

Marine Macrolides to Tackle Antimicrobial Resistance of Mycobacterium tuberculosis

 ,
,  ,
,  ,
,  ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
3. Structure–Activity Relationship of Macrolides
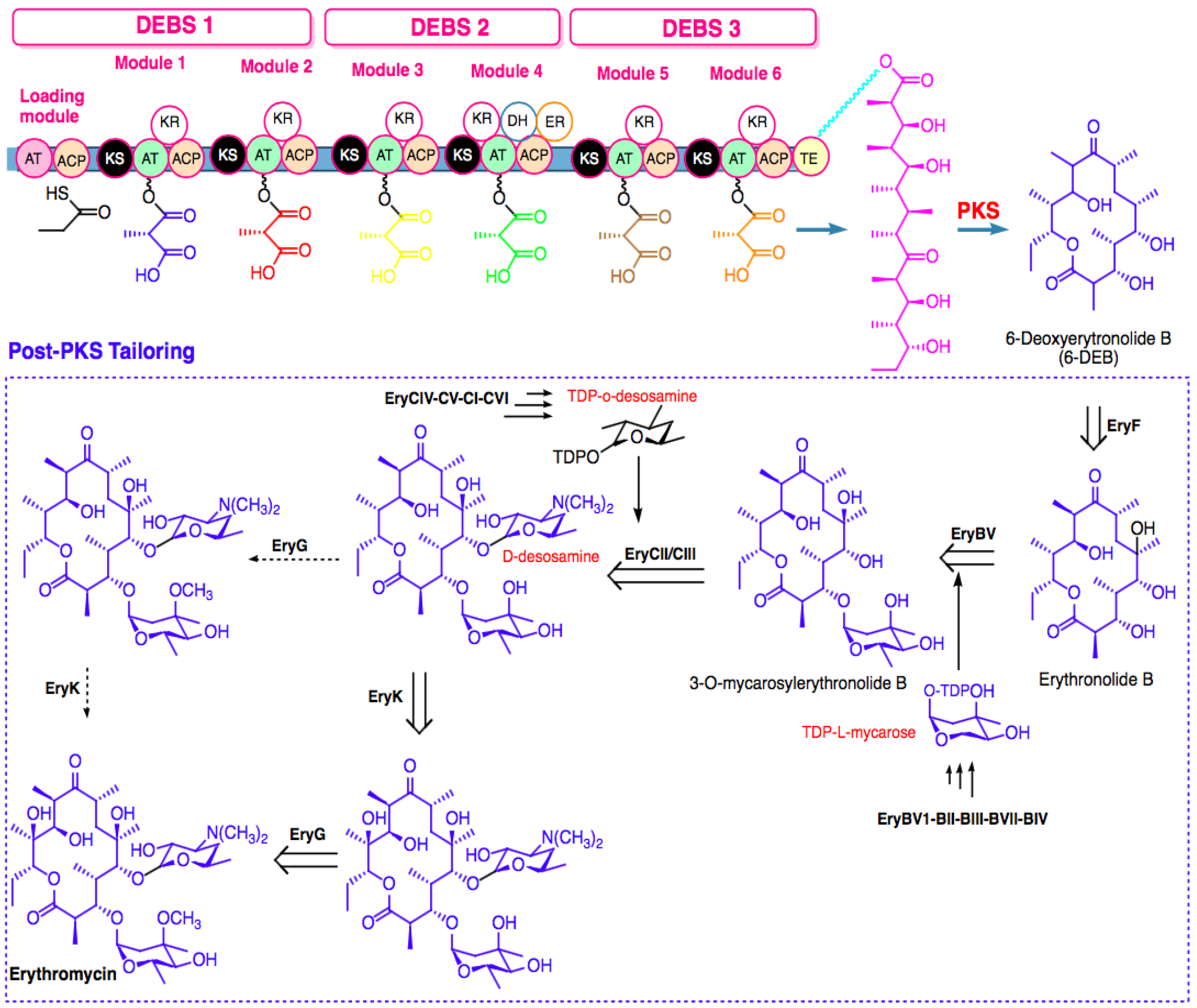
4. Biosynthesis of Marine Macrolides
4.1. Biosynthesis Challenges
4.2. Metabolic Engineering
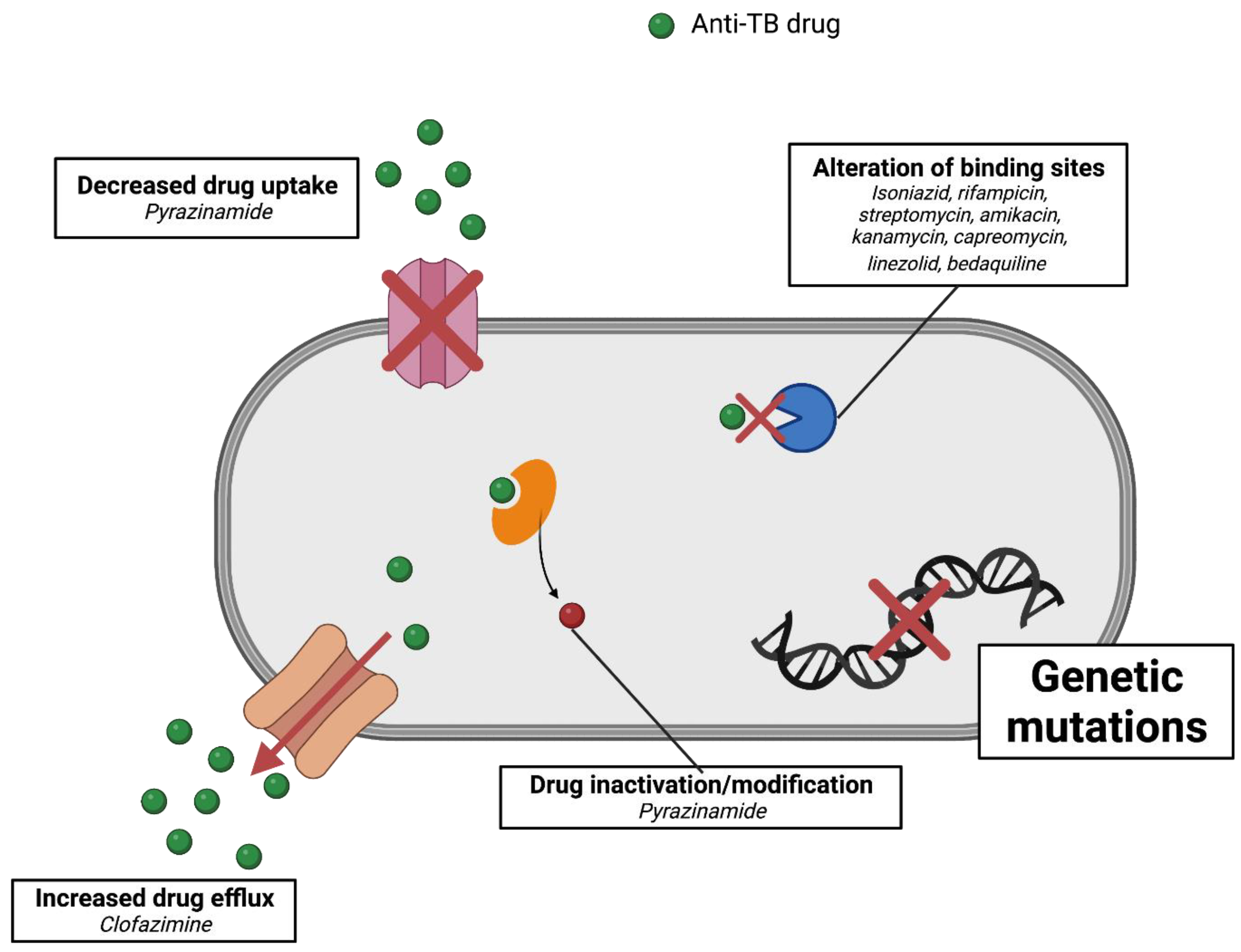
5. Mechanism of Resistance of Mycobacterium tuberculosis
5.1. Isoniazid
5.2. Rifampicin
5.3. Ethambutol
5.4. Pyrazinamide
5.5. Streptomycin
5.6. Fluoroquinolones
5.7. Second-Line Injectables
5.8. Para-Amino Aalicylic Acid (PAS)
5.9. Novel and Repurposed Drugs
6. Marine Macrolides to Counter Mycobacterium tuberculosis Resistance
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- WHO. Global Tuberculosis Report 2020; WHO: Geneva, Switzerland, 2021. [Google Scholar]
- Almeida Da Silva, P.E.; Palomino, J.C. Molecular basis and mechanisms of drug resistance in Mycobacterium tuberculosis: Classical and new drugs. J. Antimicrob. Chemother. 2011, 66, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Dookie, N.; Rambaran, S.; Padayatchi, N.; Mahomed, S.; Naidoo, K. Evolution of drug resistance in Mycobacterium tuberculosis: A review on the molecular determinants of resistance and implications for personalized care. J. Antimicrob. Chemother. 2018, 73, 1138–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palomino, J.C.; Martin, A. Drug resistance mechanisms in Mycobacterium tuberculosis. Antibiotics 2014, 3, 317–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsen, S.M.; Karlsson, M.; Johansson, L.B.; Vester, B. The pleuromutilin drugs tiamulin and valnemulin bind to the RNA at the peptidyl transferase centre on the ribosome. Mol. Microbiol. 2001, 41, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Muxfeldt, H.; Shrader, S.; Hansen, P.; Brockmann, H. The structure of pikromycin. J. Am. Chem. Soc. 1968, 90, 4748–4749. [Google Scholar] [CrossRef]
- Berry, M. The macrolide antibiotics. Q. Rev. Chem. Soc. 1963, 17, 343–361. [Google Scholar] [CrossRef]
- van der Paardt, A.-F.; Wilffert, B.; Akkerman, O.W.; de Lange, W.C.M.; van Soolingen, D.; Sinha, B.; van der Werf, T.S.; Kosterink, J.G.W.; Alffenaar, J.-W.C. Evaluation of macrolides for possible use against multidrug-resistant Mycobacterium tuberculosis. Eur. Respir. J. 2015, 46, 444. [Google Scholar] [CrossRef] [Green Version]
- Fonseca, J.; Knight, G.; McHugh, T. The complex evolution of antibiotic resistance in Mycobacterium tuberculosis. Int. J. Infect. Dis. 2015, 32, 94–100. [Google Scholar] [CrossRef] [Green Version]
- Safi, H.; Lingaraju, S.; Amin, A.; Kim, S.; Jones, M.; Holmes, M.; McNeil, M.; Peterson, S.N.; Chatterjee, D.; Fleischmann, R. Evolution of high-level ethambutol-resistant tuberculosis through interacting mutations in decaprenylphosphoryl-β-D-arabinose biosynthetic and utilization pathway genes. Nat. Genet. 2013, 45, 1190–1197. [Google Scholar] [CrossRef]
- Somoskovi, A.; Parsons, L.M.; Salfinger, M. The molecular basis of resistance to isoniazid, rifampin, and pyrazinamide in Mycobacterium tuberculosis. Respir. Res. 2001, 2, 1–5. [Google Scholar] [CrossRef]
