Abstract
Chemical investigation of the psychrophilic fungus Pseudogymnoascus sp. HDN17-933 derived from Antarctica led to the discovery of six new tetrapeptides psegymamides A–F (1–6), whose planar structures were elucidated by extensive NMR and MS spectrometric analyses. Structurally, psegymamides D–F (4–6) possess unique backbones bearing a tetrahydropyridoindoles unit, which make them the first examples discovered in naturally occurring peptides. The absolute configurations of structures were unambiguously determined using solid-phase total synthesis assisted by Marfey’s method, and all compounds were evaluated for their inhibition of human (h) nicotinic acetylcholine receptor subtypes. Compound 2 showed significant inhibitory activity. A preliminary structure–activity relationship investigation revealed that the tryptophan residue and the C-terminal with methoxy group were important to the inhibitory activity. Further, the high binding affinity of compound 2 to hα4β2 was explained by molecular docking studies.
1. Introduction
Psychrophilic fungi are a group of cold-adapted fungi residing in polar regions, alpine permafrost, glaciers, deep oceans, and other habitats [1,2,3,4,5,6], which are known for long-term low temperature, strong ultraviolet radiation, low nutrient and water availability, and frequent freeze and thaw cycles [7,8,9]. In order to adapt to such harsh conditions, these fungi have evolved special strategies in their metabolism and physiology [7,10,11,12], which endows the ability to produce diversified secondary metabolites, and makes psychrophilic fungi competitive microorganism group to serve structurally novel and bioactive natural products for drug development [13,14,15,16].
So far, a large number of novel natural products have been isolated from psychrophilic fungi, such as trisorbicillone A, a novel sorbicillin trimer that showed cytotoxic against HL60 cell lines (IC50 3.14 μM) from deep-sea fungal strain Phialocephala sp. [17]; brevione A, the first breviane spiroterpenoid family from Penicillium brevicompactum [18]; penilactones A and B, novel polyketides with NF-kB inhibitory activity (inhibitory rate of 40% at 10 Mm) from Penicillium crustosum PRB-2 [19]; 16-membered trichobotryside A inhibiting the larvae settlement of Balamus amphitrite (EC50 2.5 μg/mL) and 18-membered trychobotrysides B-C from Trichobotrys effuse [20]; and cytotoxic diterpenes conidiogenones B–G isolated from Penicillium sp, and conidiogenone C displayed significant cytotoxicity against HL60 and BEL-7047 cell lines with IC50 values of 0.038 mM and 0.9 mM respectively [21].
During our ongoing research on searching bioactive structures from Antarctic-derived fungi, a psychrophilic strain Pseudogymnoascus sp. HDN17-933, isolated from sand samples collected from Fildes Peninsula, was chosen for study based on its unique HPLC-UV profile. Detailed chemical investigation on its fermentation products afforded six tetrapeptides psegynamides A–F with human nicotinic acetylcholine receptors (nAChRs) targeting activity. Among those structures, psegynamides D–F (4–6) represented the first group of naturally occurring tetrapeptides carrying tetrahydropyridoindole units in the backbones. Herein, we will describe the isolation, structural elucidation, solid-phase total synthesis, and biological activity evaluation of these new compounds.
2. Results and Discussion
The psychrophilic fungal strain Pseudogymnoascus sp. HDN17-933 was isolated from Antarctica. They can grow in an environment of −5 °C, and the optimum growth temperature is 15–18 °C. The studies of their secondary metabolites are infrequent, and only 44 secondary metabolites have been reported until the time of writing. In this paper, the strain was cultured under static conditions at 15 °C for 30 days in a rice medium. The EtOAc extract of strain Pseudogymnoascus sp. HDN17-933 (Figure S2) was fractionated and purified by vacuum chromatography on silica gel, octadecyl-silica (ODS), Sephadex LH-20, and HPLC to afford six tetrapeptides psegynamides A–F (1–6) (Figure 1).
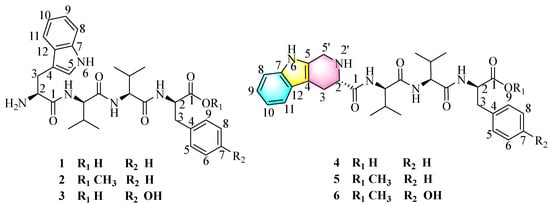
Figure 1.
The structures of compounds 1–6 isolated from Pseudogymnoascus sp. HDN17-933.
Compound 1 named psegynamide A was obtained as a white amorphous powder. Its molecular formula was determined to be C30H39N5O5 based on the molecular ion peak at m/z 550.3025 [M + H]+ (calcd 550.3024) in the HRESIMS analysis, indicating 14 degrees of hydrogen deficiency. In the 1H-NMR spectrum of 1 (Table 1), the characteristic signals of NH were displayed at low-field δH 8.68 (1H, d, J = 9.5 Hz), 8.37 (1H, d, J = 7.8 Hz), and 8.13 (1H, d, J = 9.1 Hz). The signals of α-proton, the characteristic for amino acid residues, were exhibited at δH 4.56 (1H, m), 4.44 (1H, m), 4.30 (1H, m), and 4.18 (1H, m). Four methyl groups were displayed at δH 0.72 (3H, d, J = 6.8 Hz), 0.68 (3H, d, J = 6.8 Hz), 0.61 (3H, d, J = 6.8 Hz), and 0.48 (3H, d, J = 6.8 Hz). Analysis of the 13C-NMR and HSQC spectra indicated the presence of four carbonyl peaks assignable to amide carbonyl groups at δC 173.6, 171.1, 170.7, and 161.9. Four groups of α-proton signals were showed at δC 57.3, 57.1, 53.9, and 52.9. The appearance of these spectra was typical of peptides. Combined analyses of 1H-1H COSY, HMBC, HSQC, NOESY, and TOCSY spectra further revealed the presence of 1 × Trp, 2 × Val, and 1 × Phe residues (Table 1), which were in accordance with the result of Marfey’s analysis (Figure S58). In the HMBC spectrum (Figure 2), NH of Val (1) has a correlation signal with C-1 on Trp; NH of Val (2) has a correlation signal with C-1 on Val (1); NH of Phe has a correlation signal with C-1 on Val (2). Meanwhile, in the NOESY spectrum, the cross-peaks of NH in Val (1) and H-2 in Trp, NH in Val (2) and H-2 in Val (1), NH in Phe and H-2 in Val (2) further confirmed the linkage of amino acid residues by HMBC spectrum. Thus, the planar structure of compound 1 was established as Trp1-Val2-Val3-Phe4.

