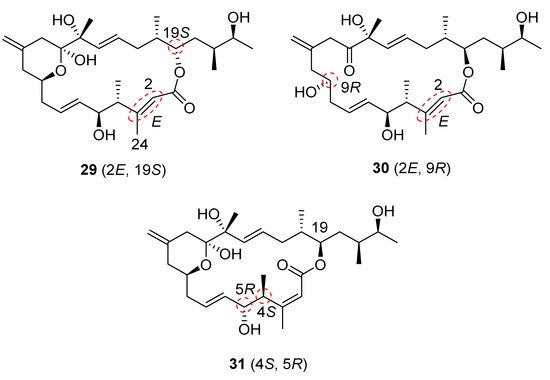
Figure 3.
Structures of diastereomers 29, 30 and 31.
2.2.2. Synthesis of Diastereomer 29
The synthesis of diastereomer 29 requires alteration of olefin geometry at C2–C3. As shown in Scheme 6, a carbocupration of alkynyl ester 21 with Gilman reagent at 0 °C in the presence of TMSCl resulted in the desired E-olefin as a major isomer (E:Z = 7:3) [38]. DIBAL-H reduction furnished the alcohol 32, which was separated from its Z-isomer by flash column chromatography over silica gel. Protection of alcohol with tert-butyldimethylsilyl chloride and oxidative cleavage of the terminal olefin provided the C1–C6 fragment aldehyde 33 for diastereomer 29. Treatment of sulfone 5 with slightly less than one equivalent of KHMDS, followed by exposure to aldehyde 33, afforded the E-olefin. However, we found that the use of more than one equivalent base led to the epimerization of the chiral center on C5. Removal of the benzyl group followed by Dess–Martin oxidation [39,40] provided the C1–C15 fragment, aldehyde 34.
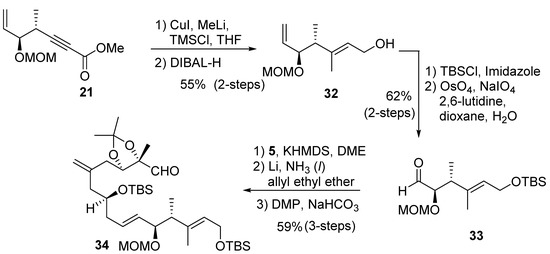
Scheme 6.
Synthesis of aldehyde 34.
The synthesis of diastereomer 29 is shown in Scheme 7. Brown asymmetric crotyllation, by utilizing (+)-B-methoxydiisopino-campheylborane and cis-2-butene with aldehyde 35 derived from 16, provided syn-alcohol 36 in good diastereoselectivity (8:1 dr). PMB protection and hydroboration-oxidation provided alcohol 37. Alcohol 37 was converted to sulfone 38, as described previously. A Julia–Kocienski olefination [24,25] between aldehyde 34 and sulfone 38 using KHMDS in 1,2-dimethoxyethane furnished (15E)-olefin in good selectivity (E:Z = 9:1). The use of THF as a solvent caused an increase in Z-olefin by-product (E:Z = 4:1). Removal of the PMB ether and primary TBS ether led to diol 39. MnO2 oxidation and Pinnick oxidation [41] gave the corresponding seco-acid. Yamaguchi esterification furnished the macrolactone 40. A protecting group exchange protocol [26] afforded the corresponding vicinal diol. A one-pot reaction with the addition of HF•Py and Parikh–Doering oxidation led to isomer 29 exclusively, without isomerization of the exomethylene group at C11.
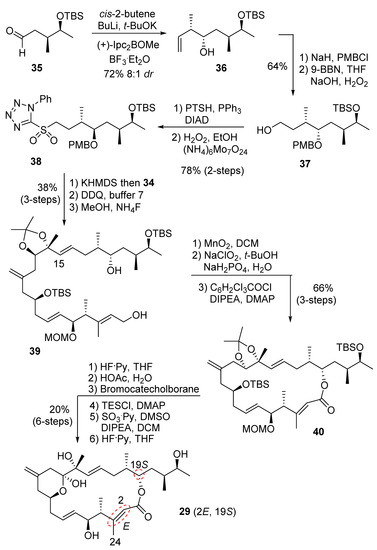
Scheme 7.
Synthesis of diastereomer 29.
2.2.3. Synthesis of Diastereomer 30
The synthesis of diastereomer 30 requires alteration of the stereochemistry at C9. Our synthesis started with commercially available (R)-D-malic acid. As shown in Scheme 8, reduction of malic acid with the borane dimethylsulfide complex furnished the corresponding triol. Selective protection of the diol with acetone and p-TsOH furnished the alcohol 41 with desired chirality at C9. Using the previous synthetic route, enantiomeric sulfone ent-8 was obtained. Sulfone ent-8 was then converted to the corresponding macrolactone, as described previously. Protecting the group exchange protocol and oxidation, followed by the removal of the TES group as described previously, provided diastereomeric structure 30, where the hydroxyl ketone stays as a δ-hydroxyl-ketone instead of cyclic hemiketal, as revealed from the analysis of its 1H NMR and 13C NMR spectral data.
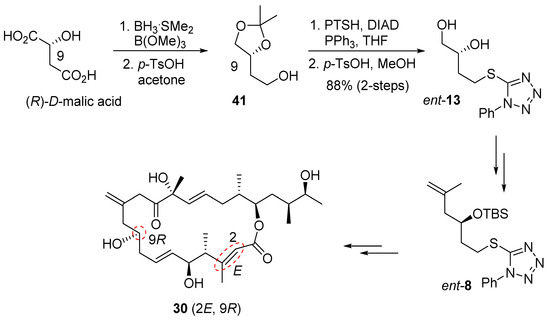
Scheme 8.
Synthesis of iriomoteolide diastereomer 30.
2.2.4. Synthesis of Diastereomer 31
Our synthesis of this diastereomeric structure of iriomoteolide-1a required altering the configurations at the C4 and C5 chiral centers. Therefore, the enantiomeric C1–C5 segment aldehyde, ent-6, was synthesized as shown in Scheme 9. Our enzymatic resolution of racemic alcohol 19 provided nearly a 1:1 mixture of (R)-19 alcohol and its acetate derivative 42 in excellent yield. Saponification of acetate by treatment of K2CO3 in MeOH at −30 °C provided (S)-19 alcohol in 92% ee. Seebach–Fráter alkylation [36] of (S)-19 with methyl iodide, as described in Scheme 3, furnished the corresponding anti-alcohol. Protection of alcohol as an MOM ether, followed by the reduction of the ester using LAH, afforded ent-20 alcohol. This was then converted to C1–C6 segment aldehyde ent-6. Aldehyde ent-6 was then exposed to Julia–Kocienski olefination with sulfone 5 to provide the corresponding trans-olefin, which was converted to the corresponding diastereomeric aldehyde, as described in Scheme 4. A second Julia–Kocienski olefination [24,25] with sulfone 4 provided the carbon framework for diastereomer 31. This was converted to the macrolactone, followed by the final target diastereomer 31 by following the steps described in Scheme 5.
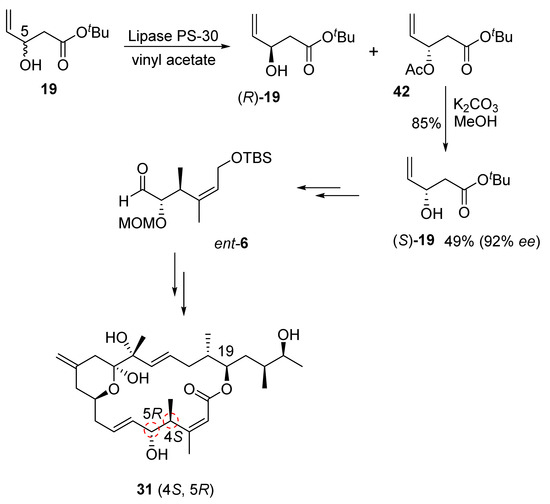
Scheme 9.
Synthesis of ent-19 alcohol and iriomoteolide-1a diastereomer 31.
NMR spectra analysis of these diastereomers revealed that some individual chemical shifts, such as H4 and H24 of 29, H19 and H26 of 31, came closer to those of the natural product. These observations suggest that there may be an E-enoate and/or C4 and C5-epimers in the natural product. However, none of these isomers match the natural product. We carried out biological evaluations of the synthetic iriomoteolide-1a (1), -1b (2) and structural variants 29, 30, and 31. However, none of these compounds show any appreciable cytotoxicity. Yang and Dai’s research groups also reported their independent synthetic approach of several other diastereomers, such as 43 [20,22], 44 [20], 45 [20], 46 [22] and 47 [20], as shown in Figure 4. Unfortunately, none of these structures match that of natural iriomoteolide-1a. The real structure of this biologically potent natural product remains veiled, waiting for collective effort in the synthetic community.
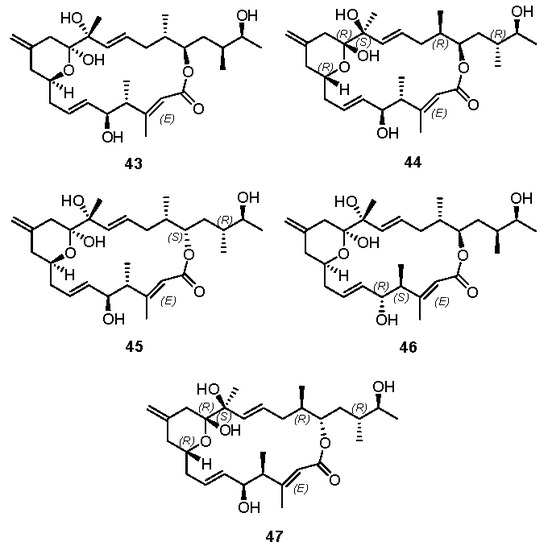
Figure 4.
Structures of diastereomers 43, 44, 45, 46, and 47.
3. Materials and Methods
With regard to the general techniques used in this study, all moisture sensitive reactions were carried out under argon atmosphere. Anhydrous solvents were obtained as follows: THF and DME distilled from sodium and benzophenone; dichloromethane, toluene, triethylamine and diisopropylamine, distilled from CaH2. Column chromatography was performed with 230–400 mesh silica gel under low pressure of 5–10 psi. TLC was carried out with silica gel 60-F-254 plates, visualized under UV light and stained with phosphomolybdic acid. In addition, 1H NMR and 13C NMR spectra were recorded on Bruker Avance ARX- 400 (400 and 100 MHz), or Bruker DRX500 (500 and 125 MHz) spectrometers. High and low resolution mass spectra were carried out by the Mass Spectroscopy Center at Purdue University. HPLC analysis and preparative HPLC were performed on Agilent 1100 Series instruments (Agilent Technologies, Santa Clara, CA, USA, Agilent 1200 Series Autosampler used for analytical work).
(S)-4-(1-phenyl-1H-tetrazol-5-ylthio)butane-1,2-diol (13): To a stirred solution of alcohol 10 (1.505 g, 10.3 mmol), 1-(4-Hydroxyphenyl)-1H-tetrazole-5-thiol (3.67 g, 20.6 mmol) and triphenylphosphine (4.05 g, 15.5 mmol) in THF (30 mL), we added DIAD (3.6 mL, 18.5 mmol) at 0 °C. The reaction mixture was warmed up to rt and stirred overnight, before it was poured into sat NaHCO3 (20 mL). The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over MgSO4 and concentrated in vacuo. Column chromatography (20% EtOAc/hexanes) provided the corresponding sulfide as colorless oil (2.68 g, 85%). Rf value (EtOAc/hexane 1:1): 0.75; [α]D20 = +12.5 (c = 1.0, CHCl3); IR (film, cm−1) 3384, 2936, 1644, 1596, 1499, 1462, 1388, 1318, 1280, 1074, 760; 1H NMR (400 MHz, CDCl3): 7.58 (brs, 5 H), 4.13–3.85 (m, 2 H), 3.81–3.68 (m, 1 H), 3.68–3.56 (m, 2 H), 3.55–3.42 (m, 1 H), 2.55 (brs, 1 H), 2.09–1.90 (m, 2 H); 13C NMR (100 MHz, CDCl3): 155.1, 133.5, 130.4, 129.9, 124.0, 69.5, 66.4, 33.7, 29.8.
To a stirred solution of the above acetonide (2.60 g, 8.49 mmol) in MeOH (100 mL), we added p-TsOH (320 mg, 1.70 mmol) at rt and stirred for 24 h. Et3N (2 mL) was added at 0 °C to quench the reaction. The mixture was concentrated in vacuo. Flash chromatography on silica gel (5% MeOH/CHCl3) resulted in diol 13 (2.06 g, 91%) as a white solid. Rf value (EtOAc/hexane/MeOH 80:20:6): 0.5; [α]D20 = +10.7 (c = 0.5, CHCl3); IR (film, cm−1) 3411, 3384, 2936, 1644, 1596, 1499, 1462, 1388, 1318, 1280, 1075, 759; 1H NMR (400 MHz, CDCl3): 7.58 (brs, 5 H), 4.13–3.85 (m, 2 H), 3.81–3.68 (m, 1 H), 3.68–3.56 (m, 2 H), 3.55–3.42 (m, 1 H), 2.55 (brs, 1 H), 2.09–1.90 (m, 2 H); 13C NMR (100 MHz, CDCl3): 155.1, 133.5, 130.4, 129.9, 124, 69.5, 66.4, 33.7, 29.8.
(S)-5-(3-(oxiran-2-yl)propyl)-1-phenyl-1H-tetrazole (14): To a stirred solution of diol 13 (2.06 g, 7.74 mmol) in DCM (60 mL), Bu2SnO (3.85 g, 15.5 mmol), triethylamine (1.3 mL, 9.29 mmol) and tosyl chloride (1.58 g, 8.12 mmol) were added at 0 °C. The reaction mixture was stirred for 6 h, followed by dilution with water (10 mL). The organic layer was separated and the aqueous layer was extracted with DCM. The combined organic extracts were dried over anhydrous MgSO4 and concentrated. The crude product was purified by flash column chromatography on silica gel (ethyl acetate–hexanes 1:1) to yield the corresponding tosylate (2.52 g, 81%). Rf value (EtOAc/hexane 1:1): 0.4; [α]D20 = +12.0 (c = 0.65, CHCl3); IR (film, cm−1): 3400, 3060, 2946, 1597, 1499, 1387, 1357, 1243; 1H NMR (400 MHz, CDCl3): 7.75 (d, J = 8.2 Hz, 2 H), 7.53 (s, 5 H), 7.30 (d, J = 8.2 Hz, 2 H), 4.03–3.92 (m, 4 H), 3.50–3.43 (m, 2 H), 2.03–1.87 (m, 2 H), 2.39 (s, 3 H); 13C NMR (100 MHz, CDCl3): 154.4, 144.9, 133.3, 132.3, 130.1, 129.8, 129.7, 127.7, 123.6, 73.1, 66.9, 32.7, 29.2, 21.4.
To a stirred solution of the above tosylate (3.54 g, 8.79 mmol) in CH3OH–DCM (9:1, 90 mL), K2CO3 (1.58 g, 11.4 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature for 1 h, concentrated, and then diluted with dichloromethane (30 mL) and water (10 mL). The organic layer was separated and the aqueous layer was extracted with dichloromethane. The combined organic extracts were dried over anhydrous MgSO4 and concentrated to provide 14 (1.58 g, 82%) as a colorless oil. Rf value (EtOAc/hexane 1:1): 0.65; [α]D20 = −15.5 (c = 1, CHCl3); IR (film, cm−1): 3056, 2991, 2924, 1596, 1500, 1461; 1H NMR (400 MHz, CDCl3): 7.56–7.49 (m, 5 H), 3.54–3.45 (m, 2 H), 3.06–3.01 (m, 1 H), 2.78–2.74 (m, 1 H), 2.52 (dd, J = 4.6, 2.7 Hz, 1 H) 2.29–2.21 (t, J = 4.6, 1 H),1.92 (td, J = 14.2, 6.9 Hz, 1 H); 13C NMR (100 MHz, CDCl3): 153.9, 133.5, 130.1, 29.8, 129.8, 123.7, 50.6, 46.8, 32.0.
(R)-5-(3-(tert-butyldimethylsilyloxy)-5-methylhex-5-enylthio)-1-phenyl-1H-tetrazole (8): To a stirred solution of epoxide 14 (1.49 g, 6 mmol) and CuCN (54 mg, 0.6 mmol) in THF (40 mL) at −78 °C, we added isopropenylmagnesium bromide (3.6 mL, 0.9 mmol). The resulting suspension was warmed up to 0 °C and stirred for 30 min. The reaction mixture was cooled again to −78 °C and more vinylmagnesium bromide (12 mL, 6 mmol) was added dropwise. The reaction mixture was warmed up to 0 °C and stirred for 1 h, before 20 mL of saturated NH4Cl and 10 mL of NH4OH were added to quench the reaction. The layers were separated and the aqueous layer was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with brine and dried over anhydrous magnesium sulfate. Filtration and concentration under reduced pressure gave a crude product. Flash chromatography on silica gel (20% EtOAc/hexanes) afforded the corresponding alcohol as a colorless oil (1.6 g, 92%). Rf value (EtOAc/hexane 1:2) 0.25; [α]D20 = +15.8 (c = 2, CHCl3); IR (film, cm−1): 3414, 2932, 1596, 1500, 1388, 1074, 761; 1H NMR (400 MHz, CDCl3) δ 7.63–7.48 (m, 1H), 4.85 (s, 1H), 4.77 (s, 1H), 3.89 (ddq, J = 12.8, 6.5, 3.3 Hz, 1H), 3.58–3.48 (m, 1H), 2.75 (s, 1H), 2.20 (d, J = 6.5 Hz, 1H), 2.09–1.97 (m, 1H), 1.89 (ddd, J = 21.4, 8.0, 6.2 Hz, 1H), 1.73 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 154.7, 142.1, 133.5, 130.1, 129.7, 123.7, 113.6, 66.8, 45.8, 36.6, 29.9, 22.4.