- Das, R.; Rauf, A.; Mitra, S.; Emran, T.B.; Hossain, M.J.; Khan, Z.; Naz, S.; Ahmad, B.; Meyyazhagan, A.; Pushparaj, K.; et al. Therapeutic potential of marine macrolides: An overview from 1990 to 2022. Chem. Biol. Interact. 2022, 365, 110072. [Google Scholar] [CrossRef]
- Gianti, E.; Zauhar, R.J. Chapter 7—Structure–activity relationships and drug design. In Remington, 23rd ed.; Adejare, A., Ed.; Academic Press: Cambridge, MA, USA, 2021; pp. 129–153. [Google Scholar]
- Ouattara, M.; Songuigama, C.; N’Guessan, D.J.-P. Pharmacochemical Aspects of the Evolution from Erythromycin to Neomacrolides, Ketolides and Neoketolides. Open J. Med. Chem. 2020, 10, 57–112. [Google Scholar] [CrossRef]
- Pavlović, D.; Mutak, S.; Andreotti, D.; Biondi, S.; Cardullo, F.; Paio, A.; Piga, E.; Donati, D.; Lociuro, S. Synthesis and Structure-Activity Relationships of α-Amino-γ-lactone Ketolides: A Novel Class of Macrolide Antibiotics. ACS Med. Chem. Lett. 2014, 5, 1133–1137. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Zou, J.; Yan, X.; Chen, J.; Cao, X.; Wu, J.; Liu, Y.; Wang, T. Marine-Derived Macrolides 1990–2020: An Overview of Chemical and Biological Diversity. Mar Drugs 2021, 19, 180. [Google Scholar] [CrossRef]
- Karpiński, T.M. Marine Macrolides with Antibacterial and/or Antifungal Activity. Mar Drugs 2019, 17, 241. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Wang, Y.; Wu, J.; Skalina, K.; Pfeifer, B.A. Complete biosynthesis of erythromycin A and designed analogs using E. coli as a heterologous host. Chem. Biol. 2010, 17, 1232–1240. [Google Scholar] [CrossRef] [Green Version]
- Mabe, S.; Eller, J.; Champney, W.S. Structure-activity relationships for three macrolide antibiotics in Haemophilus influenzae. Curr. Microbiol. 2004, 49, 248–254. [Google Scholar] [CrossRef]
- Zhu, Z.J.; Krasnykh, O.; Pan, D.; Petukhova, V.; Yu, G.; Liu, Y.; Liu, H.; Hong, S.; Wang, Y.; Wan, B.; et al. Structure-activity relationships of macrolides against Mycobacterium tuberculosis. Tuberculosis 2008, 88, S49–S63. [Google Scholar] [CrossRef]
- Zhang, W.; Li, Z.; Sun, Y.; Cui, P.; Liang, J.; Xing, Q.; Wu, J.; Xu, Y.; Zhang, W.; Zhang, Y.; et al. Cryo-EM structure of Mycobacterium tuberculosis 50S ribosomal subunit bound with clarithromycin reveals dynamic and specific interactions with macrolides. Emerg Microbes Infect 2022, 11, 293–305. [Google Scholar] [CrossRef]
- Dhakal, D.; Sohng, J.K.; Pandey, R.P. Engineering actinomycetes for biosynthesis of macrolactone polyketides. Microb. Cell Factories 2019, 18, 137. [Google Scholar] [CrossRef]
- Dutta, S.; Whicher, J.R.; Hansen, D.A.; Hale, W.A.; Chemler, J.A.; Congdon, G.R.; Narayan, A.R.; Håkansson, K.; Sherman, D.H.; Smith, J.L.; et al. Structure of a modular polyketide synthase. Nature 2014, 510, 512–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whicher, J.R.; Dutta, S.; Hansen, D.A.; Hale, W.A.; Chemler, J.A.; Dosey, A.M.; Narayan, A.R.H.; Håkansson, K.; Sherman, D.H.; Smith, J.L.; et al. Structural rearrangements of a polyketide synthase module during its catalytic cycle. Nature 2014, 510, 560–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keatinge-Clay, A.T. Polyketide Synthase Modules Redefined. Angew. Chem. Int. Ed. Engl. 2017, 56, 4658–4660. [Google Scholar] [CrossRef] [PubMed]