Table 1.
1H and 13C NMR data in DMSO-d6 for compounds 1–3 (a measured at 600 MHz; b measured at 150 MHz).
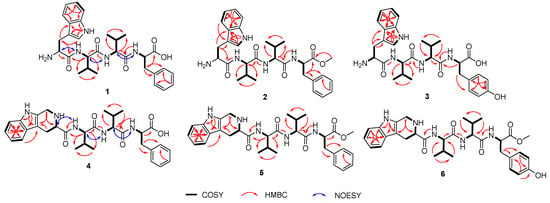
Figure 2.
Key COSY, HMBC, and NOESY correlations of 1–6.
The advanced Marfey’s acidolytic method was considered to determine the absolute configurations of compound 1 [22]. After FDAA derivatization and assisted by HPLC analysis, the presence of L-Val, D-Val, and D-Phe in compound 1 (Figure S58) was confirmed. However, due to the destruction of Trp residue during the strong acid environment, we failed to detect the Trp-FDAA derivative from the hydrolytic mixture. Therefore, we turned to using alkaline hydrolytic to determine the absolute configuration of Trp residue. With the alkaline hydrolytic condition of 5 M LiOH at 110 °C for 16 h was finally adopted, compound 1 was successfully hydrolyzed, and the absolute configuration of Trp was determined as L. As analyzed from the HPLC profile, both L-Val and D-Val derivatives were detected in the acidolytic mixture of compound 1, which makes it another challenge to determine the accurate sequence of the tetrapeptide. Hence, two possible stereoisomers, 1a (L-Trp1-L-Val2-D-Val3-D-Phe4) and 1b (L-Trp1-D-Val2-L-Val3-D-Phe4), were prepared using solid-phase total synthesis (Scheme 1A). By comparing the experimental NMR spectrum with the synthesized NMR data, the absolute configuration of compound 1 (Scheme 1B, Figure S64) was determined to be L-Trp1-D-Val2-L-Val3-D-Phe4, which was inconsistent with the synthesized stereoisomer 1b.
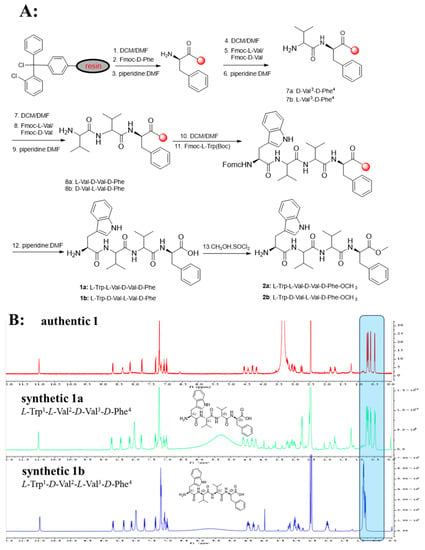
Scheme 1.
(A): Solid-Phase Synthesis of 1a, 1b and 2a, 2b. (B): Comparison of 1H-NMR spectra of authentic 1, synthetic 1a, and 1b.
Compound 2 named psegynamide B was obtained as a white amorphous powder. Its molecular formula was determined to be C31H41N5O5 based on the molecular ion peak at m/z 564.3172 [M + H]+ (calcd 564.3180) in the HRESIMS analysis, which showed 14 Da molecular weight (MW) surplus to compound 1. The 1H and 13C NMR of 2 were similar to those of 1 (Table 1), except for the presence of a methoxy group. The 1H, 13C NMR, and HSQC spectra showed a signal at δC 52.4/δH 3.56, which was assignable to a methoxy group. In addition, the methoxy site was established unambiguously by an HMBC experiment, in which a long-range correlation between CH3 (δH 3.56) and C-1 (δC 172.6) in the Phe unit was observed. Combined analyses of 1H-1H COSY, HMBC, HSQC, and TOCSY spectra assigned the linkage of amino acid residues as Trp1-Val2-Val3-Phe4-OCH3. Marfey’s acidolytic analysis confirmed the presence of L-Val, D-Val, and D-Phe in compound 2, which was a similar case to compound 1. Then, the solid-phase total synthesis and Marfey’s alkaline hydrolytic method were also used to determine the absolute configurations of compound 2. Finally, the absolute configuration of 2 was assigned to be L-Trp1-D-Val2-L-Val3-D-Phe4-OCH3.
Compound 3 named psegynamide C was isolated as a white amorphous powder. The HRESIMS analysis of compound 3 gave a hydrogen adduct [M + H]+ at m/z 566.2977 (calcd 566.2973), corresponding to the molecular formula of C30H39N5O6, which showed one more oxygen than compound 1. In the 1H NMR spectrum (Table 1), two aromatic H-atom signals appearing at δH 6.97 (2H, overlap, m) and 6.59 (2H, overlap, m) were attributable to a 1,4-substituted phenyl. In addition, a characteristic signal of OH was observed at low-field δH 9.18 (1H, s). Careful analysis of NMR of compound 3 revealed a similar structure as compound 1, except for the presence of Tyr residue instead of Phe residue. Thus, the planar structure of compound 3 was established as Trp1-Val2-Val3-Tyr4. Based on Marfey’s acid/alkaline hydrolytic analysis, as well as solid-phase total synthesis, the absolute configuration of 3 was assigned to be L-Trp1- D-Val2-L-Val3-D-Tyr4.