To a stirred solution of the above alcohol (942mg, 3.24 mmol) in DMF (6 mL), we added imidazole (353 mg, 5.18 mmol) and TBSCl (538 mg, 3.57 mmol), respectively, at 0 °C. The reaction mixture was stirred at 23 °C for 12 h. A solution of saturated NaHCO3 (aq) was added and the aqueous layer was extracted by diethyl ether. The combined organic extracts were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography (4% EtOAc/hexanes) gave the silyl ether 8 (1.25 g, 95%) as a colorless oil. Rf value (EtOAc/hexane 1:10) 0.45; [α]D20 = +12.5 (c = 1, CHCl3); IR (film, cm−1): 2953, 2929, 2857, 1598, 1500, 1387, 1074, 821, 775; 1H NMR (400 MHz, CDCl3) δ 7.81–7.37 (m, 5H), 4.77 (s, 1H), 4.70 (s, 1H), 3.96 (tdd, J = 7.5, 5.4, 4.0 Hz, 1H), 3.55–3.34 (m, 2H), 2.27 (dd, J = 13.6, 5.3 Hz, 1H), 2.17 (dd, J = 13.6, 7.6 Hz, 1H), 2.09–1.95 (m, 1H), 1.93–1.79 (m, 1H), 1.71 (s, 3H), 0.88 (s, 9H), 0.06 (d, J = 1.0 Hz, 6H); 13C NMR (100 MHz, CDCl3) δ 154.3, 141.8, 133.6, 129.9, 129.7, 123.7, 113.5, 69.3, 45.8, 35.6, 29.5, 25.8, 22.8, 17.9, −4.4, −4.7. MS (ESI, m/z) [M+Na]+ 427.2.
Acetonide 15: To a stirred solution of olefin 8 (1.23 g, 3 mmol) and aldehyde 7 (645 mg, 3.3 mmol) in DCM (30 mL), we added SnCl4 (4.5 mL, 1 M soln in DCM, 4.5 mmol) at −78 °C; the reaction mixture was warmed up to 0 °C over 1 h and stirred for 4 h. The reaction mixture was then poured into sat NaHCO3 (20 mL) with crushed ice. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:3) to give the corresponding diol (1.4 g, 78% yield). To a stirred solution of the diol (1.4 g, 2.34 mmol) and 2-methoxypropene (0.66 mL, 7 mmol) in DCM (40 mL), we added PPTS (50 mg, 0.2 mmol) at 0 °C. The reaction mixture was stirred at 0 °C for 1 h before Et3N (1.0 mL) was added. The solvents were removed in vacuo and the crude product was purified by flash chromatography (EtOAc/hexane 1:10) to give the acetonide 12 (1.47 g, 98%) as a colorless oil. Rf value (EtOAc/hexane 1:4): 0.55; [α]D20 = −1.8 (c = 1, CHCl3); IR (film, cm−1) 3068, 2886, 1500, 1410, 1097; 1H NMR (400 MHz, CDCl3) δ7.56–7.53 (m, 5H), 7.32–7.25 (m, 5H), 4.90 (s, 1H), 4.85 (s, 1H), 4.55 (AB, JAB = 12.0 Hz, ΔυAB = 20.7 Hz, 2H), 4.13–4.10 (m, 1H), 4.0–3.97 (m 1H), 3.52–3.40 (m, 2H), 3.43–3.38 (m, 2H), 2.50–2.45 (m, 1H), 2.22–2.13 (m, 3H), 2.02–1.99 (m, 1H), 1.86–1.81 (m, 1H), 1.46 (s, 3H), 1.35 (s, 3H), 1.11 (s, 3H), 0.88 (s, 9H), 0.06 (s, 6H); 13C NMR (400 MHz, CDCl3) δ 154.3, 143.0, 138.2, 133..6, 130.0, 129.7, 128.2, 127.5, 127.3, 123.7, 114.8, 107.2, 81.6, 78.7, 74.8, 73.5, 69.3, 44.2, 36.4, 35.4, 29.4, 28.6, 26.5, 25.8, 19.2, 18.0, −4.3, −4.8; MS (ESI, m/z) [M + Na]+ 661.3.
Sulfone 5: To a stirred solution of thus obtained sulfide 15 (502 mg, 0.79 mmol) in ethanol (13 mL), we added a soln of ammonium molybdate (320 mg, 0.26 mmol) in hydrogen peroxide (1.6 mL) and water (0.8 mL) at rt. The reaction mixture was stirred for 12 h and poured into a mixture of sat NaHCO3 (10 mL) and sodium thiosulfate (10 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:10) to give the sulfone 5 as a colorless oil (519 mg, 98% yield). Rf value (EtOAc/hexane 1:4): 0.5; [α]D20 = −2.6 (c = 1, CHCl3); IR (film, cm−1) 3038, 2887, 1512, 1215, 1097; 1H NMR (400 MHz, CDCl3) δ7.69–7.58 (m, 5H), 7.33–7.27 (m, 5H), 4.94 (s, 1H), 4.87 (s, 1H), 4.56 (AB, JAB = 12.0 Hz, ΔυAB = 20.7 Hz, 2H), 4.12–4.05 (m, 1H), 4.07–4.03 (m 1H), 3.90–3.73 (m, 2H), 3.47–3.41 (m, 2H), 2.54–2.50 (m, 1H), 2.22–2.17 (m, 2H), 2.15–2.10 (m, 2H), 2.0–1.92 (m, 1H), 1.46 (s, 3H), 1.36 (s, 3H), 1.14 (s, 3H), 0.90 (s, 9H), 0.09 (s, 3H), 0.08 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 153.3, 142.8, 138.1, 133, 131.3, 129.6, 128.3, 127.5, 127.4, 125, 115.2, 107.3, 81.5, 79.1, 77.4, 76.7, 75, 73.5, 68.2, 52.3, 43.9, 36.4, 28.6, 28, 26.5, 25.8, 19.2, 17.9, −4.4, −4.9; MS (ESI, m/z) [M + Na]+ 693.3. HRMS (ESI) [M + Na]+ calcd for C34H50N4O6SSiNa 693.3118, found 693.3111.
(2R,3S)-3-(methoxymethoxy)-2-methylpent-4-en-1-ol (20): To a stirred solution of racemic alcohol 19 (15.2 g, 88.4 mmol) in vinyl acetate (60 mL) and pentane (120 mL), we added immobilized lipase PS 30 (20% on celite, 15.2 g); the suspension was stirred at 23 °C for 30 h, monitored by 1H NMR, until a half conversion was obtained. Suction filtration furnished a crude product, which was purified by flash chromatography (EtOAc/hexane 1:1) to give the (+)-alcohol as a colorless oil (7.70 g, 44%), along with corresponding acetate (9.3 g, 49%). Rf value (EtOAc/hexane 1:5) 0.45; [α]D20 = +8.8 (c = 1.7, CHCl3); IR (film, cm−1): 3422, 2981, 1744, 1372, 1236, 1026, 947; 1H NMR (400 MHz, CDCl3) δ 5.90–5.68 (m, 1H), 5.22 (d, J = 17.3 Hz, 1H), 5.11–4.95 (m, 1H), 4.40 (s, 1H), 3.38 (s, 1H), 2.54–2.18 (m, 2H), 1.38 (d, J = 1.5 Hz, 9H); 13C NMR (100 MHz, CDCl3) δ 171.4, 139.0, 114.9, 81.1, 68.9, 42.1, 27.9.
To a stirred solution of diisopropylamine (15.8 mL, 0.112 mol) in THF (80 mL) at −78 °C, we added n-BuLi (1.6 M in hexane, 71.8 mL, 0.107 mol) dropwise. The mixture was kept at −78 °C for 20 min before a solution of the above alcohol (7.4 g, 42.9 mmol) was added dropwise. After another 20 min of stirring, MeI (6.7 mL, 0.107 mol) was added to the reaction mixture in a dropwise manner. The reaction was stirred at −10 °C for 4 h before 50 mL sat NH4Cl was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 50 mL). The combined organic solution was washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (10% EtOAc/hexanes) afforded the anti-methyl alcohol (5.84 g, 73%) as a colorless oil. Rf value (EtOAc/hexane 1:5) 0.5; [α]D20 = −9.9 (c = 1.26, CHCl3); IR (film, cm−1): 3414, 2982, 2936, 1744, 1394, 1370, 1235, 1024, 991; 1H NMR (400 MHz, CDCl3) δ 5.79 (ddd, J = 17.0, 10.4, 6.2 Hz, 1H), 5.24 (dd, J = 17.2, 1.0 Hz, 1H), 5.13 (dd, J = 10.4, 1.1 Hz, 1H), 4.10 (q, J = 6.5 Hz, 1H), 3.01 (d, J = 5.9 Hz, 1H), 2.41 (p, J = 7.1 Hz, 1H), 1.40 (s, 9H), 1.09 (dd, J = 7.2, 1.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 174.7, 138.2, 116.2, 80.9, 74.6, 45.7, 27.9.
To a stirred solution of the above alcohol (5.41 g, 29.1 mmol) in DMF (20 mL), we added DIPEA (12.6 mL, 72.6 mmol), MOMCl (4.4 mL, 58.2 mmol), and the catalytic amount of TBAI at 0 °C, respectively. The reaction mixture was stirred for 12 h before poured into saturated NaHCO3 (aq) and the aqueous layer was extracted by Et2O (3 × 50 mL). The combined organic extracts were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (5% EtOAc/hexanes) afforded the MOM ether (6.42 g, 96%) as a colorless oil. Rf value (EtOAc/hexane 1:5) 0.65; [α]D20 = +70 (c = 1.34, CHCl3); IR (film, cm−1): 2946, 2936, 1745, 1394, 1371, 1291, 1160, 1026, 991, 845; 1H NMR (400 MHz, CDCl3) δ 5.79 (ddd, J = 17.0, 10.4, 6.2 Hz, 1H), 5.24 (dd, J = 17.2, 1.0 Hz, 1H), 5.13 (dd, J = 10.4, 1.1 Hz, 1H), 4.65 (d, J = 6.7 Hz, 1H), 4.47 (d, J = 6.7 Hz, 1H), 4.10 (q, J = 6.5 Hz, 1H), 3.34 (s, 3H), 3.01 (d, J = 5.9 Hz, 1H), 2.41 (p, J = 7.1 Hz, 1H), 1.40 (s, 9H), 1.09 (dd, J = 7.2, 1.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 174.7, 138.2, 116.2, 94.5, 80.9, 74.6, 55.6, 45.7, 27.9.
To a stirred suspension of LiAlH4 (1.54 g, 38.5 mmol) in Et2O (40 mL), a solution of the above ester (4.44 g, 19.3 mmol) in Et2O (10 mL) was transferred in at 0 °C. The reaction mixture was stirred at 0 °C for 1 h before being quenched by adding 20 mL saturated NH4Cl (aq) and 20 mL 25% Rochelle salt solution. The mixture was stirred at rt for 4 h and extracted by Et2O (3 × 50 mL). The combined organic extracts were washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (25% Et2O/hexanes) afforded alcohol 20 (3.06 g, 99%). Rf value (EtOAc/hexane 1:2) 0.4; [α]D20 = +150 (c = 2.04, CHCl3); IR (film, cm−1): 3440, 3090, 1612, 1394, 1291, 1156, 1025; 1H NMR (400 MHz, CDCl3) δ 5.75 (ddd, J = 17.0, 10.7, 7.3 Hz, 1H), 5.29–5.19 (m, 2H), 4.66 (d, J = 6.6 Hz, 1H), 4.54 (d, J = 6.7 Hz, 1H), 4.19–4.11 (m, 1H), 3.69–3.59 (m, 1H), 3.55–3.48 (m, 1H), 3.37 (s, 3H), 2.57 (s, 1H), 2.00–1.87 (m, 1H), 0.88 (d, J = 7.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 135.5, 118, 94.2, 79.6, 65.2, 55.6, 39.6, 11.7. MS (ESI, m/z) [M + Na]+ 183.1.
(4R,5S)-methyl 5-(methoxymethoxy)-4-methylhept-6-en-2-ynoate (21): To a stirred solution of DMSO (3.7 mL, 52.3 mmol) in DCM (50 mL), we added (COCl)2 (2.7 mL, 31.4 mmol) at −78 °C. The mixture was stirred for 5 min, before a solution of alcohol 14 (3.35 g, 20.9 mmol) in DCM (10 mL) was added dropwise. The resulting suspension was stirred at −78 °C for 30 min. Et3N (14.6 mL, 0.105 mol) was added slowly. The reaction mixture was stirred at −78 °C for 1 h and allowed to warm up to room temperature and stirred for 30 min, before pouring it into 1M NaHSO4 solution. The organic layer was separated and the aqueous layer was extracted with DCM. The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo to give crude aldehyde (3.35 g), which was used in the next step without further purification. Rf value (EtOAc/hexane 1:2) 0.7.
To a stirred solution of the CBr4 (13.86 g, 41.8 mmol) in DCM (50 mL) at 0 °C, we added PPh3 (21.93 g, 83.6 mmol). The reaction mixture was stirred at 0 °C for 10 min. A solution of the above aldehyde in DCM (10 mL) was added dropwise. The mixture was warmed up to 23 °C and stirred for 30 min. The reaction was poured into saturated NaHCO3 (aq). The aqueous layer was extracted by DCM. The combined organic extracts were washed with brine and dried over anhydrous Na2SO4, concentrated in vacuo. Flash chromatography on silica gel (5% EtOAc/hexanes) gave the dibromide (5.12 g, 78% for two steps) as a colorless oil. Rf value (EtOAc/hexane 1:4) 0.8; 1H NMR (400 MHz, CDCl3) δ 7.26 (s, 1H), 6.35 (d, J = 9.4 Hz, 1H), 5.64 (ddd, J = 17.3, 10.4, 7.9 Hz, 1H), 5.38–5.11 (m, 2H), 4.67 (d, J = 6.8 Hz, 1H), 4.50 (d, J = 6.8 Hz, 1H), 3.89 (dd, J = 7.7, 5.9 Hz, 1H), 3.37 (s, 3H), 2.72–2.55 (m, 1H), 1.32–0.82 (m, 3H); 13C NMR (100 MHz, CDCl3) δ 140.4, 135.7, 119.0, 93.6, 88.8, 79.6, 55.5, 42.9, 15.2.
To a stirred solution of the above dibromide (3.88 g, 10.1 mmol) in THF (30 mL), we added n-BuLi (1.6 M in hexanes, 19.0 mL, 30.3 mmol) at −78 °C. The mixture was stirred at −78 °C for 15 min before methyl chloroformate (2.34 mL, 30.3 mmol) was added. The reaction mixture was stirred at −78 °C for 1 h before pouring it into saturate NH4Cl (aq). The organic layer was separated and the aqueous layer was extracted by Et2O (3 × 50 mL). The organic extracts were combined, washed by water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (5% EtOAc/hexanes) produced alkynyl ester 21 (2.14 g, 99%) as a pale yellow oil. Rf value (EtOAc/hexane 1:10) 0.45; [α]D20 = +82 (c = 1.22, CHCl3); IR (film, cm−1): 2950, 2888, 1716, 1644, 1435, 1225, 1156, 1031, 918; 1H NMR (400 MHz, CDCl3) δ 5.64 (ddd, J = 17.0, 10.6, 7.7 Hz, 1H), 5.37–5.13 (m, 2H), 4.63 (d, J = 6.9 Hz, 1H), 4.49 (d, J = 6.9 Hz, 1H), 4.03–3.87 (m, 1H), 3.67 (s, 3H), 3.33 (s, 3H), 2.79–2.63 (m, 1H), 1.16 (d, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 153.9, 134.7, 119.8, 93.5, 90.3, 78.5, 73.8, 55.5, 52.3, 31.3, 15.6.
(4R,5S,2Z)-methyl-5-(methoxymethoxy)-3,4-dimethylhepta-2,6-dienoate (22): To a suspension of CuI (5.42 g, 28.4 mmol) in THF (60 mL), we added MeLi (28.5 mL, 45.6 mmol) at −60 °C. The mixture was slowly warmed up to 0 °C to obtain a clear solution. A soln of alkynyl ester 21 (2.41 g, 11.4 mmol) in THF (5 mL) was added slowly at −60 °C and stirred at −40 °C for 2 h. AcOH (2.74 mL, 47.9 mmol) was added to quench the reaction, followed by sat NH4Cl (50 mL). The organic layer was separated and the aqueous layer was extracted by Et2O (3 × 50 mL). The combined organic extracts were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (5% EtOAc/hexanes) produced Z-enoate 22 (2.50 g, 96%) as a colorless oil. Rf value (EtOAc/hexane 1:10) 0.45; [α]D20 = +56 (c = 0.3, CHCl3); IR (film, cm−1): 2951, 2889, 1718, 1646, 1227, 1157, 1031, 1093, 919, 859; 1H NMR (300 MHz, CDCl3) δ 5.71 (brs, 1H), 5.28–5.19 (m, 1H), 4.66 (d, J = 7 Hz, 1H), 4.47 (d, J = 7 Hz, 1H), 4.13 (dq, J = 8.8, 7 Hz, 1H), 3.95 (t, J = 8.5 Hz, 1H), 3.65 (s, 3H), 3.33 (s, 3H), 1.88 (s, 3H), 0.98 (d, J = 7 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 166.5, 161.8, 136.6, 119.2, 93.5, 80.1, 55.7, 50.7, 38.3, 30, 19.9, 15.1.