- Park, J.W.; Yoon, Y.J. Recent advances in the discovery and combinatorial biosynthesis of microbial 14-membered macrolides and macrolactones. J. Ind. Microbiol. Biotechnol. 2019, 46, 445–458. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, M.A.; Walsh, C.T. Assembly-line enzymology for polyketide and nonribosomal Peptide antibiotics: Logic, machinery, and mechanisms. Chem. Rev. 2006, 106, 3468–3496. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.Y.; Schnarr, N.A.; Kim, C.Y.; Cane, D.E.; Khosla, C. Extender unit and acyl carrier protein specificity of ketosynthase domains of the 6-deoxyerythronolide B synthase. J. Am. Chem. Soc. 2006, 128, 3067–3074. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Qiao, K.; Tang, Y. Structural analysis of protein-protein interactions in type I polyketide synthases. Crit. Rev. Biochem. Mol. Biol. 2013, 48, 98–122. [Google Scholar] [CrossRef] [Green Version]
- Cummings, M.; Breitling, R.; Takano, E. Steps towards the synthetic biology of polyketide biosynthesis. FEMS Microbiol. Lett. 2014, 351, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Koryakina, I.; Kasey, C.; McArthur, J.B.; Lowell, A.N.; Chemler, J.A.; Li, S.; Hansen, D.A.; Sherman, D.H.; Williams, G.J. Inversion of Extender Unit Selectivity in the Erythromycin Polyketide Synthase by Acyltransferase Domain Engineering. ACS Chem. Biol. 2017, 12, 114–123. [Google Scholar] [CrossRef] [Green Version]
- Nepal, K.K.; Wang, G. Streptomycetes: Surrogate hosts for the genetic manipulation of biosynthetic gene clusters and production of natural products. Biotechnol. Adv. 2019, 37, 1–20. [Google Scholar] [CrossRef]
- Weissman, K.J.; Leadlay, P.F. Combinatorial biosynthesis of reduced polyketides. Nat. Rev. Microbiol. 2005, 3, 925–936. [Google Scholar] [CrossRef]
- Shinde, P.B.; Han, A.R.; Cho, J.; Lee, S.R.; Ban, Y.H.; Yoo, Y.J.; Kim, E.J.; Kim, E.; Song, M.C.; Park, J.W.; et al. Combinatorial biosynthesis and antibacterial evaluation of glycosylated derivatives of 12-membered macrolide antibiotic YC-17. J. Biotechnol. 2013, 168, 142–148. [Google Scholar] [CrossRef]
- Jung, W.S.; Han, A.R.; Hong, J.S.; Park, S.R.; Choi, C.Y.; Park, J.W.; Yoon, Y.J. Bioconversion of 12-, 14-, and 16-membered ring aglycones to glycosylated macrolides in an engineered strain of Streptomyces venezuelae. Appl. Microbiol. Biotechnol. 2007, 76, 1373–1381. [Google Scholar] [CrossRef]
- Ye, Y.; Anwar, N.; Mao, X.; Wu, S.; Yan, C.; Zhao, Z.; Zhang, R.; Nie, Y.; Zhang, J.; Wang, J.; et al. Discovery of Three 22-Membered Macrolides by Deciphering the Streamlined Genome of Mangrove-Derived Streptomyces sp. HM190. Front. Microbiol. 2020, 11, 1464. [Google Scholar] [CrossRef]
- Kumar, C.N.S.S.P. Total Synthesis of Macrolides. In Organic Synthesis; Belakatte Parameshwarappa, N., Ed.; IntechOpen: Rijeka, Croatia, 2019. [Google Scholar]
- Nagamitsu, T.; Takano, D.; Marumoto, K.; Fukuda, T.; Furuya, K.; Otoguro, K.; Takeda, K.; Kuwajima, I.; Harigaya, Y.; Ōmura, S. Total Synthesis of Borrelidin. J. Org. Chem. 2007, 72, 2744–2756. [Google Scholar] [CrossRef]
- Terwilliger, D.W.; Trauner, D. Selective Synthesis of Divergolide I. J. Am. Chem. Soc. 2018, 140, 2748–2751. [Google Scholar] [CrossRef]