Compound 4 was obtained as a white amorphous powder. Its molecular formula was determined to be C31H39N5O5 on the basis of the molecular ion peak at m/z 562.3032 [M + H]+ (calcd 562.3024) in the HRESIMS analysis, indicating 15 degrees of hydrogen deficiency. Compared the 1D-NMR data with compound 1, compound 4 has an additional methylene (δC 40.8) and the CH-5 of Trp in compound 1 was changed as a quaternary carbon. In addition, one more degree of unsaturation indicated a cyclic structure for compound 4. With the assistance of COSY correlations of H2-5′/NH-2′, H-2/NH-2′ and H-2/H2-3, and the HMBC correlations from H2-5′ to C-4/C-2, the tetrahydropyridoindole residue was deduced. Compound 4 represented the first example naturally occurring peptide with a tetrahydropyridoindole moiety. On the basis of Marfey’s acid hydrolytic analysis and solid-phase total synthesis, the absolute configuration of 4 was assigned to be as alternating LDLD chirality and named psegynamide D.
Psegymamides E-F (5–6) were obtained as white amorphous powders with the molecular formulas of C32H41N5O5 and C32H41N5O6 by HRESIMS, respectively. The 1D NMR spectra of 5 and 6 (Table 2) indicated a skeleton similar to psegynamide D (4). The difference between 4 and 5 was the replacement of the hydroxide group by a methoxy group (δC 52.3/δH 3.57) in the Phe unit, which was confirmed by HMBC correlation from OCH3 (δH 3.57) and C-1 (δC 172.7). The difference between 4 and 6 was the presence of Tyr-OCH3 residue instead of Phe residue. Thus, the planar structures of 5–6 were established. Similarly, the absolute configuration of 5–6 was assigned as alternating LDLD chirality by Marfey’s method and solid-phase total synthesis and finally named psegynamides E-F, respectively.
Table 2.
1H and 13C NMR data in DMSO-d6 for compounds 4–6 (a measured at 600 MHz; b measured at 150 MHz).
Nicotinic acetylcholine receptors (nAChRs) belong to the ligand-gated ion channel superfamily [23,24]. There are a large number of nAChR subunits, such as α1-10, β1-4, γ, δ, and ε, which may mediate analgesic effects and adverse reactions [25,26]. Targeting specific nAChRs subtypes may reduce adverse reactions while maintaining efficient analgesia and becoming a real new analgesic target. All new compounds (1–6) were evaluated for their activity at ACh-evoked currents mediated by human (h) α1β1εδ, α1β1γδ, α3β2, α3β4, α4β2, α7, and α9α10 nAChRs, among which compound 2 showed significant inhibitory activity at all subtypes (>70% inhibition) except α7 (Figure 3, Table S1). Interestingly, compound 2 selectively inhibited (>98% inhibition, n = 8–11) the α1β1εδ and α1β1γδ subtypes. For comparison, the α-conotoxin GI peptide from the marine cone snail Conus geographus venom antagonizes ACh-evoked currents mediated by hα1β1εδ with a half-maximal inhibitory concentration (IC50) of 20 nM [27]. According to the structural features of 1–6, the preliminary structure–activity relationship (SAR) of inhibitory activities was tentatively discussed. Generally, compounds 1–3 exhibited stronger activities than compounds 4–6, indicating that the presence of tetrahydropyridoindoles unit decreases inhibitory activities. Psegynamide B (2) showed stronger activities than psegynamide A (1), suggesting that C-terminal replacement with the methoxy group was beneficial to the activity.
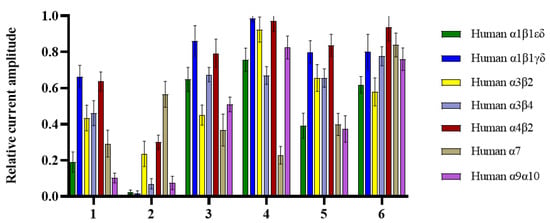
Figure 3.
Bar graph of compounds (1–6) (100 µM) inhibition of ACh-evoked peak current amplitude mediated by human (h) α1β1εδ, α1β1γδ, α3β2, α3β4, α4β2, α7 and α9α10 nAChRs. Whole-cell currents at hα1β1εδ and hα1β1γδ were activated by 5 μM ACh, hα3β2 and hα9α10 were activated by 6 μM ACh, and hα3β4, hα4β2, and hα7 were activated by 300, 3, and 100 μM ACh, respectively (mean ± SD, n = 6–11).
To explain the different inhibitory activities and obtain further insight into the mechanism of nAChRs inhibition, molecular docking studies were carried out to explore the possible binding modes of compounds 1–6 and key interactions with nAChRs. The crystal structure of hα4β2 (PDB code: 5KXI) was used for further docking. The results in the docking study matched well with the inhibition activities (Table S2), compounds with lower calculated docking scores are considered to have higher binding affinities with the target. Among them, compound 2 showed the highest binding affinity to hα4β2 with the most negative free binding energy (−4.2 kcal/mol). Analysis for optimized binding conformation of compound 2 displayed that the hydrogen at N-6 in Trp interacts with Ser A180 through a hydrogen bond with a distance of 2.2 Å, and the Trp formed H-pi stacked bonds with Gln B50 with a distance of 4.0 Å (Figure 4). While the Trp residue was replaced by the tetrahydropyridoindoles unit, the H-H and H-pi bond was lost. In addition, the NH in Val (2) of compound 2 interacts with Asp A49 through a hydrogen bond with a distance of 2.5 Å. It is interesting to find that the Arg B45 may be important for the activity because of that NH2 in Trp and the C=O in Phe-OCH3 of compound 2 simultaneously interact with Arg B45 through a hydrogen bond with distance of 2.2 Å and 3.3 Å, respectively. Furthermore, considering non-steroidal anti-inflammatory drugs and narcotics (opioids) are currently the most commonly used analgesic drugs, these drugs exhibit limitations in efficacy, unwanted side effects, and the problem of drug abuse [28,29]. The above findings provided that compound 2 might be helpful in developing different analgesic drugs by inhibiting nicotinic acetylcholine receptors.