(2R,3R,Z)-6-(tert-butyldimethylsilyloxy)-2-(methoxymethoxy)-3,4-dimethylhex-4-enal (6): To a stirred soln of ester 22 (2.43 g, 10.6 mmol) in DCM (50 mL), we added DIBAL-H (31.9 mL, 31.9 mmol) at −78 °C. The reaction mixture was stirred 1 h, before the addition of 20 mL saturated NH4Cl (aq) and 20 mL 25% Rochelle salt solution. The mixture was stirred at rt for 4 h and extracted by Et2O (3 × 50 mL). The combined organic extracts were washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (25% Et2O/hexanes) afforded the allyl alcohol (2.02 g, 95%). Rf value (EtOAc/hexane 1:4) 0.25; [α]D20 = +106 (c = 2, CHCl3); IR (film, cm−1): 3349, 2962, 2823, 1613, 1444, 1227, 1152, 1028, 916; 1H NMR (400 MHz, CDCl3): 5.69 (t, J = 7.1 Hz, 1H), 5.62–5.50 (m, 1H), 5.26 (d, J = 10.1 Hz, 1H), 5.20 (d, J = 17.1 Hz, 1H), 4.63 (d, J = 6.9 Hz, 1H), 4.35 (d, J = 6.9 Hz, 1H), 4.18 (dd, J = 8.8, 7.0 Hz, 1H), 3.82–3.75 (m, 2H), 3.26 (s, 3H), 2.85–2.75 (m, 2H). 1.67 (s, 3H), 0.88 (d, J = 7.0 Hz, 3H); 13C NMR (75 MHz, CDCl3) δ 141.9, 136.3, 126.4, 119.7, 92.8, 78.4, 57, 55.5, 38.1, 18.1, 15.1.
To a stirred solution of the above alcohol (1.45 g, 7.24 mmol) in DCM (50 mL), we added imidazole (739 mg, 10.9 mmol) and TBSCl (1.2 g, 7.96 mmol) at 0 °C. The reaction was warmed up to rt and stirred for 1 h, before pouring it into a mixture of sat NaHCO3 (50 mL) and crushed ice. The mixture was extracted with ethers (3 × 60 mL) and the organic layer was washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo to give the crude TBS ether as a clear oil, which was used for the next step without further purification. Flash chromatography on silica gel (25% EtOAc/hexanes) afforded the silyl ether (2.28 g, 99%). Rf value (EtOAc/hexane 1:4) 0.85; [α]D20 = +34 (c = 1.05, CHCl3); IR (film, cm−1): 2960 2821, 1607, 1227, 1152, 1091, 916; 1H NMR (300 MHz, CDCl3): 5.62–5.53 (m, 1H), 5.38 (t, J = 7.1 Hz, 1H), 5.25–5.15 (m, 2H), 4.62 (d, J = 6.9 Hz, 1H), 4.39 (d, J = 6.9 Hz, 1H), 4.32–4.23 (m, 1H), 4.2–4.1 (m, 1H), 3.82 (t, J = 8.5 Hz, 1H), 3.30 (s, 3H), 2.72–2.62 (m, 1H). 1.69 (d, J = 1 Hz, 3H), 0.91 (d, J = 7.0 Hz, 3H), 0.88 (s, 9H), 0.05 (s, 6H); 13C NMR (75 MHz, CDCl3) δ 137.7, 137, 127.3, 118.7, 93.2, 79.8, 60, 55.3, 38.8, 25.9, 18.7, 18.3, 15.1, −5.2.
To a stirred solution of above olefin (1.09 g, 3.47 mmol) in dioxane (24 mL) and water (8 mL), we added 2,6-lutidine (2.02 mL, 17.4 mmol), OsO4 (2.5% in t-BuOH, 1.74 mL, 0.14 mmol) and NaIO4 (2.97 g, 12.9 mmol) at 0 °C. The mixture was stirred at 0 °C for 18 h before saturated NaHCO3 (10 mL) and NaS2O3 (10 mL) were added. The mixture was stirred for another 30 min, extracted by EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (25% EtOAc/hexanes) produced the aldehyde 6 (736 mg, 67%). Rf value (EtOAc/hexane 1:10) 0.65; [α]D20 = +28 (c = 0.5, CHCl3); 1H NMR (400 MHz, CDCl3) δ 9.53 (dd, J = 3.1, 0.5 Hz, 1H), 5.41 (t, J = 6.2 Hz, 1H), 4.61 (dd, J = 22.5, 6.9 Hz, 3H), 4.23 (dd, J = 13.0, 7.0 Hz, 1H), 4.17–4.04 (m, 1H), 3.70 (dd, J = 8.2, 3.3 Hz, 1H), 3.35 (d, J = 0.6 Hz, 4H), 3.00 (p, J = 7.2 Hz, 1H), 1.69 (s, 4H), 1.01 (d, J = 7.0 Hz, 4H), 0.90 (s, 3H), 0.88 (s, 9H), 0.05 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 202.2, 136.0, 128.2, 96.8, 84.7, 59.3, 55.9, 34.9, 25.9, 18.9, 18.3, 14.3, −5.3.
Coupling product (23): To a stirred solution of sulfone 5 (628 mg, 0.94 mmol) in DME (30 mL), we added KHMDS (1.85 mL, 0.5 M soln in toluene, 0.93 mmol) at −78 °C. The reaction mixture was stirred for 30 min, before a soln of aldehyde 6 (357 mg, 1.13 mmol) in DME (5 mL) was transferred in. The reaction mixture was stirred for another 30 min, before it was warmed up to rt. The reaction was quenched by sat NH4Cl (10 mL) at −78 °C. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhydrous MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give coupling product 23 as a colorless oil (544 mg, 76% yield). Rf value (EtOAc/hexane 1:10): 0.5; [α]D20 = +14 (c = 0.6, CHCl3); IR (film, cm−1) 2928, 1455, 1248, 1108; 1H NMR (400 MHz, CDCl3) δ 5.73–5.54 (m, 1H), 5.40 (t, J = 6.1 Hz, 1H), 5.23 (dd, J = 15.4, 8.5 Hz, 1H), 4.88 (d, J = 32.2 Hz, 2H), 4.68 (d, J = 6.8 Hz, 1H), 4.56 (d, J = 5.1 Hz, 2H), 4.41 (d, J = 6.8 Hz, 1H), 4.21 (d, J = 6.1 Hz, 3H), 4.14 (dd, J = 8.7, 3.7 Hz, 1H), 3.86 (t, J = 8.3 Hz, 2H), 3.41 (q, J = 9.8 Hz, 2H), 3.30 (s, 3H), 2.35–2.09 (m, 8H), 1.63 (s, 3H), 1.44 (s, 3H), 1.35 (s, 3H), 1.13 (s, 3H), 0.95 (d, J = 7.0 Hz, 3H), 0.9 (s, 9H), 0.87 (s, 9H), 0.12–−0.07 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 143.50, 138.19, 131.28, 130.98, 128.21, 127.41, 127.29, 125.83, 114.25, 107.15, 93.11, 81.53, 79.29, 78.34, 74.98, 73.45, 70.52, 60.11, 55.31, 46.97, 43.68, 39.56, 36.49, 28.56, 26.44, 25.90, 25.80, 19.16, 18.27, 17.97, 15.76, 13.52, −4.59, −4.65, −5.15. MS (ESI, m/z) [M + Na]+ 783.5.
Aldehyde (3): To a stirred solution of benzyl ether 23 (550 mg, 0.72 mmol) in THF (10 mL) and allyl ethyl ether (1 mL), we transferred a soln of lithium metal (50 mg, 7.2 mmol) in liquid ammonia (12 mL) in portions at −78 °C. The reaction was carefully monitored by TLC and stopped immediately after the solution became slightly blue. Ammonium chloride (2 g) was added to quench the reaction. The mixture was allowed to warm up to rt to evaporate the ammonia, before water (10 mL) was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:10) to give the alcohol as a colorless oil (402 mg, 83% yield), along with the recovered starting material. Rf value (EtOAc/hexane 1:4): 0.43; [α]D20 = +36.0 (c = 1.2, CHCl3); IR (film, cm−1) 3410, 2954, 1253, 1096; 1H NMR (400 MHz, CDCl3) δ5.69–5.62 (m, 1H), 5.37 (t, J = 4.2 Hz, 1H), 5.25 (dd, J = 15.5, 8.6 Hz, 1H), 4.95 (s, 1H), 4.87 (s, 1H), 4.62 (d, J = 6.8 Hz, 1H), 4.36 (d, J = 6.8 Hz, 1H), 4.28 (dd, J = 8.0, 5.2 Hz, 1H), 4.22–4.18 (m 1H), 4.2–4.12 (m, 1H), 3.9–3.82 (m, 1H), 3.80 (t, J = 8.7 Hz, 1H), 3.53 (dd, J = 12.0, 9.8 Hz, 1H), 3.38–3.33 (m, 1H), 3.30 (s, 3H), 2.58–2.50 (m, 1H), 2.32–2.04 (m, 6H), 1.68 (s, 3H), 1.45 (s, 3H), 1.36 (s, 3H), 1.06 (s, 3H), 0.92 (d, J = 6.8 Hz, 3H), 0.90 (s, 9H), 0.87 (s, 9H), 0.06–0.03 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 143.0, 138.0, 131.6, 131.4, 127.2, 114.6, 107.0, 93.0, 82.6, 79.1, 75.9, 70.8, 65.4, 59.8, 55.3, 43.5, 39.7, 39.1, 36.1, 28.6, 26.6, 25.9, 25.8, 18.6, 18.3, 18.0, 15.4, −4.6, −5.2.
To a suspension of the above alcohol (352 mg, 0.53 mmol) and sodium bicarbonate (265 mg, 3.2 mmol) in DCM (20 mL), we added Dess–Martin periodinane (445 mg, 1.05 mmol) at rt. The reaction mixture was stirred for 1 h before it was poured into a mixture of sat NaHCO3 (10 mL) and sodium thiosulfate (10 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo to give the crude aldehyde 3 (352 mg, quantitative), which was used directly in the next step without further purification. 1H NMR (400 MHz, CDCl3) δ 9.61 (s, 1H), 5.70–5.65 (m, 1H), 5.41 (t, J = 5.3 Hz, 1H), 5.28 (dd, J = 15.5, 8.6 Hz, 1H), 4.97 (s, 1H), 4.91 (s, 1H), 4.66 (d, J = 6.8 Hz, 1H), 4.37 (d, J = 6.8 Hz, 1H), 4.30 (dd, J = 8.0, 5.2 Hz, 1H), 4.21–4.17 (m 2H), 3.90–3.82 (m, 2H), 3.30 (s, 3H), 2.7–2.65 (m, 1H), 2.25–2.12 (m, 6H), 1.69 (s, 3H), 1.50 (s, 3H), 1.46 (s, 3H), 1.20 (s, 3H), 0.92-0.87 (m, 21H), 0.1–0.05 (m, 12H).
(3S,4S)-4-(tert-butyldimethylsilyloxy)-3-methylpentanal (16): To a stirred solution of alcohol 12 (7.31 g, 73 mmol) in DMF (70 mL), we added imidazole (5.96 g, 87.6 mmol) and TBSCl (11 g, 73 mmol) at 0 °C. The reaction was warmed up to rt and stirred for 6 h. The reaction mixture was then poured into a mixture of sat NaHCO3 (50 mL) and crushed ice. The mixture was extracted with ethers (3 × 60 mL) and the organic layer was washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo to give the crude TBS ether as a clear oil, which was used for the next step without further purification. To a stirred solution of thus obtained olefin in THF (60 mL), we added the BH3·THF complex (73 mL, 1 M soln in THF, 73 mmol) at 0 °C. The reaction mixture was allowed to warm up to rt and stirred for 6 h. NaOH (10 mL) and H2O2 (15 mL, 70% soln) were added and the mixture was refluxed for 1 h. The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:20) provided corresponding alcohol 16 as a colorless oil (11.4 g, 67% for two steps). Rf value (EtOAc/hexane 1:10): 0.53; [α]D20 = +2.3 (c = 1.0, CHCl3); IR (film, cm−1) 3340, 2932, 2858, 1463, 1254, 1053; 1H NMR (400 MHz, CDCl3) δ3.77–3.71 (m, 1H), 3.69–3.62 (m, 1H), 3.59–3.53 (m, 1H), 3.19 (m, 1H), 1.71–1.66 (m, 2H), 1.39–1.34 (m, 1H), 1.1 (d, J = 6.4 Hz, 3H), 0.87 (s, 9H), 0.81 (d, J = 6.7 Hz, 3H), 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 61.8, 38.5, 27.3, 35.2, 25.7, 18.2, 17.9, 17.2, −4.7, −5.1.
(3S,4R,6S,7S)-7-(tert-butyldimethylsilyloxy)-3,6-dimethyloct-1-en-4-ol (17): To a stirred solution of DMSO (2.7 mL, 37.8 mmol) in DCM (70 mL), we added oxalyl chloride (2 mL, 22.7 mmol) at −78 °C. After 10 min of stirring, a solution of alcohol 16 (3.5 g, 15.1 mmol) in DCM (10 mL) was transferred in at the same temperature. The reaction mixture was stirred for 30 min before Et3N (10.5 mL, 75.5 mmol) was added. After stirring at −78 °C for 1 h, the reaction mixture was allowed to warm up to 0 °C for 30 min, before it was poured into sat NaHCO3 soln (30 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give the aldehyde as a colorless oil (3.03 g, 87% yield), which was used in the next step immediately. Rf value (EtOAc/hexane 1:10): 0.85.
To a stirred mixture of potassium tert-butoxide (8.7 mL, 1.0 M soln in THF, 8.7 mmol) and trans-2-butene (1.4 mL, 14.5 mmol) in THF (30 mL), we added n-butyllithium (5.5 mL, 1.6 M soln in THF, 8.7 mmol) at −78 °C. After complete addition of n-butyllithium, the mixture was stirred at −45 °C for 10 min. The resulting orange solution was cooled to −78 °C again and a solution of (−)-Ipc2BOMe (3.3 g, 10.4 mmol) in THF (10 mL) was added dropwise. After 30 min of stirring, boron trifluoride etherate (1.5 mL, 11.6 mmol) was added dropwise. Then, the above aldehyde (1.34 g, 5.8 mmol) in THF (5 mL) was transferred in. The mixture was stirred at −78 °C for 3 h before NaOH (6.8 mL, 3 M soln) and H2O2 (4.7 mL, 70% soln) were added. The contents were refluxed for 1 h. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give the alcohol 17 as a colorless oil (1.28 g, 77% yield). Rf value (EtOAc/hexane 1:10): 0.63; [α]D20 = +6.3 (c = 0.67, CHCl3); IR (film, cm−1) 3411, 2959, 1462, 1045; 1H NMR (400 MHz, CDCl3) δ5.86–5.77 (m, 1H), 5.1–5.03 (m, 2H), 3.84–3.77 (m, 1H), 3.6–3.54 (m, 1H), 2.21–2.18 (m, 1H), 2.13 (d, J = 4.9 Hz, 1H), 1.82–1.74 (m, 1H), 1.67–1.6 (m, 1H), 1.28–1.2 (m, 1H), 1.06 (d, J = 6.9 Hz, 3H), 1.02 (d, J = 6.4 Hz, 3H), 0.88 (s, 9H), 0.92 (d, J = 9.1 Hz, 3H); 0.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 140.4, 115.5, 72.8, 71.2, 43.7, 37.0, 36.4, 25.7, 19.1, 17.9, 16.5, 16.2, −4.4, −4.9; MS (ESI, m/z) [M + Na]+ 309.
(3S,4R,6S,7S)-7-(tert-butyldimethylsilyloxy)-4-(4-methoxybenzyloxy)-3,6-dimethyloctan-1-ol (18): To a stirred solution of alcohol 17 (710 mg, 2.5 mmol) in DMF (15 mL), we added NaH (60%, 150 mg, 3.7 mmol) at 0 °C. The mixture was stirred for 30 min before PMBCl (0.5 mL, 3.7 mmol) was added at 0 °C. After stirring at rt overnight, water (4 mL) and Et2NH (2 mL) were added and the mixture was stirred for 1h, before it was poured into sat NaHCO3 (aq). The mixture was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give the PMB ether as a colorless oil (854 mg, 84% yield). Rf value (EtOAc/hexane 1:10): 0.75; [α]D20 = +5.0 (c = 1, CHCl3); IR (film, cm−1) 2959, 1645, 1059; 1H NMR (500 MHz, CDCl3) δ 7.30 (d, J = 8.5 Hz, 1H), 6.91 (d, J = 8.5 Hz, 1H), 5.89–5.82 (m, 1H), 5.08 (d, J = 18.0 Hz, 1H), 5.06 (d, J = 9.4 Hz, 1H), 4.41 (AB, JAB = 11.2 Hz, ΔυAB = 33.1 Hz, 2H), 3.84 (s, 3H), 3.69–3.64 (m, 1H), 3.43–3.4 (m, 1H), 2.55–2.5 (m, 1H), 1.62–1.56 (m, 2H), 1.32–1.29 (m, 1H), 1.08 (d, J = 6.9 Hz, 3H), 1.05 (d, J = 6.4 Hz, 3H), 0.96 (s, 9H), 0.92 (d, J = 9.1 Hz, 3H); 0.06 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.0, 140.7, 131.1, 129.4, 129.3, 114.7, 113.7, 80.8, 70.9, 55.4, 37.2, 34.6, 33.5, 25.9, 20.7, 18.1, 15.2, 15.0, −4.1, −4.7.