- Zhang, M.M.; Wang, Y.; Ang, E.L.; Zhao, H. Engineering microbial hosts for production of bacterial natural products. Nat. Prod. Rep. 2016, 33, 963–987. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Li, B.Z.; Liu, D.; Zhang, L.; Chen, Y.; Jia, B.; Zeng, B.X.; Zhao, H.; Yuan, Y.J. Engineered biosynthesis of natural products in heterologous hosts. Chem. Soc. Rev. 2015, 44, 5265–5290. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Deng, Z.; Liu, T. Streptomyces species: Ideal chassis for natural product discovery and overproduction. Metab. Eng. 2018, 50, 74–84. [Google Scholar] [CrossRef]
- Chaudhary, A.K.; Dhakal, D.; Sohng, J.K. An insight into the “-omics” based engineering of streptomycetes for secondary metabolite overproduction. Biomed. Res. Int. 2013, 2013, 968518. [Google Scholar] [CrossRef]
- Lechner, A.; Brunk, E.; Keasling, J.D. The Need for Integrated Approaches in Metabolic Engineering. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Hong, M.; Chu, J.; Huang, M.; Ouyang, L.; Tian, X.; Zhuang, Y. Blocking the flow of propionate into TCA cycle through a mutB knockout leads to a significant increase of erythromycin production by an industrial strain of Saccharopolyspora erythraea. Bioprocess Biosyst. Eng. 2017, 40, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Reeves, A.R.; Brikun, I.A.; Cernota, W.H.; Leach, B.I.; Gonzalez, M.C.; Weber, J.M. Engineering of the methylmalonyl-CoA metabolite node of Saccharopolyspora erythraea for increased erythromycin production. Metab. Eng. 2007, 9, 293–303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirm, B.; Magdevska, V.; Tome, M.; Horvat, M.; Karničar, K.; Petek, M.; Vidmar, R.; Baebler, S.; Jamnik, P.; Fujs, Š.; et al. SACE_5599, a putative regulatory protein, is involved in morphological differentiation and erythromycin production in Saccharopolyspora erythraea. Microb. Cell. Fact. 2013, 12, 126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, G.; Li, Y.; Fang, L.; Pfeifer, B.A. Tailoring pathway modularity in the biosynthesis of erythromycin analogs heterologously engineered in E. coli. Sci. Adv. 2015, 1, e1500077. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, B.; Micklefield, J. Mining and engineering natural-product biosynthetic pathways. Nat. Chem. Biol. 2007, 3, 379–386. [Google Scholar] [CrossRef]
- Zhang, L.; Awakawa, T.; Abe, I. Understanding and Manipulating Assembly Line Biosynthesis by Heterologous Expression in Streptomyces. In Engineering Natural Product Biosynthesis; Springer: Berlin/Heidelberg, Germany, 2022; pp. 223–238. [Google Scholar]
- Wu, H.; Chen, M.; Mao, Y.; Li, W.; Liu, J.; Huang, X.; Zhou, Y.; Ye, B.C.; Zhang, L.; Weaver, D.T.; et al. Dissecting and engineering of the TetR family regulator SACE_7301 for enhanced erythromycin production in Saccharopolyspora erythraea. Microb. Cell Fact 2014, 13, 158. [Google Scholar] [CrossRef]
- Yi, J.S.; Kim, M.; Kim, E.J.; Kim, B.G. Production of pikromycin using branched chain amino acid catabolism in Streptomyces venezuelae ATCC 15439. J. Ind. Microbiol. Biotechnol. 2018, 45, 293–303. [Google Scholar] [CrossRef]
- Dhakal, D.; Le, T.T.; Pandey, R.P.; Jha, A.K.; Gurung, R.; Parajuli, P.; Pokhrel, A.R.; Yoo, J.C.; Sohng, J.K. Enhanced production of nargenicin A(1) and generation of novel glycosylated derivatives. Appl. Biochem. Biotechnol. 2015, 175, 2934–2949. [Google Scholar] [CrossRef]