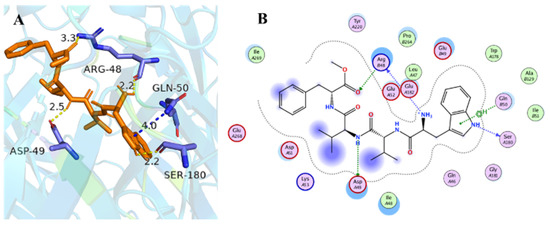
Figure 4.
Docking models of compound 2 with the crystal structure of hα4β2 (PDB code: 5kxi). (A) H-bonding interactions of compound 2 with hα4β2. (B) 2D schematic diagram of compound 2 with hα4β2. The H-bonds and H-pi stacked bonds are shown in yellow dashed lines and blue dashed lines, respectively.
3. Materials and Methods
3.1. General Experimental Procedures
NMR spectra were obtained on JEOLJN M-ECP 600 MHz spectrometers, of which TMS was an internal standard. The optical rotation of new compounds was calculated in MeOH on a JASCOP-1020 digital polarimeter. By using an LTQ Orbitrap XL (Thermo Fisher Scientific, Waltham, MA, USA) mass spectrometer, HRESIMS data were obtained. The spray voltage, capillary voltage, and tube lens were 4.0 kv, 16 v, and 35 v, respectively. The capillary temperature was 275 °C with a sheath gas flow rate of 10 arb. unit, and FT full mass spectra were acquired in the positive ionization mode at a resolution of 30,000 with 100–1500 Da mass range. The crude extract of Pseudogymnoascus sp. HDN17-933 were analyzed by reversed-phase HPLC (5 × 250 mm YMC C18 column, 5 µm) with a linear gradient of MeOH (A) and 0.1% aqueous TFA (B) from 5% to 100% A over 60 min at a flow rate of 1 mL/min. Column chromatography was carried out using the following chromatographic substrates: silica gel (300–400 mesh; Qingdao Marine Chemical Industrials, Qingdao, China), Sephadex LH-20 (GE Healthcare, Bio-Sinences Corp, Piscataway, NJ, USA). HPLC of the Waters company equipped with a 2998 PDA detector was performed on an ODS column (YMC-Pack ODS-A, 10 × 250 mm, 5 μm, 3 mL/min). UV spectra were carried out on Waters 2487 developed by Waters Corporation, Milford, USA. All Fmoc-amino acids were purchased from GL Biochem Ltd. (Shanghai, China). 2-chlorotrityl chloride resin at 1 mmol scale was purchased from Tianjin Nankai Hecheng S&T Co., Ltd (Tianjin, China).
3.2. Fungal Material and Fermentation
The fungal strain HDN17-933 was isolated from Fildes Peninsula, Antarctica, and identified as Pseudogymnoascus sp. based on internal transcribed spacer DNA sequencing. The sequence is available with the accession number MZ268166 at Genbank. It has been submitted to the Key Laboratory of Marine Drugs, working under the Ministry of Education of China, School of Medicine and Pharmacy, Ocean University of China. The fungus was cultured under the static condition at 15 °C in winter for 30 days in 1 L Erlenmeyer flasks, each containing rice (80 g) and naturally collected seawater (Huiquan Bay, Yellow Sea) (120 mL).
3.3. Extraction and Purification
All fermentation broth (40 L) was extracted with MeOH, filtered, concentrated, and partitioned between EtOAc and H2O. The EtOAc extract was evaporated under reduced pressure to give a crude gum (18.5 g). Moreover, the extract was subjected vacuum chromatography on silica gel (200–300 mesh) and eluted with stepped gradient elution via DCM:MeOH (100:1–1:1) to yield eight combined fractions (Fr.1 to Fr.8). Fr.6 was further separated using ODS (MeOH: H2O; 30:80–100:0) to obtain Fr.6-1 to Fr.6-6. Then Fr.6-3 and Fr.6-4 were further subjected to a Sephadex LH-20 column and eluted with MeOH to provide subfractions (from Fr.6-3-1 to Fr.6-3-4 and from Fr.6-4-1 to Fr.6-4-5). Fr.6-4-2 was purified by HPLC eluted with MeCN-H2O (38:62) to give compounds 1 (4.2 mg, tR = 11.5 min), 2 (3.4 mg, tR = 7.2 min), and 5 (3.5 mg, tR = 8.0 min), respectively. Fr.6-3-2 was purified by HPLC eluted with MeOH-H2O (65:35) to give compounds 3 (5.1 mg, tR = 10.2 min), 6 (4.3 mg, tR = 11.0 min), and 4 (3.2 mg, tR = 13.5 min), respectively.
3.4. Physical and Chemical Data
Psegynamide A (1): white amorphous powder; [α]20D + 30.2 (c 0.2, MeOH); UV (DAD) λmax 218 nm, 283 nm; 1H and 13C NMR (DMSO-d6), see Table 1; HREIMS m/z 550.3025 [M + H]+ (cacld for C30H40N5O5, 550.3024).
Psegynamide B (2): white amorphous powder; [α]20D + 23.8 (c 0.3, MeOH); UV (DAD) λmax 217 nm, 283 nm; 1H and 13C NMR (DMSO-d6), see Table 1; HREIMS m/z 564.3172 [M + H]+ (cacld for C31H42N5O5, 564.3180).