To a stirred solution of the above olefin (1.45 g, 3.6 mmol) in THF (30 mL), we added 9-BBN (14.2 mL, 0.5 M soln in THF, 7.2 mmol) at 0 °C. The reaction mixture was allowed to warm up to rt and stirred for 3 h. NaOH (0.6 mL) and H2O2 (4 mL) were added and the mixture was refluxed for 1 h. The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over Na2SO4 and concentrated in vacuo. Column chromatography provided alcohol 18 as a colorless oil (1.21 g, 79%). Rf value (EtOAc/hexane 1:1): 0.68; [α]D20 = +3.7 (c = 1, CHCl3); IR (film, cm−1) 3403, 2931, 1613, 1040; 1H NMR (400 MHz, CDCl3) δ 7.21 (d, J = 8.6 Hz, 1H), 6.86 (d, J = 8.6 Hz, 1H), 4.45 (s, 2H), 3.81 (s, 3H), 3.75–3.65 (m, 2H), 3.61–3.55 (m, 1H), 3.38–3.33 (m, 1H), 1.88–1.80 (m, 1H), 1.7–1.6 (m, 2H), 1.55–1.45 (m, 2H), 1.4–1.32 (m, 1H), 1.02 (d, J = 6.9 Hz, 3H), 0.95 (d, J = 6.4 Hz, 3H), 0.92 (s, 9H), 0.85 (d, J = 9.1 Hz, 3H), 0.03 (s, 3H), 0.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 129.3, 113.6, 81.2, 71.2, 70.9, 60.1, 55.1, 37.3, 34.6, 34.3, 32.8, 32.1, 27.3, 25.8, 22.6, 18, 15.5, 15.1, −4.8; MS (ESI, m/z) [M+Na]+ 447.4.
Sulfone (4): To a stirred solution of alcohol 18 (880 mg, 2.1 mmol), 1-(4-Hydroxyphenyl)-1H-tetrazole-5-thiol (739 mg, 4.1 mmol) and triphenylphosphine (814 mg, 3.11 mmol) in THF (20 mL), we added DIAD (0.72 mL, 3.7 mmol) at 0 °C. The reaction mixture was warmed up to rt and stirred overnight, before it was poured into sat NaHCO3 (20 mL). The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over MgSO4 and concentrated in vacuo. Column chromatography provided the sulfide as a colorless oil (1.03 g, 85%). Rf value (EtOAc/hexane 1:4): 0.56; [α]D20 = +1.2 (c = 0.5, CHCl3); IR (film, cm−1) 2956, 2886, 1614, 1074; 1H NMR (400 MHz, CDCl3) δ 7.53–7.57 (m, 5H), 7.23 (d, J = 8.6 Hz, 2H), 6.85 (d, J = 8.6 Hz, 2H), 4.45 (AB, JAB = 11.1 Hz, ΔυAB = 18 Hz, 2H), 3.78 (s, 3H), 3.66–3.63 (m, 1H), 3.55–3.49 (m, 1H), 3.37–3.31 (m, 2H), 1.93–1.89 (m, 2H), 1.7–1.62 (m, 1H), 1.62–1.55 (m, 1H), 1.52–1.48 (m, 1H), 1.31–1.25 (m, 1H), 1.02 (d, J = 6.8 Hz, 3H), 1 (d, J = 7 Hz, 3H), 0.86 (d, J = 6.8 Hz, 1H), 0.84 (s, 9H), 0.01 (s, 3H), 0 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 129.3, 113.6, 81.2, 71.2, 70.9, 60.1, 55.1, 37.3, 34.6, 34.3, 32.8, 32.1, 27.3, 25.8, 22.6, 18.0, 15.5, 15.1, −4.8. To a stirred solution of thus obtained sulfide (353 mg, 0.57 mmol) in ethanol (9 mL), we added a solution of ammonium molybdate (320 mg, 0.26 mmol) in hydrogen peroxide (1.6 mL) and water (0.8 mL) at rt. The reaction mixture was stirred for 12 h and poured into a mixture of sat NaHCO3 (10 mL) and sodium thiosulfate (10 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:12) to give the sulfone 4 as a colorless oil (352 mg, 95% yield). Rf value (EtOAc/hexane 1:4): 0.5; [α]D20 = +1.8 (c = 1, CHCl3); IR (film, cm−1) 2931, 2857, 1612, 1513, 1249, 1074; 1H NMR (500 MHz, CDCl3) δ7.72–7.7 (m, 2H), 7.64–7.6 (m, 3H), 7.27 (d, J = 8.5 Hz, 1H), 6.90 (d, J = 8.5 Hz, 1H), 4.47 (AB, JAB = 11.2 Hz, ΔυAB = 21.8 Hz, 2H), 3.93–3.87 (m, 1H), 3.83 (s, 3H), 3.79–3.71 (m, 2H), 3.4–3.37 (m, 1H), 2.1–2.04 (m, 1H), 2.02–1.86 (m, 2H), 1.66–1.61 (m, 1H), 1.58–1.54 (m, 1H), 1.42–1.38 (m, 1H), 1.10 (d, J = 6.9 Hz, 3H), 1.05 (d, J = 6.4 Hz, 3H), 0.92 (d, J = 3.5 Hz, 3H), 0.90 (s, 9H), 0.07 (s, 3H), 0.04 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.3, 153.4, 133.1, 131.4, 130.6, 129.7, 129.4, 125.1, 113.8, 80.8, 71.2, 71.0, 55.3, 54.6, 37.2, 36.6, 34.6, 33.4, 25.8, 24.2, 20.4, 18.1, 15.5, 15.2, −4.1, −4.7; MS (ESI, m/z) [M + Na]+ 639.3. HRMS (ESI) [M+Na]+ calcd for C31H48N4O5SSiNa 639.3012, found 639.3008.
Coupling product (24): To a stirred solution of aldehyde 3 (422 mg, 0.64 mmol) and sulfone 4 (324 mg, 0.53 mmol) in DME (15 mL), we added KHMDS (1.6 mL, 0.5 M soln in toluene, 0.8 mmol) at −65 °C. The reaction mixture was stirred for 1 h at −65 °C, before it was warmed up to rt. The reaction was quenched by sat NH4Cl (5 mL) at −65 °C and the organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:20) to give the olefin 24 as a colorless oil (376 mg, 67% yield). Rf value (EtOAc/hexane 1:10): 0.48; [α]D20 = +22.5 (c = 0.55, CHCl3); IR (film, cm−1) 2857, 1729, 1612, 1513, 1257, 1081; 1H NMR (500 MHz, CDCl3) δ 7.28 (d, J = 8.5 Hz, 1H), 6.89 (d, J = 8.5 Hz, 1H), 5.75–5.67 (m, 2H), 5.48 (d, J = 15.5 Hz, 1H), 5.41 (t, J = 3.9 Hz, 1H), 5.3–5.26 (dd, J = 15.5, 8.5 Hz, 1H), 4.96 (s, 1H), 4.89 (s, 1H), 4.68 (d, J = 6.8 Hz, 1H), 4.44 (AB, JAB = 11.1 Hz, ΔυAB = 20.6 Hz, 2H), 4.4 (d, J = 6.8 Hz, 1H), 4.34 (dd, J = 12.8, 7.1 Hz, 1H), 4.2 (dd, J = 12.8, 3.9 Hz, 1H), 3.94–3.87 (m, 2H), 3.86–3.83 (m, 1H), 3.83 (s, 3H), 3.7–3.66 (m, 1H), 3.4–3.32 (m, 1H), 3.34 (s, 3H), 2.69–2.65 (m, 1H), 2.34–2.28 (m, 2H), 2.25–2.18 (m, 3H), 2.14–2.1 (m, 1H), 1.9–1.83 (m, 2H), 1.74–1.7 (m, 1H), 1.72 (s, 3H), 1.65–1.6 (m, 1H), 1.58–1.53 (m, 1H), 1.49 (s, 3H), 1.4 (s, 3H), 1.21 (s, 3H), 1.08 (d, J = 9.7 Hz, 3H), 0.95–0.85 (m, 36H), 0.1–0.04 (m, 18H); 13C NMR (125 MHz, CDCl3) δ 159, 143.6, 138.0, 134, 131.6, 131.4, 131.1, 130, 129.3, 127.3, 114.3, 113.7, 107.2, 93.1, 82.1, 81.5, 81.2, 79.2, 71.1, 70.8, 70.7, 59.8, 55.4, 55.2, 43.8, 39.7, 39.2, 37.5, 35.7, 35.6, 32.8, 29.7, 28.5, 26.6, 26, 25.9, 20.6, 20.4, 18.7, 18.4, 18, 15.5, 15.4, 15, −4.1, −4.4, −4.5, −4.6, −4.7, −5.1; MS (ESI, m/z) [M + Na]+ 1081.7. HRMS (ESI) [M + Na]+ calcd for C60H110O9Si3Na 1081.7355, found 1081.7367.
Diol product (25): To a stirred solution of PMB ether 24 (204 mg, 0.2 mmol) in DCM (20 mL) and pH 7.0 buffer (1.6 mL), we added DDQ (88 mg, 0.4 mmol) at 0 °C and the reaction mixture was stirred at that temperature for 1 h. The reaction was quenched by sat NaHCO3 (5 mL) and the organic layer was separated. The aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with sat NaHCO3, water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:15) to give the corresponding alcohol as a colorless oil (138 mg, 76% yield). Rf value (EtOAc/hexane 1:4): 0.67; [α]D20 = +30.1 (c = 0.55, CHCl3); IR (film, cm−1) 3429, 2931, 1619, 1252, 1079; 1H NMR (500 MHz, CDCl3) δ 5.77–5.67 (m, 2H), 5.48 (d, J = 15.5 Hz, 1H), 5.41 (t, J = 5.2 Hz, 1H), 5.28 (dd, J = 15.5, 8.3 Hz, 1H), 4.99 (s, 1H), 4.88 (s, 1H), 4.68 (d, J = 6.8 Hz, 1H), 4.4 (d, J = 6.8 Hz, 1H), 4.34 (dd, J = 12.9, 7.3 Hz, 1H), 4.2 (dd, J = 12.9, 4.1 Hz, 1H), 3.92–3.86 (m, 2H), 3.86–3.72 (m, 2H), 3.56–3.52 (m, 1H), 3.34 (s, 3H), 2.7–2.65 (m, 1H), 2.45–2.4 (m, 1H), 2.32–2.28 (m, 2H), 2.28–2.2 (m, 3H), 2.14–2.1 (m, 1H), 1.92–1.88 (m, 1H), 1.71 (s, 3H), 1.68–1.55 (m, 2H), 1.48 (s, 3H), 1.4 (s, 3H), 1.2 (s, 3H), 1.12 (d, J = 6.3 Hz, 3H), 1.04–0.87 (m, 36H), 0.11–0.06 (m, 18H); 13C NMR (125 MHz, CDCl3) δ 143.6, 138.1, 134.1, 131.7, 131.4, 129.9, 127.3, 114.3, 107.2, 93.1, 82.1, 81.5, 79.2, 73.2, 72.3, 70.8, 59.9, 55.4, 43.8, 39.7, 39.2, 38.8, 36.3, 36.0, 35.5, 35.4, 29.7, 28.5, 26.6, 26, 25.9, 20.5, 18.7, 18.4, 18, 15.5, 15.4, −4.4, −4.5, −4.8, −5.1.
To a stirred solution of the above TBS ether (115 mg, 0.12 mmol) in methanol (4 mL), we added ammonium fluoride (125 mg, 3.37 mmol) at rt and the reaction mixture was stirred at that temperature for 8 h. Et2O (30 mL) was added to precipitate the ammonium fluoride, which was removed by suction filtration. The crude product was purified by flash chromatography (EtOAc/hexane 1:4) to give the alcohol 25 as a colorless oil (94 mg, 95% yield). Rf value (EtOAc/hexane 2:1): 0.36; [α]D20 = +37.4 (c = 0.5, CHCl3); IR (film, cm−1) 3434, 2956, 2931, 1644, 1374, 1253, 1077; 1H NMR (500 MHz, CDCl3) δ5.77–5.68 (m, 3H), 5.45 (d, J = 15.5 Hz, 1H), 5.24 (dd, J = 15.5, 8.4 Hz, 1H), 4.94 (s, 1H), 4.85 (s, 1H), 4.70 (d, J = 6.8 Hz, 1H), 4.37 (d, J = 6.8 Hz, 1H), 4.23 (dd, J = 11.6, 8.4 Hz, 1H), 3.9–3.82 (m, 2H), 3.82–3.75 (m, 2H), 3.51–3.49 (m, 1H), 3.29 (s, 3H), 2.85 (brs, 1H), 2.84–2.80 (m, 1H), 2.4–2.35 (m, 1H), 2.3–2.12 (m, 5H), 2.1–2.05 (m, 1H), 1.9–1.84 (m, 2H), 1.71 (s, 3H), 1.65–1.55 (m, 2H), 1.44 (s, 3H), 1.38 (s, 3H), 1.18 (s, 3H), 1.08 (d, J = 6.1 Hz, 3H), 0.94–0.82 (m, 27H), 0.1–0.02 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 143.4, 142.6, 134.0, 132.8, 130.6, 129.8, 126.1, 114.3, 107.1, 92.5, 82.0, 81.3, 77.8, 73.1, 72.2, 70.6, 65.8, 57.0, 55.5, 43.6, 39.5, 38.7, 38.4, 36.2, 36.0, 35.4, 35.3, 28.4, 26.5, 25.8, 20.4, 18.1, 18.0, 15.5, 15.2, −4.5, −4.7, −4.9, −5.1; MS (ESI, m/z) [M + Na]+ 845.5. HRMS (ESI) [M + Na]+ calcd for C46H88O8Si2Na 847.5916, found 847.5922.
Macrolactone(26): To a stirred solution of allyl alcohol 25 (107 mg, 0.13 mmol) in DCM (10 mL), we added activated MnO2 (126 mg, 90%, 1.3 mmol) at rt, and the reaction was stirred at rt for 5 h. Suction filtration gave a crude aldehyde (105 mg) that was used for the next step without further purification. Rf value (EtOAc/hexane 1:2): 0.73.
To a stirred solution of the above aldehyde (105 mg, 0.12 mmol) and 2-methyl-2-butene (2 mL) in tert-butanol (8 mL), we added a solution of NaH2PO4.H2O (200 mg) and NaClO2 (200 mg), dropwise, in H2O (2 mL) at 0 °C. In addition, the reaction mixture was allowed to warm up to rt and stirred for 30 min. The reaction was poured into water (5 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhyd Na2SO4 and evaporated in vacuo. The crude product was purified by flash chromatography (MeOH/chloroform 3:100) to give the seco-acid as a colorless oil (97 mg, 89% yield for two steps). Rf value (EtOAc/hexane 1:2): 0.68.
To the solution of thus obtained seco-acid in THF (4 mL), we added DIPEA (0.33 mL, 1.91 mmol) and 2,4,6-trichlorobenzoyl chloride (0.2 mL, 1.27 mmol) at rt. The reaction was stirred for 3 h at that temperature, before the THF solvent was removed by vacuo. The the residue toluene (10 mL) was added and the solution was transferred to a stirred solution of DMAP (388 mg, 3.18 mmol) in toluene (150 mL) at rt over 16 h, through a syringe pump. The resulting mixture was stirred at rt for 36 h and poured into sat NaHCO3 (20 mL). The organic layer was separated and the aq was extracted with diethyl ether (3 × 20 mL). The combined organic phase was washed with water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:40) provided macrolactone 26 as a colorless oil (65 mg, 69%). Rf value (EtOAc/hexane 1:10): 0.48; [α]D20 = −18 (c = 0.2, CHCl3); IR (film, cm−1) 2927, 1711, 1155, 1033; 1H NMR (500 MHz, CDCl3) δ 5.75 (d, J = 0.6 Hz, 1H), 5.62–5.55 (m, 2H), 5.48 (d, J = 15.6 Hz, 1H), 5.32 (dd, J = 15.6, 8.3 Hz, 1H), 4.99 (s, 1H), 4.89 (s, 1H), 4.86–4.88 (m, 1H), 4.67 (d, J = 6.8 Hz, 1H), 4.53 (d, J = 6.8 Hz, 1H), 4.15–4.10 (m, 1H), 3.96 (dd, J = 7.9, 5.8 Hz, 1H), 3.88 (dd, J = 7.9, 4.6 Hz, 1H), 3.84–3.79 (m, 1H), 3.79–3.76 (m, 1H), 3.38 (s, 3H), 2.22–2.05 (m, 8H), 1.91–1.85 (m, 1H), 1.9 (d, J = 0.6 Hz, 3H), 1.84–1.72 (m, 2H), 1.62–1.56 (m, 1H), 1.48 (s, 3H), 1.40 (s, 3H), 1.22 (s, 3H), 1.12 (d, J = 7.2 Hz, 3H), 1.05 (d, J = 6.9 Hz, 3H), 0.93–0.78 (m, 24H), 0.07–0.03 (m, 12H); 13C NMR (125 MHz, CDCl3) δ 166.2, 160, 142.1, 134.9, 130.8, 130.1, 129.6, 118.3, 114.5, 107.2, 93.7, 81.9, 79.9, 79.8, 75.8, 70.9, 69.8, 55.5, 42.8, 40.6, 38.8, 38.3, 37.3, 36.6, 35.8, 35.6, 34.7, 29.7, 28.5, 26.6, 25.8, 24.7, 23.3, 22.7, 21.8, 20.7, 19.5, 18.1, 15.8, 15.1, 14.0, 7.9, −4, −4.4, −4.5, −4.7. MS (ESI, m/z) [M + Na]+ 843.5. HRMS (ESI) [M + Na]+ calcd for C46H84O8Si2Na 843.5603, found 843.5611.