- Bian, X.; Tang, B.; Yu, Y.; Tu, Q.; Gross, F.; Wang, H.; Li, A.; Fu, J.; Shen, Y.; Li, Y.Z.; et al. Heterologous Production and Yield Improvement of Epothilones in Burkholderiales Strain DSM 7029. ACS Chem. Biol. 2017, 12, 1805–1812. [Google Scholar] [CrossRef]
- Liu, C.; Zhang, J.; Lu, C.; Shen, Y. Heterologous expression of galbonolide biosynthetic genes in Streptomyces coelicolor. Antonie Leeuwenhoek 2015, 107, 1359–1366. [Google Scholar] [CrossRef]
- Hashimoto, T.; Hashimoto, J.; Kozone, I.; Amagai, K.; Kawahara, T.; Takahashi, S.; Ikeda, H.; Shin-Ya, K. Biosynthesis of Quinolidomicin, the Largest Known Macrolide of Terrestrial Origin: Identification and Heterologous Expression of a Biosynthetic Gene Cluster over 200 kb. Org. Lett. 2018, 20, 7996–7999. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention, C. Emergence of Mycobacterium tuberculosis with extensive resistance to second-line drugs--worldwide, 2000–2004. MMWR Morb. Mortal. Wkly. Rep. 2006, 55, 301–305. [Google Scholar]
- Gandhi, N.R.; Moll, A.; Sturm, A.W.; Pawinski, R.; Govender, T.; Lalloo, U.; Zeller, K.; Andrews, J.; Friedland, G. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet 2006, 368, 1575–1580. [Google Scholar] [CrossRef]
- Udwadia, Z.F.; Amale, R.A.; Ajbani, K.K.; Rodrigues, C. Totally drug-resistant tuberculosis in India. Clin. Infect. Dis. 2012, 54, 579–581. [Google Scholar] [CrossRef] [Green Version]
- Hong, B.-L.; D’Cunha, R.; Li, P.; Al-Shaer, M.H.; Alghamdi, W.A.; An, G.; Peloquin, C. A systematic review and meta-analysis of isoniazid pharmacokinetics in healthy volunteers and patients with tuberculosis. Clin. Ther. 2020, 42, e220–e241. [Google Scholar] [CrossRef]
- Barry III, C.E.; Slayden, R.A.; Mdluli, K. Mechanisms of isoniazid resistance in Mycobacterium tuberculosis. Drug Resist. Updates 1998, 1, 128–134. [Google Scholar] [CrossRef]
- Zhang, Y.; Heym, B.; Allen, B.; Young, D.; Cole, S. The catalase—Peroxidase gene and isoniazid resistance of Mycobacterium tuberculosis. Nature 1992, 358, 591–593. [Google Scholar] [CrossRef]
- Hazbón, M.H.; Brimacombe, M.; Bobadilla del Valle, M.; Cavatore, M.; Guerrero, M.I.; Varma-Basil, M.; Billman-Jacobe, H.; Lavender, C.; Fyfe, J.; García-García, L. Population genetics study of isoniazid resistance mutations and evolution of multidrug-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2006, 50, 2640–2649. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaswamy, S.V.; Reich, R.; Dou, S.-J.; Jasperse, L.; Pan, X.; Wanger, A.; Quitugua, T.; Graviss, E.A. Single nucleotide polymorphisms in genes associated with isoniazid resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2003, 47, 1241–1250. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, A.; Dubnau, E.; Quemard, A.; Balasubramanian, V.; Um, K.S.; Wilson, T.; Collins, D.; De Lisle, G.; Jacobs Jr, W.R. hA, a gene encoding a target for isoniazid and ethionamide in Mycobacterium tuberculosis. Science 1994, 263, 227–230. [Google Scholar] [CrossRef]
- Sherman, D.R.; Mdluli, K.; Hickey, M.J.; Arain, T.M.; Morris, S.L.; Barry III, C.E.; Stover, C.K. Compensatory ahp C gene expression in isoniazid-resistant Mycobacterium tuberculosis. Science 1996, 272, 1641–1643. [Google Scholar] [CrossRef] [PubMed]