Psegynamide C (3): white amorphous powder; [α]20D + 25.3 (c 0.3, MeOH); UV (DAD) λmax 217 nm, 282 nm; 1H and 13C NMR (DMSO-d6), see Table 1; HREIMS m/z 566.2977 [M + H]+ (cacld for C30H40N5O6, 566.2973).
Psegynamide D (4): white amorphous powder; [α]20D − 14.7 (c 0.3, MeOH); UV (DAD) λmax 217 nm, 282 nm; 1H and 13C NMR (DMSO-d6), see Table 2; HREIMS m/z 562.3032 [M + H]+ (cacld for C31H40N5O5, 562.3024).
Psegynamide E (5): white amorphous powder; [α]20D − 17.8 (c 0.2, MeOH); UV (DAD) λmax 216 nm, 282 nm; 1H and 13C NMR (DMSO-d6), see Table 2; HREIMS m/z 576.3186 [M + H]+(cacld for C32H42N5O5, 576.3180).
Psegynamide F (6): white amorphous powder; [α]20D − 20.3 (c 0.2, MeOH); UV (DAD) λmax 216 nm, 283 nm; 1H and 13C NMR (DMSO-d6), see Table 2; HREIMS m/z 592.3133 [M + H]+ (cacld for C32H42N5O6, 592.3130).
3.5. Advanced Marfey’s Analysis of Acid Hydrolytic for Val, Phe, Tyr
Compounds 1–6 (1.0 mg each) were reacted with 6 M HCl (1.5 mL) at 110 °C for 12 h; the hydrolysates were concentrated to dryness. The hydrolysates and standard amino acids (150 μM) were then successively treated with water (300 μL), FDAA (10 mg/mL solution in acetone, 100 μL), acetone (300 μL), and NaHCO3 (1 M, 150 μL) at 45 °C water bath heating for 2.0 h. Then, the reaction was stopped with HCl (2 M, 75 μL) prior to HPLC analysis. Amino acid standards were similarly derivatized with FDAA. The resulting FDAA derivatives of compounds 1–6, L- and D-Val, L- and D- Phe, L- and D- Tyr were analyzed by HPLC eluted with a linear gradient of MeOH (A) and 0.10% aqueous TFA (B) from 40% to 100% in an over 45 min with UV detection at 340 nm.
3.6. Modified Marfey’s Analysis of Alkaline Hydrolytic for Trp
Considering that Trp FDAA derivatives were not detected in the resulting FDAA derivatives of compounds 1–3, it might result from that Trp amino acid was hydrolytic and destroyed in strong hydrochloric acid. Therefore, Marfey’s alkaline hydrolytic method was considered to determine the absolute configuration of Trp residue. Through continuous attempts, the alkaline hydrolytic condition of 5 M LiOH at 110 °C for 16 h was finally adopted, and the absolute configuration of Trp was determined as L.
3.7. Solid-Phase Total Synthesis of 1a, 1b
Compounds 1a, 1b were synthesized by 2-chlorotrityl chloride resin (loading 1 mmol/g) as described in Scheme 1. Then, 4 equiv of Fmoc-D-Phe (2 mmol) was added to a suspension of 1 equiv of 2-chlorotrityl chloride resin (0.50 g, 0.5 mmol), 4 equiv of HCTU (2 mmol), 8 equiv of DIEA (4 mmol), and DCM/DMF (v:v = 1:1). After stirring for 2 h, the resin was filtered and washed with DCM/DMF (1:1) for three times. The reaction mixture was treated with piperidine:DMF (v:v = 1:4) for stirring 30 min to deprotect the Fmoc group. After filtering and washing, the resin was used as resin-D-Phe4-NH2 for the next coupling reaction.
A solution of HCTU (2 mmol) and DIEA (4 mmol) in DCM/DMF (1:1) was added to a mixture of resin-D-Phe4-NH2, Fmoc-L-Val/ Fmoc-D-Val (2 mmol). After the reaction mixture was stirred for 60 min, the resin was filtered and washed. To remove the Fmoc group, the same procedure was repeated as above. The resin was used as resin-D-Phe4-L-Val3-NH2/D-Phe4-D-Val3-NH2 for the subsequent next coupling reaction, respectively. A solution of HCTU (2 mmol) and DIEA (4 mmol) in DCM/DMF (1:1) was added to a mixture of resin-D-Phe4-L/D-Val3-NH2, Fmoc-D-Val/ Fmoc-L-Val (2 mmol). After the reaction mixture was stirred for 60 min, the resin was filtered and washed. To remove the Fmoc group, the same procedure was repeated as above. The resin was used as resin-D-Phe4-L-Val3-D-Val2-NH2/D-Phe4-D-Val3-L-Val2-NH2 for the next coupling reaction, respectively.
A solution of HCTU (2 mmol) and DIEA (4 mmol) in DCM/DMF (1:1) was added to a mixture of resin-D-Phe4-L/D-Val3-D/L-Val2-NH2, Fmoc-L-Trp (Boc)-OH (2 mmol). After the reaction mixture was stirred for 60 min, the resin was filtered and washed. To remove the Fmoc group, the same procedure was repeated as above. The resin was used as resin-D-Phe4-L-Val3-D-Val2-L-Trp1(Boc)-NH2/D-Phe4-D-Val3-L-Val2-L-Trp1(Boc)-NH2, respectively. Subsequent to TFA cleavage (TFA: TIPS: H2O, 90:5:5, 3 h), the reaction solution was treated with cold ether to obtain precipitation. The precipitate was furtherly purified by HPLC to give the compounds 1a (L-Trp1-L-Val2-D-Val3-D-Phe4) and 1b (L-Trp1-D-Val2-L-Val3-D-Phe4), respectively.