Macrolactone alcohol (27): To a stirred solution of macrolactone 26 (52 mg, 0.063 mmol) in THF (4 mL), we added pyridine (1 mL) followed by HF.pyridine complex (70%, 0.5 mL) at 0 °C, and the reaction was warmed up to rt and stirred for 10 h. The reaction mixture was cooled to 0 °C again and sat NaHCO3 (20 mL) was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 10 mL). The combined organic phase was washed with sat NaHCO3, water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:2) provided diol as a colorless oil (33 mg, 89%). Rf value (EtOAc/hexane 2:1): 0.55; [α]D20 = −12 (c = 0.18, CHCl3); IR (film, cm−1) 3456, 2926, 1707, 1155, 1032; 1H NMR (500 MHz, CDCl3) δ 5.75 (s, 1H), 5.66–5.55 (m, 2H), 5.53 (d, J = 15.6 Hz, 1H), 5.44 (dd, J = 15.6, 8.3 Hz, 1H), 5.11 (s, 1H), 4.99–4.95 (m, 1H), 4.96 (s, 1H), 4.69 (d, J = 6.8 Hz, 1H), 4.55 (d, J = 6.8 Hz, 1H), 4.12–4.08 (m, 1H), 4.07–4.03 (m, 2H), 3.87–3.83 (m, 1H), 3.77–3.72 (m, 1H), 3.4 (s, 3H), 2.4 (brs, 1H), 2.36 (dd, J = 14.4, 2.8 Hz, 1H), 2.29–2.25 (m, 2H), 2.23–2.2 (m, 2H), 2.14–2.10 (m, 1H), 2.08–2.04 (m, 1H), 1.92 (d, J = 1.1 Hz, 3H), 1.9–1.87 (m, 1H), 1.8–1.72 (m, 2H), 1.56–1.52 (m, 1H), 1.49 (s, 3H), 1.40 (s, 3H), 1.22 (s, 3H), 1.17 (d, J = 7.0 Hz, 3H), 1.12 (d, J = 6.9 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 7.0 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.5, 160.7, 143.5, 134.5, 131.7, 129.9, 129.2, 118.2, 114.8, 107.3, 93.9, 81.9, 80.3, 79.6, 74.9, 69.5, 69.2, 55.6, 43.1, 40.1, 39.1, 37.4, 36.0, 35.9, 35.4, 32.9, 28.5, 26.7, 21.9, 20.2, 20.0, 15.2, 15.1, 14.9.
A solution of the above acetonide (12 mg, 0.020 mmol) in HOAc (0.8 mL) and water (0.2 mL) was heated at 55 °C for 3 h, before the solvents were removed in vacuo. Column chromatography (MeOH/chloroform 1:20) provided alcohol 27 as a colorless oil (11 mg, 81%). Rf value (EtOAc/hexane 4:1): 0.22; [α]D20 = −8 (c = 0.2, CHCl3); IR (film, cm−1) 3500, 2956, 1712, 1034; 1H NMR (500 MHz, CDCl3) δ 5.75 (s, 1H), 5.78–5.62 (m, 2H), 5.58–5.5 (m, 2H), 5.07 (s, 1H), 5.01 (s, 1H), 4.97–4.93 (m, 1H), 4.69 (d, J = 6.8 Hz, 1H), 4.52 (d, J = 6.8 Hz, 1H), 4.08 (t, J = 6.4 Hz, 1H), 4.01–3.97 (m, 1H), 3.86–3.83 (m, 1H), 3.82–3.76 (m, 1H), 3.72–3.7 (m, 2H), 3.4 (s, 3H), 2.58 (brs, 1H), 2.55 (d, J = 14.1 Hz, 1H), 2.42–2.3 (m, 4H), 2.23–1.98 (m, 1H), 1.95–1.9 (m, 1H), 2.05–2.02 (m, 1H), 2–1.95 (m, 1H), 1.98 (d, J = 1.1 Hz, 3H), 1.89–1.85 (m, 1H), 1.81–1.75 (m, 1H), 1.5–1.45 (m, 1H), 1.21 (s, 3H), 1.15 (d, J = 6.5 Hz, 3H), 1.1 (d, J = 6.4 Hz, 3H), 0.98 (d, J = 6.8 Hz, 3H), 0.87 (d, J = 7.2 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.5, 163.7, 144, 138.5, 132.4, 128.9, 125.8, 117.5, 116.3, 94.2, 79.1, 74.8, 73.8, 73.2, 69.2, 68.9, 55.9, 41.8, 40.4, 40.5 37.2, 37, 35.5, 35.2, 32.9, 22.5, 21, 19.9, 16.1, 15, 14.1. MS (ESI, m/z) [M + Na]+ 575.3. HRMS (ESI) [M + Na]+ calcd for C31H52O8Na 575.3560, found 575.3555.
TES ether derivative (28): To a stirred solution of the above diol 27 (7 mg, 0.013 mmol) in DCM (2 mL), we added bromocatechol borane (0.65 mL, 0.1 M soln in DCM, 0.065 mmol) at −78 °C, and the mixture was stirred at that temperature for 1h, before it was quenched with sat NaHCO3 (5 mL). The organic layer was separated and the aq layer was extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over anhyd Na2SO4 and concentrated in vacuo. Column chromatography (MeOH/chloroform 1:20) provided the corresponding alcohol as a colorless oil (4.8 mg, 72%). Rf value (EtOAc/hexane/MeOH 8:2:1): 0.33; [α]D20 = −21 (c = 0.074, CHCl3); IR (film, cm−1) 3430, 2956, 1706; 1H NMR (500 MHz, CDCl3) δ 5.9–5.85 (m, 1H), 5.84–5.78 (m, 1H), 5.82 (s, 1H), 5.75–5.69 (m, 1H), 5.58 (d, J = 15.8 Hz, 1H), 5.05 (s, 1H), 5.05–5.02 (m, 1H), 5.02 (s, 1H), 3.96–3.92 (m, 1H), 3.81–3.73 (m, 2H), 3.62–3.58 (m, 2H), 2.65–2.55 (m, 2H), 2.5–2.44 (m, 1H), 2.42–2.38 (m, 2H), 2.18–2.1 (m, 2H), 2.02–1.98 (m, 1H), 1.92 (s, 3H), 1.7–1.64 (m, 2H), 1.43–1.40 (m, 1H), 1.19 (s, 3H), 1.07 (d, J = 6.9 Hz, 3H), 1.05 (d, J = 7.1 Hz, 3H), 0.95 (d, J = 6.6 Hz, 3H), 0.88 (d, J = 6.9 Hz, 3H).
To a stirred solution of the above alcohol (3.2 mg, 0.006 mmol) and DMAP (18 mg, 0.14 mmol) in DCM (2 mL), we added TESCl (16 µL, 0.1 mmol) at 0 °C, and the reaction was stirred at 0 °C for 30 min, before sat NaHCO3 (5 mL) was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 10 mL). The combined organic phase was washed with water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:20) provided tris-TES ether 28 as a colorless oil (4.9 mg, 92%). Rf value (EtOAc/hexane 1:4): 0.5; [α]D20 = +11 (c = 0.22, CHCl3); IR (film, cm−1) 3430, 2896, 1079; 1H NMR (500 MHz, CDCl3) δ 5.84-5.78 (m, 1H), 5.64 -5.60 (m, 3H), 5.42 (d, J = 15.5 Hz, 1H), 4.91 (brs, 2H), 4.76 (dt, J = 5.0, 2.5 Hz, 1H), 4.1–4.06 (m, 1H), 4.02–3.98 (m, 1H), 3.88–3.84 (m, 1H), 3.75–3.70 (m, 1H), 3.6–3.56 (m, 1H), 3.52 (brs, 1H), 2.53–2.49 (m, 1H), 2.4–2.36 (m, 2H), 2.3–2.25 (m, 1H), 2.18–2.08 (m, 2H), 2.07–2.01 (m, 4H), 1.85 (brs, 3H), 1.85–1.8 (m, 1H), 1.4–1.35 (m, 1H), 1.18 (s, 3H), 1.11 (d, J = 6.4 Hz, 3H), 0.96–0.9 (m, 33H), 0.8 (d, J = 6.9 Hz, 3H), 0.6–0.53 (m, 18H); MS (ESI, m/z) [M+Na]+ 873.33. HRMS (ESI) [M + Na]+ calcd for C47H90O7Si3Na 873.5892, found 873.5888.
Iriomoteolide-1a (1): To a stirred solution of diol 28 (3 mg, 3.5 μmol) in DCM (1 mL), we added Dess–Martin periodinane (0.12 mL, 0.3 M soln in DCM, 0.035 mmol) at rt, and the reaction was stirred at rt for 1 h. Direct column chromatography (EtOAc/hexane 1:15) provided the corresponding ketone as a colorless oil (1.9 mg, 65%, 90% BRSM), along with the recovered starting material (1 mg). Rf value (EtOAc/hexane 1:4): 0.75; 1H NMR (500 MHz, CDCl3) δ 5.85–5.80 (m, 1H), 5.63 (s, 1H), 5.61–5.55 (m, 1H), 5.46 (d, J = 15.6 Hz, 1H), 5.37 (dd, J = 15.6, 8.0 Hz, 1H), 5.01 (s, 1H), 4.91–4.87 (m, 1H), 4.85 (s, 1H), 4.18–4.13 (m, 2H), 4.08 (s, 1H), 3.79–3.75 (m, 1H), 3.69–3.66 (m, 1H), 3.42 (d, J = 15.0 Hz, 1H), 3.12 (d, J = 15.0 Hz, 1H), 2.2–2.16 (m, 2H), 2.1–2 (m, 4H), 1.97–1.92 (m, 2H), 1.87 (brs, 3H), 1.85–1.81 (m, 1H), 1.46 (s, 3H), 1.1 (d, J = 6.4 Hz, 3H), 1.04 (d, J = 6.6 Hz, 3H), 0.98–0.92 (m, 30H), 0.85 (d, J = 7.0 Hz, 3H), 0.61–0.54 (m, 18H). To a stirred solution of the above ketone (3.0 mg, 0.0035 mmol) in THF (0.6 mL), we added a HF·Py solution (0.1 mL containing 1 mL 70% HF·Py: 1.1 mL pyridine: 2.4 mL THF) at rt. After 1 h, the reaction was quenched with sat NaHCO3 and extracted with diethyl ether (3 × 10 mL). The combined organic layer was washed with water and brine, dried over MgSO4 and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (MeOH/chloroform 1:25) to give iriomoteolide-1a (1) (1.0 mg, 56%) as a colorless oil, along with the isomerized product, iriomoteolide-1b (2) (0.3 mg, 17%), as a colorless oil.
Iriomoteolide-1a (1) Rf value (EtOAc/hexane 2:1): 0.4. [α]D20 = −12.0 (c = 0.10, CHCl3); IR (film, cm−1) 3456, 2926, 1707, 1032; 1H NMR (500 MHz, CDCl3) δ5.9–5.88 (m, 1H), 5.88–5.83 (m, 1H), 5.83–5.8 (m, 1H), 5.78 (s, 1H), 5.68 (dd, J = 15.4, 6.8 Hz, 1H), 5.05–4.99 (m, 1H), 4.88 (d, J = 1.6 Hz, 1H), 4.86 (d, J = 1.4 Hz, 1H), 4.07 (dd, J1 = J2 = 6.8 Hz, 1H), 4.03–3.97 (m, 1H), 4–3.95 (m, 1H), 3.89–3.83 (m, 1H), 3.29 (brs, 1H), 2.67 (brs, 1H), 2.37–2.3 (m, 1H), 2.32–2.28 (m, 1H), 2.25–2.2 (m, 3H), 2.18–2.13 (m, 2H), 2.05–2 (m, 1H), 1.96 (s, 3H), 2–1.92 (m, 1H), 1.84–1.78 (m, 1H), 1.56–1.5 (m, 1H), 1.4–1.35 (m, 1H), 1.33 (s, 3H), 1.12 (d, J = 6.4 Hz, 3H), 1.1 (d, J = 7 Hz, 3H), 1.01 (d, J = 7 Hz, 3H), 0.9 (d, J = 6.9 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 166.3, 160, 141.7, 134.8, 132.7, 129.6, 127.2, 118.8, 110.8, 99.1, 77.2, 74.9, 74.4, 70.3, 69.3, 40.6, 39.7, 37.8, 37.3, 36.7, 35.5, 34.2, 20.8, 20.6, 20, 15.8, 15.7, 14.9; MS (ESI, m/z) [M + Na]+ 529.24. HRMS (ESI) [M + Na]+ calcd for C29H46O7Na 529.3141, found 529.3139.
Iriomoteolide-1b (2) Rf value (EtOAc/hexane 2:1): 0.32; [α]D20 = −78 (c = 0.03, CHCl3); IR (film, cm−1) 3703, 2965, 1810, 1694; 1H NMR (500 MHz, CDCl3) δ 6.32 (s, 1H), 5.82 (s, 1H), 5.83–5.78 (m, 1H), 5.73–5.68 (m, 2H), 5.54 (d, J = 15.7 Hz, 1H), 4.98 (dt, J = 7.5, 3.0 Hz, 1H), 4.55 (s, 1H), 4.17–4.1 (m, 1H), 3.90 (m, 1H), 3.82–3.75 (m, 1H), 3.75–3.7 (m, 1H), 2.35–2.3 (m, 2H), 2.3–2.22 (m, 2H), 2.27 (s, 3H), 2.12–2.08 (m, 2H), 1.99 (s, 3H), 1.98–1.9 (m, 2H), 1.66–1.6 (m, 1H), 1.48 (s, 3H), 1.42–1.35 (m, 1H), 1.18 (d, J = 7.1 Hz, 3H), 1.12 (d, J = 6.8 Hz, 3H), 0.92 (d, J = 7.0 Hz, 3H), 0.89 (d, J = 6.4 Hz, 3H); 13C NMR (125 MHz, CDCl3) δ 200.7, 167.2, 160.1, 159.3, 136, 132.9, 129.8, 126.5, 120.5, 118.6, 77.6, 76.9, 75.5, 69.4, 68.2, 48.3, 40.9, 40.4, 36.2, 35.6, 31.5, 30.9, 22.6, 21.6, 20.3, 20.1, 15.7, 15, 13.7; MS (ESI, m/z) [M + Na]+ 529.33. HRMS (ESI) [M + Na]+ calcd for C29H46O7Na 529.3141, found 529.3135.
Alcohol (32): To a suspension of CuI (2.18 g, 11.4 mmol) in THF (30 mL), we added MeLi (14.3 mL, 22.8 mmol) at −60 °C. The mixture was slowly warmed up to 0 °C to obtain a clear solution and cooled back to −60 °C. TMSCl (1.5 mL, 11.4 mmol) was added dropwise and stirred at this temperature for 5 min. A soln of alkynyl ester 21 (1.2 g, 5.7 mmol) in THF (5 mL) was added dropwise at −60 °C. After addtion, the reaction mixture was warmed up to 0 °C slowly and stirred for 30 min. Then, the reaction mixture was poured into sat NH4Cl (30 mL) and crushed ice. The organic layer was separated and the aqueous layer was extracted by Et2O (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhydrous MgSO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (5% EtOAc/hexanes) produced an inseparable mixture of E-enoate and Z-isomer (1.2 g, 92%) as a colorless oil. Rf value (EtOAc/hexane 1:10) 0.45.
To the above stirred enoate (1.15 g, 5.04 mmol) in DCM (50 mL), we added DIBAL-H (20 mL, 20 mmol) at −78 °C. The reaction mixture was stirred 1h before addition of 10 mL saturated NH4Cl (aq) and 20 mL 25% Rochelle salt solution. The mixture was stirred at rt, until a clear soln was obtained. The organic phase was separated and the aq phase was extracted with Et2O (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (15% Et2O/hexanes) afforded the E-allyl alcohol 32 (0.71 g, 68%) and Z-isomer (0.3 g, 29%). Rf value (EtOAc/hexane 1:4) 0.2; [α]D20 = +27 (c = 0.2, CHCl3); IR (film, cm−1): 3350, 2965, 1624, 1456, 1210, 1008, 914; 1H NMR (400 MHz, CDCl3) δ 5.55 (ddd, J = 17.2, 10.3, 8.1 Hz, 1H), 5.44 (t, J = 6.7 Hz, 1H), 5.26–5.09 (m, 2H), 4.62 (d, J = 6.9 Hz, 1H), 4.41 (d, J = 6.9 Hz, 1H), 4.11 (d, J = 6.7 Hz, 2H), 3.86 (t, J = 8.2 Hz, 1H), 3.27 (s, 3H), 2.35–2.16 (m, 1H), 2.10 (s, 1H), 1.63 (s, 3H), 0.92 (d, J = 7.1 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 140.28, 136.49, 125.11, 118.63, 93.38, 79.84, 77.19 58.99, 55.37, 46.73, 15.5, 13.36. MS (ESI, m/z) [M+Na]+ 223.1.