- Slayden, R.; Barry 3rd, C. The role of KasA and KasB in the biosynthesis of meromycolic acids and isoniazid resistance in Mycobacterium tuberculosis. Tuberculosis 2002, 82, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Fox, W. The chemotherapy of pulmonary tuberculosis: A review. Chest 1979, 76, 785–796. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.A.; Korzheva, N.; Mustaev, A.; Murakami, K.; Nair, S.; Goldfarb, A.; Darst, S.A. Structural mechanism for rifampicin inhibition of bacterial RNA polymerase. Cell 2001, 104, 901–912. [Google Scholar] [CrossRef]
- Telenti, A.; Imboden, P.; Marchesi, F.; Schmidheini, T.; Bodmer, T. Direct, automated detection of rifampin-resistant Mycobacterium tuberculosis by polymerase chain reaction and single-strand conformation polymorphism analysis. Antimicrob. Agents Chemother. 1993, 37, 2054–2058. [Google Scholar] [CrossRef] [Green Version]
- Imperiale, B.R.; Di Giulio, Á.B.; Adrián Cataldi, Á.; Morcillo, N.S. Evaluation of Mycobacterium tuberculosis cross-resistance to isoniazid, rifampicin and levofloxacin with their respective structural analogs. J. Antibiot. 2014, 67, 749–754. [Google Scholar] [CrossRef]
- Takayama, K.; Kilburn, J.O. Inhibition of synthesis of arabinogalactan by ethambutol in Mycobacterium smegmatis. Antimicrob. Agents Chemother. 1989, 33, 1493–1499. [Google Scholar] [CrossRef] [Green Version]
- Sreevatsan, S.; Stockbauer, K.E.; Pan, X.; Kreiswirth, B.N.; Moghazeh, S.L.; Jacobs Jr, W.R.; Telenti, A.; Musser, J.M. Ethambutol resistance in Mycobacterium tuberculosis: Critical role of embB mutations. Antimicrob. Agents Chemother. 1997, 41, 1677–1681. [Google Scholar] [CrossRef] [Green Version]
- Tulyaprawat, O.; Chaiprasert, A.; Chongtrakool, P.; Suwannakarn, K.; Ngamskulrungroj, P. Association of ubiA mutations and high-level of ethambutol resistance among Mycobacterium tuberculosis Thai clinical isolates. Tuberculosis 2019, 114, 42–46. [Google Scholar] [CrossRef]
- Mitchison, D. The action of antituberculosis drugs in short-course chemotherapy. Tubercle 1985, 66, 219–225. [Google Scholar] [CrossRef]
- Konno, K.; Feldmann, F.M.; McDermott, W. Pyrazinamide susceptibility and amidase activity of tubercle bacilli. Am. Rev. Respir. Dis. 1967, 95, 461–469. [Google Scholar]
- Zhang, Y.; Mitchison, D. The curious characteristics of pyrazinamide: A review. Int. J. Tuberc. Lung Dis. 2003, 7, 6–21. [Google Scholar]
- Juréen, P.; Werngren, J.; Toro, J.-C.; Hoffner, S. Pyrazinamide resistance and pncA gene mutations in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2008, 52, 1852–1854. [Google Scholar] [CrossRef] [Green Version]
- Cheng, S.-J.; Thibert, L.; Sanchez, T.; Heifets, L.; Zhang, Y. pncA mutations as a major mechanism of pyrazinamide resistance in Mycobacterium tuberculosis: Spread of a monoresistant strain in Quebec, Canada. Antimicrob. Agents Chemother. 2000, 44, 528–532. [Google Scholar] [CrossRef] [Green Version]
- Moazed, D.; Noller, H.F. Interaction of antibiotics with functional sites in 16S ribosomal RNA. Nature 1987, 327, 389–394. [Google Scholar] [CrossRef]
- Crofton, J.; Mitchison, D. Streptomycin resistance in pulmonary tuberculosis. Br. Med. J. 1948, 2, 1009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, S.H. Evolution of drug resistance in Mycobacterium tuberculosis: Clinical and molecular perspective. Antimicrob. Agents Chemother. 2002, 46, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Berning, S.E. The role of fluoroquinolones in tuberculosis today. Drugs 2001, 61, 9–18. [Google Scholar] [CrossRef]