3.8. Xenopus Laevis Oocyte Preparation and Microinjection
All procedures were approved by the University of Wollongong Animal Ethics Committees (project number: AE2003). Female Xenopus laevis were sourced from Nasco (Fort Atkinson, WI, USA), and a maximum of four frogs were kept in a 15 L aquarium at 20–26 °C with 12 h light/dark cycle. Oocytes were obtained from five-year-old frogs anesthetized with 1.7 mg/mL ethyl 3-aminobenzoate methanesulfonate (pH 7.4 with NaHCO3). Stage V-VI oocytes (Dumont’s classification; 1200–1300 μm diameter) were defolliculated with 1.5 mg/mL collagenase Type II (Worthington Biochemical Corp., Lakewood, NJ, USA) at room temperature for 1–2 h in OR-2 solution containing (in mM): 82.5 NaCl, 2 KCl, 1 MgCl2 and 5 HEPES at pH 7.4.
The human muscle nAChR clones (α1, β1, γ, δ and ε) were purchased from Integrated DNA Technologies (Coralville, IA, USA), whereas the human α3, α9, α10, β2, and β4 clones were purchased from OriGene (Rockville, MD, USA), and all were subsequently inserted into the pT7TS vector. The human α4 and α7 clones were obtained from Prof. Jon Lindstrom (University of Pennsylvania, Philadelphia, PA, USA). Plasmid constructs of the human nAChR clones were linearized for in vitro mRNA synthesis using mMessage mMachine transcription kit (AMBION, Forster City, CA, USA).
Oocytes were injected with 5 ng cRNA for hα1β1γδ, hα1β1εδ, hα3β2, hα3β4 and hα4β2, 10 ng cRNA for hα7 nAChR, and 35 ng cRNA for hα9α10 nAChR (concentration confirmed spectrophotometrically and by gel electrophoresis). The muscle subunit cRNA ratio was 2:1:1:1 (α1:β1: γ/ε:δ), whereas the heteromeric α and β subunit cRNA ratio was 1:1, injected using glass pipettes pulled from glass capillaries (3-000-203 GX, Drummond Scientific Co., Broomall, PA, USA). Oocytes were incubated at 18 °C in sterile ND96 solution composed of (in mM): 96 NaCl, 2 KCl, 1 CaCl2, 1 MgCl2, and 5 HEPES at pH 7.4, supplemented with 5% fetal bovine serum, 50 mg/L gentamicin (GIBCO, Grand Island, NY, USA) and 10000 U/mL penicillin-streptomycin (GIBCO).
3.9. Oocyte Two-Electrode Voltage Clamp Recording and Data Analysis
Electrophysiological recordings were carried out 2–5 days post cRNA microinjection. Two-electrode voltage clamp recordings of X. laevis oocytes expressing human nAChRs were performed at room temperature (21–24 °C) using a GeneClamp 500B amplifier and pClamp9 software interface (Molecular Devices, Sunnyvale, CA, USA) at a holding potential −80 mV. Voltage-recording and current-injecting electrodes were pulled from GC150T-7.5 borosilicate glass (Harvard Apparatus, Holliston, MA) and filled with 3 M KCl, giving resistances of 0.3–1 MΩ. Due to the Ca2+ permeability of α9α10 nAChRs, 100 µM BAPTA-AM incubation was carried out before recording to prevent the activation of X. laevis oocyte endogenous Ca2+-activated chloride channels. Oocytes expressing hα9α10 nAChRs were perfused with ND115 solution containing (in mM): 115 NaCl, 2.5 KCl, 1.8 CaCl2, and 10 HEPES at pH 7.4, whereas oocytes expressing all other nAChR subtypes were perfused with ND96 solution using a continuous Legato 270 push/pull syringe pump perfusion system (KD Scientific, Holliston, MA, USA) at a rate of 2 mL/min in an OPC-1 perfusion chamber of <20 µL volume (Automate Scientific, Berkeley, CA, USA).
Initially, oocytes were briefly washed with ND115/ND96 solution, followed by 3 applications of ACh at a half-maximal excitatory ACh concentration (EC50) for the nAChR subtypes (3 µM for hα4β2, 5 µM for hα1β1γδ and hα1β1εδ, 6 µM for hα3β2 and hα9α10, 100 µM for hα7 and 300 µM for hα3β4) [27]. Washout with bath solution was performed for 3 min between ACh applications. Oocytes were incubated with compounds for 5 min with the perfusion system turned off, followed by co-application of ACh and compound with flowing bath solution. All compound solutions were prepared in ND115/ND96 + 0.1% bovine serum albumin (BSA). Incubation with 0.1% BSA was performed to ensure that the BSA and the pressure of the perfusion system had no effect on nAChRs. Peak current amplitudes before (ACh alone) and after (ACh + compound) compound incubation were measured using Clampfit version 10.7.0.3 software (Molecular Devices, Sunnyvale, CA, USA), where the ratio of ACh + compound-evoked current amplitude to ACh alone-evoked current amplitude was used to assess the activity of the compounds at the nAChRs. All electrophysiological data were pooled (n = 6–11) and represent means ± standard deviation (SD). Data analysis was performed using GraphPad Prism 5 (GraphPad Software, La Jolla, CA, USA).
3.10. Molecular Docking
Molecular docking studies were performed using MOE 2016. The crystal structure of human α4β2 nAChR (PDB identifier: 5kxi) was obtained from the Protein Data Bank (http://www.rcsb.org, accessed on 28 September 2016). Prior to docking, heteroatoms and water molecules in the protein were removed. In the meantime, compounds were minimized. The result of each ligand was furtherly analyzed by using PyMOL.