Aldehyde (33): To a stirred solution of the above alcohol (0.7 g, 3.5 mmol) (1.45 g, 7.24 mmol) in DCM (30 mL), we added imidazole (357 mg, 5.26 mmol) and TBSCl (0.58 g, 3.82 mmol) at 0 °C. The reaction was warmed up to rt and stirred for 1 h, before pouring into a mixture of sat NaHCO3 (50 mL) and crushed ice. The mixture was extracted with ethers (3 × 30 mL) and the organic layer was washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo to give the crude TBS ether as a clear oil. The resulting crude product was used for the next step without further purification. Flash chromatography on silica gel (25% EtOAc/hexanes) afforded the silyl ether (1.1 g, 100%). Rf value (EtOAc/hexane 1:4) 0.8; [α]D20 = +12 (c = 0.55, CHCl3); IR (film, cm−1): 2970, 2811, 1610, 1230, 1155, 925; 1H NMR (400 MHz, CDCl3) δ 5.59 (ddd, J = 17.2, 10.4, 8.0 Hz, 1H), 5.39 (t, J = 6.0 Hz, 1H), 5.26–5.10 (m, 2H), 4.65 (d, J = 6.8 Hz, 1H), 4.44 (d, J = 6.8 Hz, 1H), 4.20 (d, J = 6.1 Hz, 2H), 3.89 (t, J = 8.1 Hz, 1H), 3.30 (s, 3H), 2.36–2.16 (m, 1H), 1.62 (d, J = 0.7 Hz, 3H), 0.95 (d, J = 7.1 Hz, 3H), 0.88 (s, 9H), 0.04 (s, 6H). 13C NMR (101 MHz, CDCl3) δ 137.81, 136.65, 126.04, 118.29, 93.45, 79.81, 60.09, 55.33, 46.64, 25.86, 18.24, 15.29, 13.66, −5.20.
To a stirred solution of the above olefin (1.05 g, 3.34 mmol) (1.09 g, 3.47 mmol) in dioxane (20 mL) and water (7 mL), we added 2,6-lutidine (1.9 mL, 16.7 mmol), OsO4 (2.5% in t-BuOH, 1.6 mL, 0.13 mmol) and NaIO4 (2.85 g, 12.4 mmol) at 0 °C. The mixture was stirred at 0 °C for 16 h before saturated NaHCO3 (10 mL) and Na2S2O3 (10 mL) were added. The mixture was stirred for another 30 min, extracted by EtOAc. The combined organic extracts were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. Flash chromatography on silica gel (25% EtOAc/hexanes) produced the aldehyde 33 (655 mg, 62%), which was used for the next step quickly.
Aldehyde (34): To a stirred solution of sulfone 5 (628 mg, 0.94 mmol) in DME (30 mL), we added KHMDS (1.85 mL, 0.5 M soln in toluene, 0.93 mmol) at −78 °C. The reaction mixture was stirred for 30 min, before a soln of aldehyde 33 (357 mg, 1.13 mmol) in DME (5 mL) was transferred in. The reaction mixture was stirred for another 30 min, before it was warmed up to rt. The reaction was quenched by sat NH4Cl (10 mL) at −78 °C. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give olefin as a colorless oil (544 mg, 76% yield). Rf value (EtOAc/hexane 1:10): 0.5; [α]D20 = +14 (c = 0.6, CHCl3); IR (film, cm−1) 2928, 1455, 1248, 1108; 1H NMR (400 MHz, CDCl3) δ 5.73–5.54 (m, 1H), 5.40 (t, J = 6.1 Hz, 1H), 5.23 (dd, J = 15.4, 8.5 Hz, 1H), 4.88 (d, J = 32.2 Hz, 2H), 4.68 (d, J = 6.8 Hz, 1H), 4.56 (d, J = 5.1 Hz, 2H), 4.41 (d, J = 6.8 Hz, 1H), 4.21 (d, J = 6.1 Hz, 3H), 4.14 (dd, J = 8.7, 3.7 Hz, 1H), 3.86 (t, J = 8.3 Hz, 2H), 3.41 (q, J = 9.8 Hz, 2H), 3.30 (s, 3H), 2.35–2.09 (m, 8H), 1.63 (s, 3H), 1.44 (s, 3H), 1.35 (s, 3H), 1.13 (s, 3H), 0.95 (d, J = 7.0 Hz, 3H), 0.9 (s, 9H), 0.87 (s, 9H), 0.12–−0.07 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 143.50, 138.19, 131.28, 130.98, 128.21, 127.41, 127.29, 125.83, 114.25, 107.15, 93.11, 81.53, 79.29, 78.34, 74.98, 73.45, 70.52, 60.11, 55.31, 46.97, 43.68, 39.56, 36.49, 28.56, 26.44, 25.90, 25.80, 19.16, 18.27, 17.97, 15.76, 13.52, −4.59, −4.65, −5.15.
To a stirred solution of the above benzyl ether (520 mg, 0.68 mmol) in THF (20 mL) and allyl ethyl ether (1 mL), we transferred a soln of lithium metal (100 mg, 14.4 mmol) in liquid ammonia (30 mL) in portions at −78 °C. The reaction was carefully monitored by TLC and stopped immediately after the solution became slightly blue. Ammonium chloride (3 g) was added to quench the reaction. The mixture was allowed to warm up to rt to evaporate ammonia, before water (10 mL) was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:10) to give the alcohol as a colorless oil (356 mg, 78% yield), along with the recovered starting material. Rf value (EtOAc/hexane 1:4): 0.4; [α]D20 = +12 (c = 1, CHCl3); IR (film, cm−1) 3430, 2965, 1255, 1087; 1H NMR (400 MHz, CDCl3) δ 5.70–5.55 (m, 1H), 5.38 (t, J = 6.0 Hz, 1H), 5.23 (dd, J = 15.5, 8.4 Hz, 1H), 4.93 (s, 1H), 4.85 (s, 1H), 4.67 (d, J = 6.8 Hz, 1H), 4.40 (d, J = 6.8 Hz, 1H), 4.19 (d, J = 5.6 Hz, 3H), 3.85 (dd, J = 11.1, 5.3 Hz, 2H), 3.51 (dd, J = 11.8, 3.6 Hz, 1H), 3.34 (dd, J = 11.5, 9.5 Hz, 1H), 3.28 (s, 3H), 2.37–2.03 (m, 10H), 1.60 (s, 3H), 1.43 (s, 3H), 1.34 (s, 3H), 1.04 (s, 3H), 0.97–0.91 (m, 3H), 0.87 (s, 9H), 0.86 (s, 9H), 0.04 (s, 12H); 13C NMR (100 MHz, CDCl3) δ 143, 138.15, 131.21, 131.01, 125.82, 114.54, 106.99, 93.07, 82.56, 79.24, 75.94, 70.70, 65.40, 60.10, 55.29, 46.92, 43.44, 39.63, 36.07, 28.59, 26.60, 25.88, 25.78, 18.60, 18.25, 17.95, 15.70, 13.52, −4.62, −4.66, −5.17.
To a suspension of the above alcohol (139 mg, 0.21 mmol) and sodium bicarbonate (106 mg, 1.26 mmol) in DCM (15 mL), we added Dess–Martin periodinane (176 mg, 0.42 mmol) at rt. The reaction mixture was stirred for 1 h, before it was poured into a mixture of sat NaHCO3 (10 mL) and sodium thiosulfate (10 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo to give the crude aldehyde 34 (138 mg, quantitative), which was used directly in the next step without further purification.
Alcohol (36): To a stirred mixture of potassium tert-butoxide (13.9 mL, 1.0 M soln in THF, 13.9 mmol) and trans-2-butene (2.2 mL, 23.2 mmol) in THF (40 mL), we added n-butyllithium (8.8 mL, 1.6 M soln in THF, 13.9 mmol) at −78 °C. After complete addition of n-butyllithium, the mixture was stirred at −45 °C for 10 min. The resulting orange solution was cooled to −78 °C again and a solution of (−)-Ipc2BOMe (5.3 g, 16.6 mmol) in THF (10 mL) was added dropwise. After 30 min of stirring, boron trifluoride etherate (2.4 mL, 18.6 mmol) was added dropwise. Then, the aldehyde 35 (2.14 g, 9.3 mmol) in THF (5 mL) was transferred in. The mixture was stirred at −78 °C for 3 h, before NaOH (11 mL, 3 M soln) and H2O2 (7.5 mL, 70% soln) were added. The contents were refluxed for 1 h. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:20) to give the alcohol 36-favored desired syn product (8:1 dr) as a colorless oil (1.92 g, 72% yield). Rf value (EtOAc/hexane 1:10): 0.6; [α]D20 = +1.5 (c = 0.33, CHCl3); IR (film, cm−1) 3410, 2965, 1456, 1044; 1H NMR (500 MHz, CDCl3) δ 5.81 (ddd, J = 17.8, 10.4, 7.6 Hz, 1H), 5.07–5.05 (m, 1H), 5.04–5.01 (m, 1H), 5.02–5.00 (m, 1H), 3.76 (qd, J = 6.5, 3.5 Hz, 1H), 3.46 (dd, J = 8.5, 3.6 Hz, 1H), 3.24 (d, J = 3.6 Hz, 1H), 2.30–2.21 (m, 1H), 1.80–1.69 (m, 1H), 1.48–1.30 (m, 3H), 1.08 (d, J = 6.3 Hz, 3H), 1.03 (d, J = 6.9 Hz, 3H), 0.88 (s, 9H), 0.84 (d, J = 7.0 Hz, 3H), 0.06 (s, 6H); 13C NMR (125 MHz, CDCl3) δ 141.36, 114.43, 73.75, 72.83, 44.16, 38.41, 36.93, 18.30, 17.99, 16.98, 15.22, −4.72, −5.09; MS (ESI, m/z) [M + Na]+ 309.
Alcohol (37): To a stirred solution of alcohol 36 (1.78 g, 6.25 mmol) in DMF (25 mL), we added NaH (60%, 375 mg, 9.25 mmol) at 0 °C. The mixture was stirred for 30 min before PMBCl (1.25 mL, 9.25 mmol) was added at 0 °C. After stirring at rt over night, water (10 mL) and Et2NH (5 mL) were added and the mixture was stirred for 1h, before it was poured into sat NaHCO3 (aq). The mixture was extracted with diethyl ether (3 × 30 mL). The combined organic extracts were washed with water and brine, dried over anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:30) to give the PMB ether as a colorless oil (2.11 g, 83% yield). Rf value (EtOAc/hexane 1:10): 0.7; [α]D20 = +3.2 (c = 0.35, CHCl3); IR (film, cm−1) 2952, 1654, 1062; 1H NMR (500 MHz, CDCl3) δ 7.27 (d, J = 8.6 Hz, 1H), 6.87 (d, J = 8.6 Hz, 1H), 5.94–5.83 (m, 1H), 5.09–4.99 (m, 1H), 4.56 (d, J = 11.0 Hz, 1H), 4.39 (d, J = 11.0 Hz, 1H), 3.81 (s, 1H), 3.67 (tt, J = 9.7, 4.9 Hz, 1H), 3.37 (ddd, J = 10.2, 5.0, 2.4 Hz, 1H), 2.60–2.52 (m, 1H), 1.65 (tt, J = 13.5, 5.0 Hz, 1H), 1.51 (ddd, J = 13.4, 10.3, 2.8 Hz, 1H), 1.27 (ddd, J = 13.7, 10.9, 2.6 Hz, 1H), 1.07 (d, J = 6.2 Hz, 3H), 1.03 (d, J = 6.9 Hz, 3H), 0.88 (brs, 9H), 0.79 (d, J = 6.7 Hz, 3H), 0.02 (m, 6H); 13C NMR (125 MHz, CDCl3) δ 158.93, 140.66, 131.08, 129.18, 114.21, 113.57, 80.37, 72.47, 71.02, 55.16, 40.42, 36.03, 35.42, 25.79, 20.94, 18.01, 15.45, 12.96, −4.35, −4.91.
To a stirred solution of the above olefin (1.89 g, 4.68 mmol) in THF (30 mL), we added 9-BBN (18.5 mL, 0.5 M soln in THF, 9.36 mmol) at 0 °C. The reaction mixture was allowed to warm up to rt and stirred for 3 h. NaOH (1 mL) and H2O2 (5.5 mL) were added and the mixture was refluxed for 1 h. The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over Na2SO4 and concentrated in vacuo. Column chromatography provided alcohol 37 as a colorless oil (1.55 g, 77%). Rf value (EtOAc/hexane 1:1): 0.65; [α]D20 = +1.5 (c = 0.2, CHCl3); IR (film, cm−1) 3411, 2926, 1609, 1038; 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 8.5 Hz, 1H), 6.86 (d, J = 8.5 Hz, 1H), 4.56 (d, J = 11.0 Hz, 1H), 4.38 (d, J = 11.0 Hz, 1H), 3.78 (d, J = 2.1 Hz, 2H), 3.73–3.63 (m, 1H), 3.59 (d, J = 12.3 Hz, 1H), 3.36 (d, J = 10.1 Hz, 1H), 2.93 (s, 1H), 2.05 (dd, J = 7.9, 5.6 Hz, 1H), 1.76 (dt, J = 21.2, 7.1 Hz, 1H), 1.69–1.51 (m, 1H), 1.45–1.31 (m, 1H), 1.18 (dd, J = 13.4, 10.9 Hz, 1H), 1.07 (d, J = 6.2 Hz, 3H), 0.88 (d, J = 8.1 Hz, 3H), 0.87 (s, 9H), 0.76 (d, J = 6.7 Hz, 3H), 0.02 (s, 3H), 0.01 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 159.05, 130.55, 129.32, 113.63, 80.86, 72.38, 71.19, 61.80, 55.12, 36.18, 34.81, 33.45, 33.11, 25.80, 20.75, 18.01, 17.32, 13.21, −4.34, −4.86. MS (ESI, m/z) [M + Na]+ 447.4.
Sulfone (38): To a stirred solution of alcohol 37 (1.5 g, 3.57 mmol), 1-(4-Hydroxyphenyl)-1H-tetrazole-5-thiol (1.26 g, 6.97 mmol) and triphenylphosphine (1.38 g, 5.29 mmol) in THF (30 mL), we added DIAD (1.2 mL, 6.29 mmol) at 0 °C. The reaction mixture was warmed up to rt and stirred overnight, before it was poured into sat NaHCO3 (30 mL). The organic layer was separated and the aq layer was extracted with Et2O; the combined organic layer was washed with water and brine, dried over MgSO4 and concentrated in vacuo. Column chromatography provided the sulfide as a colorless oil (1.7 g, 83%). Rf value (EtOAc/hexane 1:4): 0.5; [α]D20 = +2.2 (c = 0.25, CHCl3); IR (film, cm−1) 2962, 2878, 1616, 1069; 1H NMR (500 MHz, CDCl3) δ 7.58–7.51 (m, 1H), 7.22 (d, J = 8.7 Hz, 1H), 6.84 (d, J = 8.7 Hz, 1H), 4.50 (d, J = 11.1 Hz, 1H), 4.36 (d, J = 11.1 Hz, 1H), 3.77 (s, 1H), 3.68 (qd, J = 6.1, 3.8 Hz, 1H), 3.53 (ddd, J = 12.9, 8.9, 5.6 Hz, 1H), 3.41–3.32 (m, 1H), 2.09–2.00 (m, 1H), 1.63 (ddt, J = 10.5, 8.9, 4.4 Hz, 1H), 1.54 (ddd, J = 13.3, 10.0, 3.0 Hz, 1H), 1.19 (ddd, J = 13.6, 10.5, 2.8 Hz, 1H), 1.06 (d, J = 6.2 Hz, 3H), 0.93 (d, J = 6.8 Hz, 3H), 0.87 (s, 9H), 0.78 (d, J = 6.7 Hz, 3H), 0.01 (s, J = 8.2 Hz, 6H); 13C NMR (125 MHz, CDCl3) δ 158.90, 154.35, 133.63, 130.92, 129.92, 129.64, 129.09, 123.71, 113.57, 80.20, 72.31, 71.08, 55.15, 36.20, 34.40, 33.69, 31.92, 30.96, 25.80, 20.68, 18.02, 15.52, 13.39, −4.32, −4.84.
To a stirred solution of thus obtained sulfide (1.41 g, 2.28 mmol) in ethanol (35 mL), we added a solution of ammonium molybdate (1.28 g, 1.04 mmol) in hydrogen peroxide (6.5 mL) and water (3 mL) at rt. The reaction mixture was stirred for 12 h and poured into a mixture of sat NaHCO3 (10 mL) and sodium thiosulfate (10 mL). The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:10) to give sulfone 38 as a colorless oil (1.4 g, 94% yield). Rf value (EtOAc/hexane 1:4): 0.5; [α]D20 = +0.6 (c = 0.3, CHCl3); IR (film, cm−1) 2928, 2860, 1609, 1511, 1252, 1068; 1H NMR (500 MHz, CDCl3) δ 7.70–7.65 (m, 1H), 7.63–7.56 (m, 2H), 7.24 (d, J = 8.5 Hz, 1H), 6.87 (d, J = 8.4 Hz, 1H), 4.49 (d, J = 11.1 Hz, 1H), 4.39 (d, J = 11.1 Hz, 1H), 3.80 (s, 3H), 3.68 (tt, J = 6.3, 3.1 Hz, 1H), 3.39 (dt, J = 9.8, 3.0 Hz, 1H), 2.21–2.11 (m, 1H), 2.07 (ddd, J = 9.7, 7.4, 3.2 Hz, 1H), 1.74 (ddd, J = 12.0, 9.2, 7.3 Hz, 1H), 1.63 (dtd, J = 9.7, 6.5, 3.1 Hz, 1H), 1.56 (ddd, J = 13.2, 9.9, 3.0 Hz, 1H), 1.18 (ddd, J = 13.5, 10.6, 3.0 Hz, 1H), 1.07 (d, J = 6.2 Hz, 3H), 0.93 (d, J = 6.9 Hz, 3H), 0.87 (s, 9H), 0.78 (d, J = 6.7 Hz, 3H), 0.03 (s, 3H), 0.01 (s, 3H); 13C NMR (125 MHz, CDCl3) δ 159.01, 153.29, 132.95, 131.29, 130.62, 129.57, 129.26, 124.99, 113.67, 79.96, 72.23, 71.19, 55.18, 54.71, 36.20, 34.18, 33.41, 25.78, 24.00, 20.63, 18.01, 15.65, 13.43, −4.33, −4.86.