- Ruan, Q.; Liu, Q.; Sun, F.; Shao, L.; Jin, J.; Yu, S.; Ai, J.; Zhang, B.; Zhang, W. Moxifloxacin and gatifloxacin for initial therapy of tuberculosis: A meta-analysis of randomized clinical trials. Emerg. Microbes Infect. 2016, 5, e12. [Google Scholar] [CrossRef] [Green Version]
- Fàbrega, A.; Madurga, S.; Giralt, E.; Vila, J. Mechanism of action of and resistance to quinolones. Microb. Biotechnol. 2009, 2, 40–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takiff, H.E.; Salazar, L.; Guerrero, C.; Philipp, W.; Huang, W.M.; Kreiswirth, B.; Cole, S.T.; Jacobs, W.R., Jr.; Telenti, A. Cloning and nucleotide sequence of Mycobacterium tuberculosis gyrA and gyrB genes and detection of quinolone resistance mutations. Antimicrob. Agents Chemother. 1994, 38, 773–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georghiou, S.B.; Magana, M.; Garfein, R.S.; Catanzaro, D.G.; Catanzaro, A.; Rodwell, T.C. Evaluation of genetic mutations associated with Mycobacterium tuberculosis resistance to amikacin, kanamycin and capreomycin: A systematic review. PLoS ONE 2012, 7, e33275. [Google Scholar] [CrossRef] [PubMed]
- Johansen, S.K.; Maus, C.E.; Plikaytis, B.B.; Douthwaite, S. Capreomycin binds across the ribosomal subunit interface using tlyA-encoded 2’-O-methylations in 16S and 23S rRNAs. Mol Cell 2006, 23, 173–182. [Google Scholar] [CrossRef]
- Jugheli, L.; Bzekalava, N.; de Rijk, P.; Fissette, K.; Portaels, F.; Rigouts, L. High level of cross-resistance between kanamycin, amikacin, and capreomycin among Mycobacterium tuberculosis isolates from Georgia and a close relation with mutations in the rrs gene. Antimicrob. Agents Chemother. 2009, 53, 5064–5068. [Google Scholar] [CrossRef] [Green Version]
- Krüüner, A.; Jureen, P.; Levina, K.; Ghebremichael, S.; Hoffner, S. Discordant resistance to kanamycin and amikacin in drug-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2003, 47, 2971–2973. [Google Scholar] [CrossRef] [Green Version]
- Mathys, V.; Wintjens, R.; Lefevre, P.; Bertout, J.; Singhal, A.; Kiass, M.; Kurepina, N.; Wang, X.-M.; Mathema, B.; Baulard, A.; et al. Molecular genetics of para-aminosalicylic acid resistance in clinical isolates and spontaneous mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2009, 53, 2100–2109. [Google Scholar] [CrossRef] [Green Version]
- Zhao, F.; Wang, X.D.; Erber, L.N.; Luo, M.; Guo, A.Z.; Yang, S.S.; Gu, J.; Turman, B.J.; Gao, Y.R.; Li, D.F.; et al. Binding pocket alterations in dihydrofolate synthase confer resistance to para-aminosalicylic acid in clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 1479–1487. [Google Scholar] [CrossRef] [Green Version]
- Singh, B.; Cocker, D.; Ryan, H.; Sloan, D.J. Linezolid for drug-resistant pulmonary tuberculosis. Cochrane Database Syst. Rev. 2019, 3, Cd012836. [Google Scholar] [CrossRef]
- Richter, E.; Rüsch-Gerdes, S.; Hillemann, D. First linezolid-resistant clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1534–1536. [Google Scholar] [CrossRef] [Green Version]
- Huitric, E.; Verhasselt, P.; Andries, K.; Hoffner, S.E. In vitro antimycobacterial spectrum of a diarylquinoline ATP synthase inhibitor. Antimicrob. Agents Chemother 2007, 51, 4202–4204. [Google Scholar] [CrossRef] [Green Version]