4. Conclusions and Discussion
In conclusion, the genus Pseudogymnoascus is a group of psychrophilic fungi that possess good potential in serving structurally unique structures but not widely investigated. In this study, chemical investigation of secondary metabolites from the psychrophilic fungus Pseudogymnoascus sp. HDN17-933 led to the discovery of six new tetrapeptides psegynamides A–F (1–6), among which psegynamides D–F (4–6) was the first naturally occurring peptide bearing a tetrahydropyridoindoles moiety. To the best of our knowledge, only a few synthetic peptides bearing the tetrahydropyridoindoles motif have been reported [30,31], for example, tetrahydropyridoindoles as cholecystokinin and gastrin antagonists in 1992 [30]. Solid synthesis techniques assisted by Marfey’s method were employed to work out the absolute configuration, especially in solving the challenges of determining the order of L-Val and D-Val isomers in the peptide sequences. Moreover, compound 2 was found to have bioactivity by inhibiting human nAChRs. Considering that non-steroidal anti-inflammatory drugs and narcotics (opioids) are currently the most commonly used analgesic drugs, these drugs exhibit limitations in efficacy, unwanted side effects, and the problem of drug abuse. To overcome these problems, the discovery of different molecular participants in the pain pathways could bring new opportunities for therapeutic intervention. Compound 2 might be helpful in developing potential natural short peptide inhibitor of nAChRs from Antarctica-derived fungus. The above findings illustrate and highlight the validity of exploiting psychrophilic fungus for the discovery of structurally novel and bioactive natural products.
Supplementary Materials
The following are available as supplementary materials https://www.mdpi.com/article/10.3390/md20100593/s1: Figure S1: 18S rRNA sequences data of Pseudogymnoascus sp. HDN17-933; Figure S2: HPLC analysis of the crude extract of HDN17-933; Figures S3–S57: 1D and 2D NMR spectra, HRESIMS spectra, IR spectra of compounds 1–6; Figures S58–S88: Advanced Marfey’s acid/alkaline hydrolytic analysis of compounds 1–6, and NMR spectra of solid-phase total synthesis. Table S1. Table of compounds (1–6) inhibition of ACh-evoked peak current amplitude mediated by human (h) α1β1εδ, α1β1γδ, α3β2, α3β4, α4β2, α7 and α9α10 nAChRs. Table S2. Free binding energy estimation and molecular interactions of compounds 1–6.
Author Contributions
The contributions of the respective authors are as follows: X.H.: conceptualization, resources, investigation, writing—original draft, and writing—review and editing. C.L., R.Z., Y.L., H.L. and Y.Z.: investigation and data curation. H.-S.T. and R.Y.: assistance of nicotinic acetylcholine receptors activity and molecular docking. D.L., G.Z., T.Z. and Q.C.: funding acquisition, and writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by Marine S&T Fund of Shandong Province for Pilot National Laboratory for Marine Science and Technology (2022QNLM030003-1, 2022QNLM030003-2), National Natural Science Foundation of China (81991522, 41976105), the NSFC-Shandong Joint Fund (U1906212), the Hainan Provincial Joint Project of Sanya Yazhou Bay Science and Technology City (2021CXLH0012), Taishan Scholar Youth Expert Program in Shandong Province (tsqn 202103153, tsqn 201812021), Major Basic Research Programs of Natural Science Foundation of Shandong Province (ZR2019ZD28) and an Australian Research Council (ARC) Discovery Project Grant (DP150103990) awarded to D.J. Adams.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
We thank Adams for the facilities and support of the functional studies carried out in Xenopus oocytes at IHMRI, University of Wollongong.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Hassan, N.; Rafiq, M.; Hayat, M.; Shah, A.A.; Hasan, F. Psychrophilic and psychrotrophic fungi: A comprehensive review. Rev. Environ. Sci. Bio/Technol. 2016, 15, 147–172. [Google Scholar] [CrossRef]
- Golubev, W.I. New species of basidiomycetous yeasts, Rhodotorula creatinovora and R. yakutica, isolated from permafrost soils of Eastern-Siberian Arctic. Mikol. Fitopatol. 1998, 32, 8–13. [Google Scholar]
- Tosi, L.; Carbognin, L.; Teatini, P.; Rosselli, R.; Stori, G.G. The ISES project subsidence monitoring of the catchment basin south of the Venice Lagoon. In Land Subsidence; La Garangola: Padova, Italy, 2000; pp. 113–126. [Google Scholar]
- Ma, L.; Catranis, C.M.; Starmer, W.T.; Rogers, S.O. Revival and characterization of fungi from ancient polar ice. Mycologist 1999, 13, 70–73. [Google Scholar] [CrossRef]
- Broady, P.A.; Weinstein, R.N. Algae, lichens and fungi in La Gorce Mountains, Antarctica. Antarct. Sci. 1998, 10, 376–385. [Google Scholar] [CrossRef]
- Tojo, M.; Newshamb, K.K. Snow moulds in polar environments. Fungal Ecol. 2012, 5, 395–402. [Google Scholar] [CrossRef]
- Robinson, C.H. Cold adaptation in Arctic and Antarctic fungi. New Phytol. 2001, 151, 341–353. [Google Scholar] [CrossRef]