Coupling product (39): To a stirred solution of the above solfone (194 mg, 0.32 mmol) and aldehyde 34 (253 mg, 0.38 mmol) in DME (15 mL), we added KHMDS (1 mL, 0.5 M soln in toluene, 0.5 mmol) at −65 °C. The reaction mixture was stirred for 1 h at −65 °C before it was warmed up to rt. The reaction was quenched by sat NH4Cl (5 mL) at −65 °C and the organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:20) to give the pure E-olefin 39 as a colorless oil (180 mg, 54% yield) and corresponding Z-isomer (21 mg, 6%). Rf value (EtOAc/hexane 1:10): 0.45; [α]D20 = +11 (c = 0.35, CHCl3); IR (film, cm−1) 2861, 1735, 1622, 1515, 1263, 1080; 1H NMR (500 MHz, CDCl3) δ 7.78 (d, J = 8.3 Hz, 1H), 7.39 (d, J = 8.4 Hz, 1H), 6.24–6.13 (m, 1H), 5.97 (d, J = 15.6 Hz, 1H), 5.93 (t, J = 6.1 Hz, 1H), 5.78 (dd, J = 15.4, 8.5 Hz, 1H), 5.42 (d, J = 39.9 Hz, 1H), 5.22 (d, J = 6.8 Hz, 1H), 5.02 (d, J = 11.1 Hz, 1H), 4.93 (dd, J = 20.6, 9.0 Hz, 1H), 4.74 (d, J = 6.0 Hz, 1H), 4.40 (t, J = 8.5 Hz, 2H), 4.33 (s, 3H), 4.25–4.17 (m, 1H), 3.88 (d, J = 5.5 Hz, 1H), 3.84 (s, 3H), 2.91–2.75 (m, 2H), 2.72 (dd, J = 12.4, 6.9 Hz, 2H), 2.63 (d, J = 12.9 Hz, 1H), 2.42–2.31 (m, 1H), 2.16 (s, 3H), 1.99 (s, 3H), 1.90 (s, 3H), 1.60 (d, J = 6.2 Hz, 3H), 1.49 (d, J = 7.0 Hz, 3H), 1.42 (m, 27H), 1.32 (d, J = 6.6 Hz, 3H), 0.58–0.53 (m, 18H).
To a stirred solution of the above PMB ether (163 mg, 0.16 mmol) in DCM (10 mL) and pH 7.0 buffer (0.8 mL), we added DDQ (70 mg, 0.32 mmol) at 0 °C and the reaction mixture was stirred at that temperature for 1 h. The reaction was quenched by sat NaHCO3 (5 mL) and the organic layer was separated. The aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic extracts were washed with sat NaHCO3, water and brine, dried on anhyd MgSO4 and evaporated in vacuo. The crude product was purified by flash chromatography (EtOAc/hexane 1:15) to give the corresponding alcohol as a colorless oil (119 mg, 79% yield). Rf value (EtOAc/hexane 1:4): 0.6; [α]D20 = +21 (c = 0.45, CHCl3); IR (film, cm−1) 3440, 2926, 1621, 1245, 1088; 1H NMR (400 MHz, CDCl3) δ 5.72–5.60 (m, 1H), 5.51–5.36 (m, 3H), 5.25 (dd, J = 15.4, 8.4 Hz, 1H), 4.97 (s, 1H), 4.88 (s, 1H), 4.68 (d, J = 6.8 Hz, 1H), 4.42 (d, J = 6.8 Hz, 1H), 4.21 (d, J = 6.1 Hz, 2H), 3.94 (dd, J = 9.2, 2.9 Hz, 1H), 3.91–3.84 (m, 2H), 3.74 (dd, J = 6.2, 3.4 Hz, 1H), 3.62 (d, J = 7.4 Hz, 1H), 3.44 (d, J = 3.3 Hz, 1H), 3.30 (s, 3H), 2.62–2.49 (m, 1H), 2.38–2.09 (m, 8H), 2.06 (dd, J = 5.3, 3.1 Hz, 1H), 2.04–2.00 (m, 1H), 1.62 (s, 3H), 1.45 (s, 3H), 1.35 (s, 3H), 1.25 (s, 3H), 1.09 (d, J = 6.2 Hz, 3H), 0.89 (s, 12H), 0.88 (s, 15H), 0.82 (d, J = 6.8 Hz, 3H), 0.13–−0.03 (m, 18H). To a stirred solution of the above TBS ether (114 mg, 0.12 mmol) in methanol (4 mL), we added ammonium fluoride (124 mg, 3.37 mmol) at rt and the reaction mixture was stirred at that temperature for 8 h. Et2O (30 mL) was added to precipitate the ammonium fluoride, which was removed by suction filtration. The crude product was purified by flash chromatography (EtOAc/hexane 1:4) to give the alcohol 39 as a colorless oil (90 mg, 90% yield). Rf value (EtOAc/hexane 2:1): 0.35; [α]D20 = +24 (c = 0.3, CHCl3); IR (film, cm−1) 3445, 2966, 2928, 1652, 1376, 1251, 1071; 1H NMR (400 MHz, CDCl3) δ 5.80–5.57 (m, 1H), 5.49 (t, J = 7.0 Hz, 1H), 5.45 (d, J = 15.6 Hz, 1H), 5.23 (dd, J = 15.5, 8.4 Hz, 1H), 4.92 (s, 1H), 4.84 (s, 1H), 4.68 (d, J = 6.8 Hz, 1H), 4.42 (d, J = 6.8 Hz, 1H), 4.16 (d, J = 6.7 Hz, 2H), 3.91–3.86 (m, 1H), 3.85 (dd, J = 8.9, 3.2 Hz, 1H), 3.77 (dd, J = 6.3, 3.2 Hz, 1H), 3.52 (d, J = 9.4 Hz, 1H), 3.30 (s, 3H), 2.35–2.11 (m, 6H), 2.08 (dd, J = 15.0, 3.1 Hz, 1H), 1.96–1.82 (m, 1H), 1.74 (d, J = 8.0 Hz, 1H), 1.68 (s, 3H), 1.59–1.48 (m, 1H), 1.44 (s, 3H), 1.35 (s, 3H), 1.17 (s, 3H), 1.09 (d, J = 6.3 Hz, 3H), 0.96 (d, J = 7.1 Hz, 3H), 0.89 (s, 9H), 0.87 (s, 9H), 0.84 (d, J = 7.0 Hz, 3H), 0.09–0.00 (m, 12H). 13C NMR (100 MHz, CDCl3) δ 143.35, 140.99, 133.85, 131.41, 130.87, 129.89, 124.82, 114.29, 107.06, 93.20, 81.99, 81.24, 79.35, 77.12, 73.18, 72.90, 70.61, 59.19, 55.36, 47.04, 43.54, 39.49, 39.41, 38.88, 37.16, 36.16, 35.43, 28.40, 26.51, 25.79, 25.71, 20.44, 17.97, 17.65, 15.82, 13.92, 13.49, −4.57, −4.66, −4.77, −5.07. MS (ESI, m/z) [M + Na]+ 845.5.
Macrolactone (40): To a stirred solution of allyl alcohol 39 (85 mg, 0.104 mmol) in DCM (10 mL), we added activated MnO2 (36 mg, 90%, 0.416 mmol) at rt, and the reaction was stirred at rt for 3 h. Another portion of MnO2 (36 mg, 90%, 0.416 mmol) was added and was stirred for two more hours. Suction filtration gave a crude aldehyde (83 mg) that was used for the next step without further purification. Rf value (EtOAc/hexane 1:2): 0.7. To a stirred solution of thus obtained aldehyde (83 mg, 0.1 mmol) and 2-methyl-2-butene (2 mL) in tert-butanol (8 mL), we added a solution of NaH2PO4.H2O (200 mg) and NaClO2 (200 mg), dropwise, in H2O (2 mL) at 0 °C. In addition, the reaction mixture was allowed to warm up to rt and stirred for 20 min. The reaction was poured into water (5 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic extracts were washed with water and brine, dried over anhyd Na2SO4 and evaporated in vacuo. The crude product was purified by flash chromatography (MeOH/chloroform 3:100) to give the seco-acid as a colorless oil (77 mg, 88% yield for two steps). Rf value (EtOAc/hexane 1:2): 0.6.
To the solution of thus obtained seco-acid in THF (5 mL), we added DIPEA (0.24 mL, 1.38 mmol) and 2,4,6-trichlorobenzoyl chloride (0.14 mL, 0.92 mmol) at rt. The reaction was stirred for 3 h at that temperature, before the THF solvent was removed by vacuo. The the residue toluene (10 mL) was added and the solution was transferred to a stirred solution of DMAP (280 mg, 2.3 mmol) in toluene (180 mL) at rt over 16 h, through a syringe pump. The resulting mixture was stirred at rt for 48 h and poured into sat NaHCO3 (20 mL). The organic layer was separated and the aq was extracted with diethyl ether (3 × 20 mL). The combined organic phase was washed with water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:40) provided macrolactone 40 as a colorless oil (56 mg, 75%). Rf value (EtOAc/hexane 1:10): 0.5; [α]D20 = +16 (c = 0.3, CHCl3); IR (film, cm−1) 2930, 1715, 1166, 1029; 1H NMR (400 MHz, CDCl3) δ 5.70–5.56 (m, 3H), 5.37 (d, J = 15.7 Hz, 1H), 5.28 (dd, J = 15.6, 7.5 Hz, 1H), 4.99 (t, J = 6.1 Hz, 1H), 4.96 (s, 1H), 4.84 (s, 1H), 4.66 (d, J = 6.7 Hz, 1H), 4.53 (d, J = 6.7 Hz, 1H), 4.11 (dd, J = 7.5, 3.4 Hz, 1H), 3.96–3.88 (m, 1H), 3.79–3.71 (m, 1H), 3.63 (dt, J = 9.1, 5.9 Hz, 1H), 3.47 (dd, J = 14.8, 7.7 Hz, 1H), 3.38 (s, 3H), 2.52 (dd, J = 7.0, 3.6 Hz, 1H), 2.36–2.23 (m, 1H), 2.19 (s, 3H), 2.18–2.11 (m, 2H), 2.12–2.05 (m, 2H), 1.93–1.77 (m, 1H), 1.68–1.58 (m, 1H), 1.45 (s, 3H), 1.37 (s, 3H), 1.22 (s, 3H), 1.18 (d, J = 7.0 Hz, 3H), 1.03 (d, J = 6.1 Hz, 3H), 0.88 (s, 9H), 0.87 (s, 9H), 0.84 (d, J = 6.6 Hz, 3H), 0.06–0 (m, 12H); 13C NMR (100 MHz, CDCl3) δ 166.21, 160.37, 141.98, 133.78, 130.36, 129.80, 129.66, 128.10, 117.71, 114.67, 107.00, 93.88, 81.80, 79.80, 78.93, 77.12, 73.77, 72.17, 71.36, 55.45, 48.73, 43.06, 39.77, 37.12, 36.09, 35.47, 34.96, 33.04, 28.34, 26.59, 25.77, 20.44, 19.81, 18.00, 17.43, 15.70, 13.68, 13.06, −4.34, −4.58, −4.70, −4.90. MS (ESI, m/z) [M + Na]+ 843.5.
Diastereomer(2E, 19S) (29): To a stirred solution of TBS ether 40 (56 mg, 0.068 mmol) in THF (4 mL), we added pyridine (1 mL) followed by HF.pyridine complex (70%, 0.5 mL) at 0 °C, and the reaction was warmed up to rt and stirred for 16 h. The reaction mixture was cooled to 0 °C again and sat NaHCO3 was added until the bubbles disappeared. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 20 mL). The combined organic phase was washed with sat NaHCO3, water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:2) provided diol as a colorless oil (31 mg, 76%). Rf value (EtOAc/hexane 2:1): 0.5. A solution of the above acetonide (31 mg, 0.052 mmol) in HOAc (2.4 mL) and water (0.6 mL) was heated at 55 °C for 3 h before the solvents were removed in vacuo. Column chromatography (MeOH/chloroform 1:20) provided the vicinal alcohol as a colorless oil (18 mg, 64%). Rf value (EtOAc/hexane 4:1): 0.2. [α]D20 = −9 (c = 0.14, CHCl3); IR (film, cm−1) 3442, 2962, 1715, 1639, 1431, 1016; 1H NMR (400 MHz, CDCl3) δ 5.76–5.65 (m, 1H), 5.60 (s, 1H), 5.54 (t, J = 7.8 Hz, 1H), 5.51 (d, J = 7.6 Hz, 1H), 5.38 (dd, J = 15.6, 7.3 Hz, 1H), 5.03 (s, 1H), 5.05-5 (m, 1H), 4.97 (s, 1H), 4.68 (d, J = 6.7 Hz, 1H), 4.57 (d, J = 6.8 Hz, 1H), 4.12 (dd, J = 7.3, 3.5 Hz, 1H), 3.74 (dd, J = 6.3, 3.7 Hz, 1H), 3.63 (d, J = 6.2 Hz, 1H), 3.58 (d, J = 10.3 Hz, 1H), 3.39 (s, 3H), 2.56 (dd, J = 6.9, 3.5 Hz, 2H), 2.36 (d, J = 14.5 Hz, 2H), 2.28 (d, J = 13.0 Hz, 1H), 2.17 (s, 3H), 2.13–1.94 (m, 4H), 1.8–1.65 (m, 1H), 1.6–1.40 (m, 2H), 1.47 (brs, 1H), 1.16 (d, J = 7.0 Hz, 3H), 1.13 (d, J = 6.4 Hz, 3H), 0.98 (d, J = 6.9 Hz, 3H), 0.89 (d, J = 6.7 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 160.79, 143.10, 135.04, 130.43, 129.83, 129.61, 117.55, 115.48, 94.16, 78.74, 77.12, 74.58, 73.59, 70.74, 68.04, 55.46, 48.29, 42.94, 39.72, 37.99, 36.48, 35.89, 35.82, 32.90, 19.87, 19.29, 18.46, 15.34, 14.09, 13.2.
To a stirred solution of MOM ether (11 mg, 0.02 mmol) in DCM (2 mL), we added B-bromocatechol borane (0.5 mL, 0.2 M soln in DCM, 0.1 mmol) at −78 °C, and the mixture was stirred at that temperature for 1h, before it was quenched with sat NaHCO3 (5 mL). The organic layer was separated and the aq layer was extracted with ethyl acetate. The combined organic phase was washed with water and brine, dried over anhyd Na2SO4 and concentrated in vacuo. Column chromatography (MeOH/chloroform 1:20) provided the corresponding alcohol as a colorless oil (6.7 mg, 66%). Rf value (EtOAc/hexane/MeOH 8:2:1): 0.3.
To a stirred solution of thus obtained alcohol (3 mg, 0.0059 mmol) and DMAP (9 mg, 0.074 mmol) in DCM (2 mL), we added TESCl (0.01 mL, 0.059 mmol) at 0 °C, and the reaction was stirred at 0 °C for 30 min before sat NaHCO3 (5 mL) was added. The organic layer was separated and the aq layer was extracted with diethyl ether (3 × 10 mL). The combined organic phase was washed with water and brine, dried over anhyd MgSO4 and concentrated in vacuo. Column chromatography (EtOAc/hexane 1:20) provided tris-TES ether as a colorless oil (4.4 mg, 88%). Rf value (EtOAc/hexane 1:4): 0.5.
To a stirred solution of thus obtained diol (2.4 mg, 2.8 μmol) in DCM (1 mL), we added DMSO (40 μL, 0.141 mmol), DIPEA (49 μL, 0.282 mmol) and SO3.Py (22.4 mg, 0.141 mmol) at 0 °C. The mixture was stirred at rt for 30 min. The solvent was removed by high vacuum and THF (1 mL) was added, followed by a HF·Py solution (0.3 mL containing 1 mL 70% HF·Py: 1.1 mL pyridine: 2.4 mL THF) at rt. The reaction mixture was stirred for 1h, before it was quenched with sat NaHCO3 and extracted with diethyl ether (3 × 10 mL). The combined organic layer was washed with water and brine, dried over MgSO4 and concentrated under reduced pressure. The resulting residue was purified by flash chromatography (MeOH/chloroform 1:25) to give diastereomer 29 (1 mg, 71%) as a colorless oil. Rf value (EtOAc/hexane 2:1): 0.3. [α]D20 = +35.7 (c = 0.042, CH2Cl2); IR (film, cm−1) 3465, 2980, 2360, 1695, 1452, 1369, 1222, 1004; 1H NMR (500 MHz, CDCl3) δ 5.77–5.70 (m, 1H), 5.67 (d, J = 18.6 Hz, 2H), 5.58–5.51 (m, 1H), 5.47 (dd, J = 16.0, 7.4 Hz, 1H), 4.86 (s, 1H), 4.84 (s, 1H), 4.83–4.79 (m, 1H), 4.21 (dd, J = 8.2, 3.8 Hz, 1H), 4.07–3.95 (m, 1H), 3.80 (dd, J = 6.3, 3.4 Hz, 1H), 3.35 (s, 1H), 3.06 (brs, 1H), 2.67 (dd, J = 6.8, 3.9 Hz, 1H), 2.50 (d, J = 14.4 Hz, 1H), 2.33 (m, 1H), 2.20 (m, 2H), 2.18 (d, J = 0.8 Hz, 3H), 2.15–2.08 (m, 2H), 2.07–1.96 (m, 2H), 1.94–1.81 (m, 1H), 1.78–1.69 (m, 1H), 1.51–1.45 (m, 1H), 1.45–1.37 (m, 1H), 1.30 (s, 3H), 1.15 (d, J = 6.4 Hz, 3H), 1.13 (d, J = 7.1 Hz, 3H), 1.02 (d, J = 6.9 Hz, 3H), 0.89 (d, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 159.2, 141.4, 134.1, 130.5, 130.3, 129.6, 118.2, 110.9, 99.2, 73.1, 69.7, 48.6, 38.3, 37.85, 37.1, 36.5, 35.7, 35.3, 33.6, 24.6, 23.2, 19.5, 18.7, 14.8, 14.3, 10.5. MS (ESI, m/z) [M + Na]+ 529.31.
Diastereomer (2E, 9R) (30): [α]D20 = +18.8 (c = 0.07, CH2Cl2); IR (film, cm−1) 3380, 2924, 1690, 1684,1541, 1508, 1458, 1212; 1H NMR (500 MHz, CDCl3) δ 9.42 (s, 1H), 6.22 (d, J = 11.0 Hz, 1H), 5.73–5.59 (m, 3H), 5.52 (dd, J = 15.6, 8.1 Hz, 1H), 5.02–4.91 (m, 2H), 4.89 (s, 1H), 4.42 (dd, J = 8.0, 3.6 Hz, 1H), 3.85 (dd, J = 6.3, 3.4 Hz, 1H), 3.56–3.50 (m, 1H), 3.46–3.38 (m, 1H), 2.63 (dd, J = 6.8, 4.0 Hz, 1H), 2.31–2.10 (m, 6H), 2.19 (d, J = 0.9 Hz, 3H), 2.04 (dd, J = 13.8, 9.3 Hz, 1H), 1.94 (dd, J = 14.2, 9.1 Hz, 1H), 1.80 (s, 3H), 1.54–1.45 (m, 1H), 1.38–1.28 (m, 1H), 1.16 (d, J = 6.9 Hz, 3H), 1.11 (d, J = 6.4 Hz, 3H), 0.90–0.80 (m, 6H); 13C NMR (126 MHz, CDCl3) δ 191.2, 167.0, 161.0, 153.3, 143.0, 135.5, 130.7, 130.5, 115.6, 115.3, 75.8, 73.7, 69.5, 48.5, 45.6, 42.7, 40.2, 35.9, 35.9, 35.7, 35.1, 32.1, 20.5, 19.9, 16.7, 16.5, 14.9. MS (ESI, m/z) [M + Na]+ 529.33.
Diastereomer(4S, 5R) (31): [α]D20 = −15 (c = 0.03, CH2Cl2); IR (film, cm−1) 3470, 2987, 2975, 2360, 1684, 1255, 1005; 1H NMR (500 MHz, CDCl3) δ 5.87 (s, 1H), 5.86–5.74 (m, 3H), 5.57 (dd, J = 15.7, 7.2 Hz, 1H), 5.12 (dd, J = 11.5, 5.8 Hz, 1H), 4.88 (s, 1H), 4.85 (s, 1H), 4.16–4.07 (m, 1H), 3.94–3.85 (m, 1H), 3.80 (dt, J = 10, 6.2 Hz, 1H), 3.42 (dq, J = 13.9, 6.9 Hz, 1H), 2.76 (brs, 1H), 2.45–2.32 (m, 3H), 2.33–2.22 (m, 3H), 2.18 (d, J = 11.2 Hz, 1H), 2.08–1.93 (m, 4H), 1.89 (s, 3H), 1.79–1.7 (m, 1H), 1.5–1.41 (m, 1H), 1.13 (d, J = 6.3 Hz, 3H), 1.06 (d, J = 6.9 Hz, 3H), 0.95 (d, J = 6.6 Hz, 3H), 0.91 (d, J = 6.8 Hz, 3H); 13C NMR (126 MHz, CDCl3) δ 167.3, 155.5, 141.5, 134.66, 132.0, 130.0, 128.4, 120.4, 111.1, 99.4, 75.1, 70.3, 69.7, 53.3, 42.6, 39.6, 36.8, 36.8, 35.8, 34.8, 34.1, 32.8, 21.9, 19.7, 19.0, 16.7, 15.8, 14.8. MS (ESI, m/z) [M + Na]+ 529.33.
4. Conclusions
In summary, we completed an efficient synthesis of the proposed structures of iriomoteolide-1a and iriomoteolide-1b. In an effort toward the assignment of correct structures, we rationally designed and synthesized three diastereomers of iriomoteolide-1a. The synthesis of the C1–C15 fragment features an ene reaction and Julia–Kocienski olefination. The trisubstituted Z-enoate or E-enoate of fragment C1–C6 was constructed from a carbocupration of the corresponding acetylene ester by utilizing a Gilman reagent. Other key reactions involved Seebach–Fráter asymmetric alkylation and enzyme kinetic resolution. Of particular interest, spectral data of synthetic iriomoteolide-1a and iriomoteolide-1b did not correlate with those reported for natural iriomoteolide-1a and -1b. Therefore, the structures of both natural iriomoteolide-1a and iriomoteolide-1b were assigned incorrectly. Our convergent synthetic routes to iriomoteolides provided efficient access to a variety of diastereomers and structural derivatives. We synthesized three rationally designed diastereomers of iriomoteolide-1a for the assignment of structures and evaluation of bioactivity. While the NMR data of these diastereomeric compounds are closer to those reported for natural iriomoteolide-1a, they did not completely match the natural product NMRs. We evaluated our synthetic iriomoteolide-1a, iriomoteolide -1b, and three diastereomers. However, none of these iriomoteolide derivatives showed any appreciable cytotoxicity in cancer cell lines. Further work towards the assignment of iriomoteolide structures is in progress.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/md20100587/s1. Table S1: 1H and 13C NMR (CDCl3) data for natural and synthetic iriomoteolide -1a (1). Table S2: 1H and 13C NMR (CDCl3) data for natural and synthetic iriomoteolide -1b (2). Figures S1–S25: Characterization data, 1H-NMR, and 13C NMR spectra.
Author Contributions
Conceptualization, A.K.G.; investigation, A.K.G. and H.Y.; software, A.K.G., H.Y.; methodology, A.K.G. and H.Y.; original draft writing, A.K.G. and H.Y.; writing and editing, all authors; funding acquisition, A.K.G. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Institutes of Health (A.K.G. A1150466).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the manuscript and in the Supplementary Materials.
Acknowledgments
Financial support by the National Institutes of Health (GM122279 and A1150466) is gratefully acknowledged. The authors would like to thank the Purdue University Center for Cancer Research, which provided the shared NMR and mass spectrometry facilities. We thank David D. Anderson (Purdue University) for his assistance with the 2D NMR studies.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Montaser, R.; Luesch, H. Marine natural products: A new wave of drugs? Future Med. Chem. 2011, 3, 1475–1489. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Bardón, M.; Pérez-Pertejo, Y.; Ordóñez, C.; Sepulveda-Crespo, D.; Carballeira, N.M.; Tekwani, B.L.; Murugesan, S.; Martinez-Valladares, M.; García-Estrada, C.; Reguera, R.M.; et al. Screening Marine Natural Products for New Drug Leads against Trypanosomatids and Malaria. Mar. Drugs 2020, 18, 187. [Google Scholar] [CrossRef] [PubMed]
- Ghareeb, M.A.; Tammam, M.A.; El-Demerdash, A.; Atanasov, A.G. Insights about clinically approved and Preclinically investigated marine natural products. Curr. Res. Biotechnol. 2020, 2, 88–102. [Google Scholar] [CrossRef]
- Shinde, P.; Banerjee, P.; Mandhare, A. Marine natural products as source of new drugs: A patent review (2015–2018). Expert Opin. Ther. Pat. 2019, 29, 283–309. [Google Scholar] [CrossRef]
- Manzo, E. Synthesis of Marine Natural Products and Molecules Inspired by Marine Substances. Mar. Drugs 2021, 19, 208. [Google Scholar] [CrossRef]
- Martins, A.; Vieira, H.; Gaspar, H.; Santos, S. Marketed Marine Natural Products in the Pharmaceutical and Cosmeceutical Industries: Tips for Success. Mar. Drugs 2014, 12, 1066–1101. [Google Scholar] [CrossRef]
- Tsuda, M.; Oguchi, K.; Iwamoto, R.; Okamoto, Y.; Kobayashi, J.; Fukushi, E.; Kawabata, J.; Ozawa, T.; Masuda, A.; Kitaya, Y.; et al. Iriomoteolide-1a, a Potent Cytotoxic 20-Membered Macrolide from a Benthic Dinoflagellate Amphidinium Species. J. Org. Chem. 2007, 72, 4469–4474. [Google Scholar] [CrossRef]
- Tsuda, M.; Oguchi, K.; Iwamoto, R.; Okamoto, Y.; Kobayashi, J.; Kawabata, J.; Fukushi, E.; Ozawa, T.; Masuda, A.; Kitaya, Y.; et al. Iriomoteolides-1b and -1c, 20-membered macrolides from a marine dinoflagellate Amphidinium species. J. Nat. Prod. 2007, 70, 1661–1663. [Google Scholar] [CrossRef]
- Fang, L.; Xue, H.; Yang, J. Synthesis of the C1−C12 Fragment of Iriomoteolide 1a by Sequential Catalytic Asymmetric Vinylogous Aldol Reactions. Org. Lett. 2008, 10, 4645–4648. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Yuan, H. Stereoselective synthesis of the C1–C12 segment of iriomoteolide-1a: A very potent macrolide antitumor agent. Tetrahedron Lett. 2009, 50, 1416–1418. [Google Scholar] [CrossRef][Green Version]
- Xie, J.; Horne, D.A. Stereoselective synthesis of iriomoteolide-1a hemiketal core. Tetrahedron Lett. 2009, 50, 4485–4487. [Google Scholar] [CrossRef]
- Chin, Y.-J.; Wang, S.-Y.; Loh, T.-P. Synthesis of Iriomoteolide-1a C13−C23 Fragment via Asymmetric Conjugate Addition and Julia−Kocienski Coupling Reaction. Org. Lett. 2009, 11, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Ye, Z.; Deng, L.; Qian, S.; Zhao, G. Synthetic Studies towards Iriomoteolide-1a: Construction of the C13–C23 Fragment. Synlett 2009, 15, 2469–2472. [Google Scholar]
- Xie, J.; Ma, Y.; Horne, D.A. Asymmetric Synthesis of the C(7)−C(23) Fragment of Iriomoteolide-1a. Org. Lett. 2009, 11, 5082–5084. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-Y.; Chin, Y.-J.; Loh, T.-P. Synthesis of C13–C23 Fragment of Iriomoteolide-1a. Synthesis 2009, 21, 3557–3564. [Google Scholar]
- Paterson, I.; Rubenbauer, P. Synthetic Studies towards Iriomoteolide 1a: Stereocontrolled Construction of C1–C9 and C11–C23 Segments Using Lactate Aldol Chemistry. Synlett 2010, 4, 571–574. [Google Scholar] [CrossRef]
- Li, S.; Chen, Z.; Xu, Z.; Ye, T. Synthesis of the macrocyclic core of iriomoteolide-1a. Chem. Commun. 2010, 46, 4773–4775. [Google Scholar] [CrossRef][Green Version]
- Xie, J.; Ma, Y.; Horne, D.A. Total synthesis of the proposed structure of iriomoteolide-1a. Tetrahedron 2011, 67, 7485–7501. [Google Scholar] [CrossRef]
- Ghosh, A.K.; Yuan, H. Enantioselective Syntheses of the Proposed Structures of Cytotoxic Macrolides Iriomoteolide-1a and -1b. Org. Lett. 2010, 12, 3120–3123. [Google Scholar] [CrossRef]
- Fang, L.; Yang, J.; Yang, F. Enantioselective Synthesis of a Diastereomer of Iriomoteolide-1a. What Is the Correct Structure of the Natural Product? Org. Lett. 2010, 12, 3124–3127. [Google Scholar] [CrossRef]
- Wang, F.; Wu, F.; Jia, X.; Xu, Z.; Guo, Y.; Ye, T. Studies toward the synthesis of iriomotelide-2a: Construction of the C(6)-C(28) fragment. Org. Lett. 2018, 20, 2213–2215. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Feng, G.; Wang, J.; Wu, J.; Dai, W.-M. Synthesis of Two Diastereomers of Iriomoteolide-1a via a Tunable Four-Module Coupling Approach Using Ring-Closing Metathesis as the Key Step. Synlett 2011, 12, 1774–1778. [Google Scholar]
- Huang, J.; Yang, J. Studies Toward Elucidating the Stereochemical Structure of Iriomoteolide 1a. Synlett 2012, 23, 737–740. [Google Scholar]
- Blakemore, P.R. The modified Julia olefination: Alkene synthesis via the condensation of metallated heteroarylalkylsulfones with carbonyl compounds. J. Chem. Soc. Perkin Trans. 2002, 23, 2563–2585. [Google Scholar] [CrossRef]
- Blakemore, P.R.; Cole, W.J.; Kocieński, P.J.; Morley, A. A Stereoselective Synthesis of trans-1,2-Disubstituted Alkenes Based on the Condensation of Aldehydes with Metallated 1-Phenyl-1H-tetrazol-5-yl Sulfones. Synlett 1998, 1, 26–28. [Google Scholar] [CrossRef]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A Rapid Esterification by Means of Mixed Anhydride and Its Application to Large-ring Lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar] [CrossRef]
- Mikami, K.; Shimizu, M. Asymmetric ene reactions in organic synthesis. Chem. Rev. 1992, 92, 1021–1052. [Google Scholar] [CrossRef]
- Snider, B.B. Comprehensive Organic Synthesis; Trost, B.M., Ed.; Pergamon: Oxford, UK, 1991; Volume 2, pp. 527–561. [Google Scholar]
- Mitsunobu, O. The Use of Diethyl Azodicarboxylate and Triphenylphosphine in Synthesis and Transformation of Natural Products. Synthesis 1981, 1, 1–28. [Google Scholar] [CrossRef]
- Gudippati, V.; Bajpai, R.; Curran, D.P. Sulfanylation of 1,3-dithiane anions by 5-(alkylsulfanyl)-1-phenyltetrazoles. Collect. Czech. Chem. Commun. 2009, 74, 771–783. [Google Scholar] [CrossRef][Green Version]
- Keck, G.E.; Boden, E.P. Chelation controlled diastereofacial selectivity in crotyltri-n-butylstannane additions to α-alkoxyaldehydes. Tetrahedron Lett. 1984, 25, 1879–1882. [Google Scholar] [CrossRef]
- Reetz, M.T. Chelation or Non-Chelation Control in Addition Reactions of Chiral α- and β- Alkoxy Carbonyl Compounds. Angew. Chem. Int. Ed. 1984, 23, 556–569. [Google Scholar] [CrossRef]
- Schlosser, M.; Stähle, M. Allylic Compounds of Magnesium, Lithium, and Potassium: σ- or π-Structures? Angew. Chem. Int. Ed. 1980, 19, 487–489. [Google Scholar] [CrossRef]
- Roush, W.R.; Ando, K.; Powers, D.B.; Palkowitz, A.D.; Halterman, R.L. Asymmetric synthesis using diisopropyl tartrate modified (E)- and (Z)-crotylboronates: Preparation of the chiral crotylboronates and reactions with achiral aldehydes. J. Am. Chem. Soc. 1990, 112, 6339–6348. [Google Scholar] [CrossRef]
- Vrielynck, S.; Vandewalle, M.; García, A.M.; Mascareñas, J.L.; Mouriño, A. A chemoenzymatic synthesis of a-ring key-intermediates for 1α,25-dihydroxyvitamin d3 and analogues. Tetrahderon Lett. 1995, 36, 9023–9026. [Google Scholar] [CrossRef]
- Züger, M.; Weller, T. Seebach, D. 2,3-Disubstituted γ-Butyrolactams from the Michael-Additions of Doubly Deprotonated, Optically Active β-Hydroxycarboxylates to Nitroolefins. Preliminary Communication. Helv. Chim. Acta 1980, 63, 2005–2009. [Google Scholar] [CrossRef]
- Corey, E.J.; Fuchs, P.L. A synthetic method for formylethynyl conversion. Tetrahedron Lett. 1972, 13, 3769–3772. [Google Scholar] [CrossRef]
- Gilman, H.; Jones, R.G.; Woods, L.A. The Preparation of Methylcopper and some Observations on the Decomposition of Organocopper Compounds. J. Org. Chem. 1952, 17, 1630–1634. [Google Scholar] [CrossRef]
- Dess, D.B.; Martin, J.C. Readily accessible 12-I-5 oxidant for the conversion of primary and secondary alcohols to aldehydes and ketones. J. Org. Chem. 1983, 48, 4155–4156. [Google Scholar] [CrossRef]
- Meyer, S.D.; Schreiber, S.L. Acceleration of the Dess-Martin Oxidation by Water. J. Org. Chem. 1994, 59, 7549–7552. [Google Scholar] [CrossRef]
- Bal, B.S.; Childers, W.E.; Pinnick, H.W. Oxidation of α,β-un saturated aldehydes. Tetrahedron 1981, 37, 2091–2096. [Google Scholar] [CrossRef]













Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).