- McKenzie, R.L.; Björn, L.O.; Bais, A.; Ilyasd, M. Changes in biologically active ultraviolet radiation reaching the Earth’s surface. Photochem. Photobiol. Sci. 2003, 2, 5–15. [Google Scholar] [CrossRef]
- Selbmann, L.; Onofri, S.; Fenice, M.; Federici, F.; Petruccioli, M. Production and structural characterization of the exopolysaccharide of the Antarctic fungus Phoma herbarum CCFEE 5080. Res. Microbiol. 2002, 153, 585–592. [Google Scholar] [CrossRef]
- Brown, A. Compatible solutes and extreme water stress in eukaryotic micro-organisms. Adv. Microb. Physiol. 1978, 17, 181–242. [Google Scholar]
- Lewis, D.; Smith, D. Sugar alcohols (polyols) in fungi and green plants. I. Distribution, physiology and metabolism. New Phytol. 1967, 66, 143–184. [Google Scholar] [CrossRef]
- Weinstein, R.N.; Montiel, P.O.; Johnstone, K. Influence of growth temperature on lipid and soluble carbohydrate synthesis by fungi isolated from fellfield soil in the maritime Antarctic. Mycologia 2000, 92, 222–229. [Google Scholar] [CrossRef]
- Margesin, R.; Schinner, F.; Marx, J.C.; Gerday, C. Bacteria in snow and glacier ice. In Psychrophiles: From Biodiversity to Biotechnology; Springer: Berlin/Heidelberg, Germany, 2008; pp. 381–387. [Google Scholar]
- Georlette, D.; Damien, B.; Blaise, V.; Depiereux, E.; Uversky, V.N. Structural and functional adaptations to extreme temperatures in psychrophilic, mesophilic, and thermophilic DNA ligases. J. Biol. Chem. 2003, 278, 37015–37023. [Google Scholar] [CrossRef]
- Krohn, D. Endophytic fungi: A source of novel biologically active secondary metabolites. Mycol. Res. 2002, 106, 996–1004. [Google Scholar]
- Singh, P.; Raghukumar, C.; Verma, P.; Shouche, Y. Fungal community analysis in the deep-sea sediments of the Central Indian Basin by culture-independent approach. Microb. Ecol. 2011, 61, 507–517. [Google Scholar] [CrossRef]
- Li, D.; Wang, F.; Xiao, X.; Fang, Y.; Zhu, T.; Gu, Q.; Zhu, W. Trisorbicillinone A, a novel sorbicillin trimer, from a deep sea fungus, Phialocephala sp. FL30. Tetrahedron Lett. 2007, 48, 5235–5238. [Google Scholar] [CrossRef]
- MacíAs, F.; Varela, R.M.; Simonet, A.M.; Cutler, H.G.; Cutler, S.J.; Ross, S.A.; Dunbar, D.C.; Dugan, F.M.; Hill, R.A. (+)-Brevione A. The first member of a novel family of bioactive spiroditerpenoids isolated from Penicillium brevicompactum Dierckx. Tetrahedron Lett. 2000, 41, 2683–2686. [Google Scholar] [CrossRef]
- Wu, G.; Ma, H.; Zhu, T.; Jing, L.; Gu, Q.; Li, D. Penilactones A and B, two novel polyketides from Antarctic deep-sea derived fungus Penicillium crustosum PRB-2. Tetrahedron 2012, 68, 9745–9749. [Google Scholar] [CrossRef]
- Sun, Y.L.; Zhang, X.Y.; Nong, X.H.; Xu, X.Y.; Qi, S.H. New antifouling macrodiolides from the deep-sea-derived fungus Trichobotrys effuse DFFSCS021. Tetrahedron Lett. 2016, 57, 366–370. [Google Scholar] [CrossRef]
- Du, L.; Li, D.; Zhu, T.; Cai, S.; Wang, F.; Xiao, X.; Gu, Q. New alkaloids and diterpenes from a deep ocean sediment derived fungus Penicillium sp. Tetrahedron 2009, 65, 1033–1039. [Google Scholar] [CrossRef]
- Fujii, K.; Ikai, Y.; Oka, H.; Suzuki, M.; Harada, K.I. A nonempirical method using LC/MS for determination of the absolute configuration of constituent amino acids in a peptide: Combination of Marfey’s method with mass spectrometry and its practical application. Anal. Chem. 1997, 69, 549–557. [Google Scholar] [CrossRef]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian nicotinic acetylcholine receptors: From structure to function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef]
- Thompson, A.J.; Lester, H.A.; Lummis, S.C.R. The structural basis of function in Cys-loop receptors. Q. Rev. Biophys. 2010, 43, 449–499. [Google Scholar] [CrossRef]
- Anand, R.; Conroy, W.G.; Schoepfer, R.; Whiting, P.; Lindstrom, J. Neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes have a pentameric quaternary structure. J. Biol. Chem. 1991, 266, 11192–11198. [Google Scholar] [CrossRef]
- Sherry, L.; Daniel, B. Neuronal nicotinic receptors: From structure to function. Nicotine Tob. Res. 2001, 3, 203–223. [Google Scholar]
- Tae, H.S.; Gao, B.; Jin, A.H.; Alewood, P.F.; Adams, D.J. Globular and ribbon isomers of Conus geographus α-conotoxins antagonize human nicotinic acetylcholine receptors. Biochem. Pharmacol. 2021, 190, 114638–114646. [Google Scholar] [CrossRef] [PubMed]
- Woodcock, J. A difficult balance-pain management, drug safety, and the FDA. N. Engl. J. Med. 2009, 361, 2105–2107. [Google Scholar] [CrossRef] [PubMed]
- Ballantyne, J.C.; Shin, N.S. Efficacy of opioids for chronic pain: A review of the evidence. Clin. J. Pain 2008, 24, 469–478. [Google Scholar] [CrossRef]
- Molino, B.F.; Darkes, P.R.; Ewing, W.R. Tetrahydro-Pyrido-Indoles as Cholecystokinin and Gastrin Antagonists. U.S. Patent 5,162,336, 10 November 1992. [Google Scholar]
- Steffen, W.; Torsten, D.; Degenhard, M.; Beate, S.; Thomas, S.; Dieter, F.; Ulrich, K.; Daniela, H.; Jörg, D.; Christiaans, J.A.M. Christiaans, 6-Benzyl-2, 3, 4, 7-Tetrahydro-Indolo [2, 3-c] Quinoline Compounds Useful as Pde5 Inhibitors. U.S. Patent 12/449,173, 13 May 2010. [Google Scholar]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).