- Huitric, E.; Verhasselt, P.; Koul, A.; Andries, K.; Hoffner, S.; Andersson, D.I. Rates and mechanisms of resistance development in Mycobacterium tuberculosis to a novel diarylquinoline ATP synthase inhibitor. Antimicrob. Agents Chemother. 2010, 54, 1022–1028. [Google Scholar] [CrossRef] [Green Version]
- Petrella, S.; Cambau, E.; Chauffour, A.; Andries, K.; Jarlier, V.; Sougakoff, W. Genetic basis for natural and acquired resistance to the diarylquinoline R207910 in mycobacteria. Antimicrob. Agents Chemother. 2006, 50, 2853–2856. [Google Scholar] [CrossRef]
- Lechartier, B.; Cole, S.T. Mode of Action of Clofazimine and Combination Therapy with Benzothiazinones against Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015, 59, 4457–4463. [Google Scholar] [CrossRef] [Green Version]
- Hartkoorn, R.C.; Uplekar, S.; Cole, S.T. Cross-resistance between clofazimine and bedaquiline through upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 2979–2981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
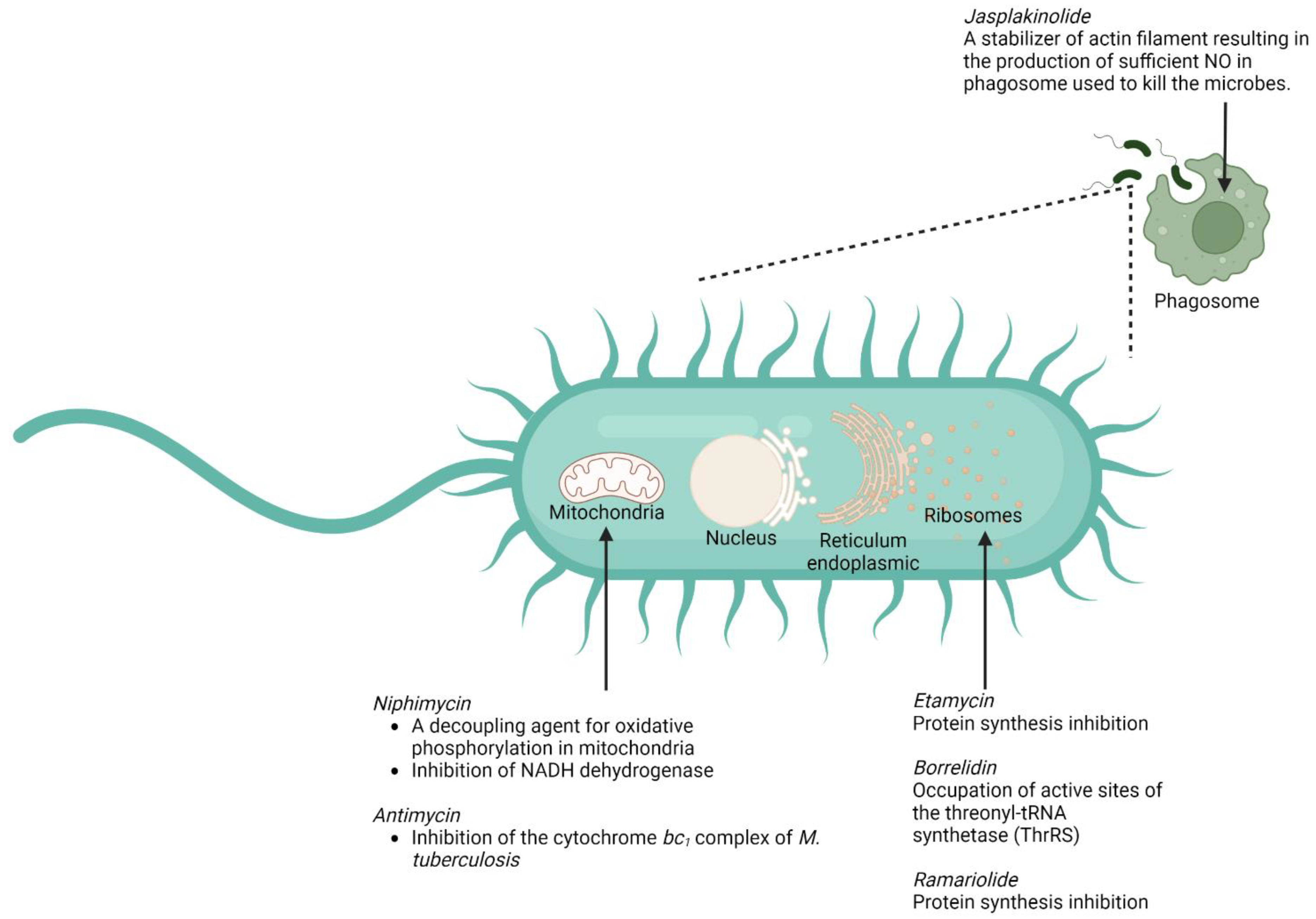
- Miller, B.H.; Fratti, R.A.; Poschet, J.F.; Timmins, G.S.; Master, S.S.; Burgos, M.; Marletta, M.A.; Deretic, V. Mycobacteria inhibit nitric oxide synthase recruitment to phagosomes during macrophage infection. Infect. Immun. 2004, 72, 2872–2878. [Google Scholar] [CrossRef] [Green Version]
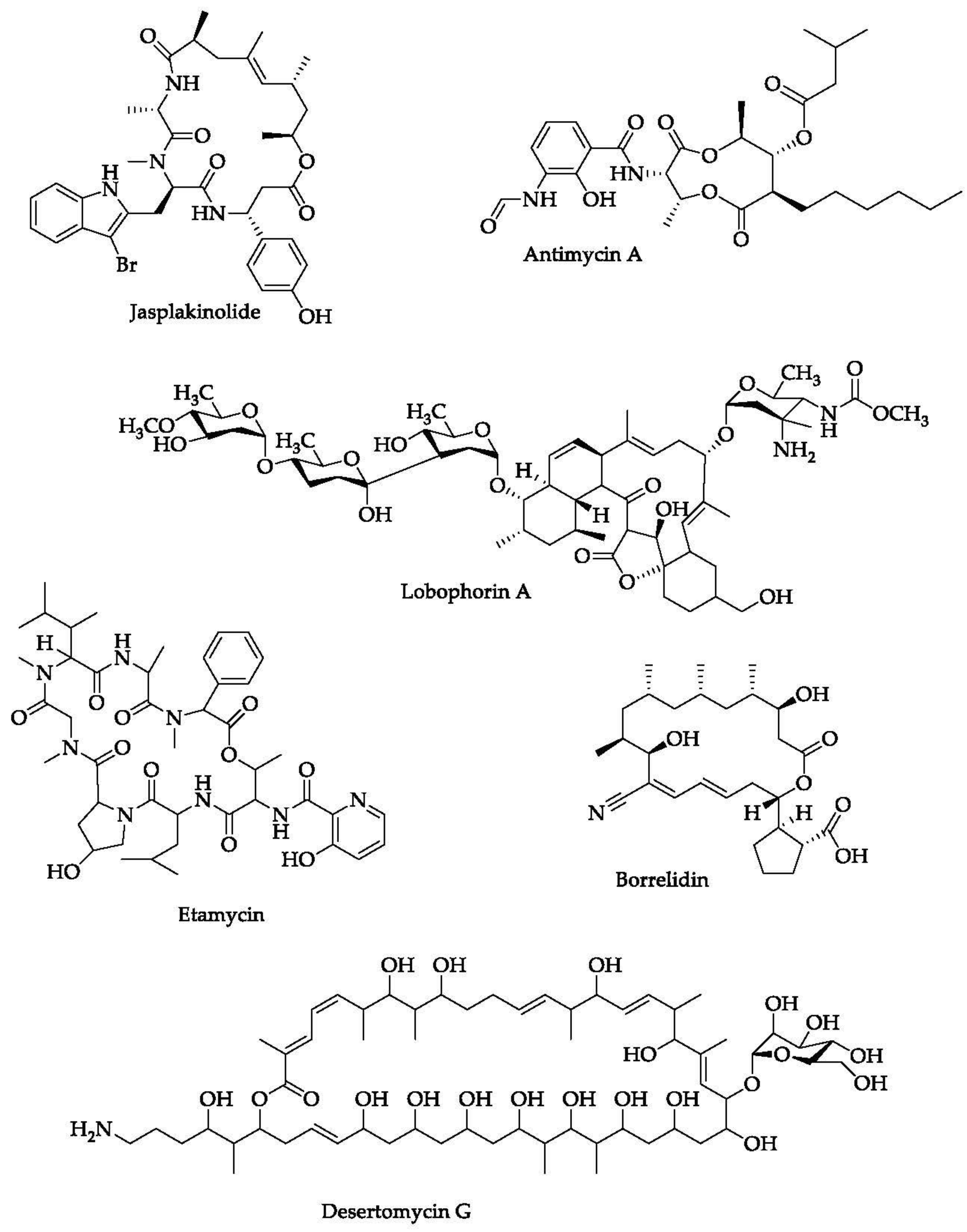
- Holzinger, A.; Blaas, K. Actin-Dynamics in Plant Cells: The Function of Actin-Perturbing Substances: Jasplakinolide, Chondramides, Phalloidin, Cytochalasins, and Latrunculins. Methods Mol. Biol. 2016, 1365, 243–261. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Wang, J.; Guo, H.; Hou, W.; Yang, N.; Ren, B.; Liu, M.; Dai, H.; Liu, X.; Song, F.; et al. Three antimycobacterial metabolites identified from a marine-derived Streptomyces sp. MS100061. Appl. Microbiol. Biotechnol. 2013, 97, 3885–3892. [Google Scholar] [CrossRef]
- Lin, Z.; Koch, M.; Pond, C.D.; Mabeza, G.; Seronay, R.A.; Concepcion, G.P.; Barrows, L.R.; Olivera, B.M.; Schmidt, E.W. Structure and activity of lobophorins from a turrid mollusk-associated Streptomyces sp. J. Antibiot. 2014, 67, 121–126. [Google Scholar] [CrossRef] [Green Version]
- Chandra, P.; He, L.; Zimmerman, M.; Yang, G.; Köster, S.; Ouimet, M.; Wang, H.; Moore, K.J.; Dartois, V.; Schilling, J.D.; et al. Inhibition of Fatty Acid Oxidation Promotes Macrophage Control of Mycobacterium tuberculosis. mBio 2020, 11. [Google Scholar] [CrossRef]
- Cumming, B.M.; Addicott, K.W.; Adamson, J.H.; Steyn, A.J. Mycobacterium tuberculosis induces decelerated bioenergetic metabolism in human macrophages. Elife 2018, 7, e39169. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.T.; Uddin, M.S.; Jeandet, P.; Emran, T.B.; Mitra, S.; Albadrani, G.M.; Sayed, A.A.; Abdel-Daim, M.M.; Simal-Gandara, J. Anti-Alzheimer’s molecules derived from marine life: Understanding molecular mechanisms and therapeutic potential. Mar. Drugs 2021, 9, 251. [Google Scholar] [CrossRef] [PubMed]
- Rotsaert, F.A.J.; Ding, M.G.; Trumpower, B.L. Differential efficacy of inhibition of mitochondrial and bacterial cytochrome bc1 complexes by center N inhibitors antimycin, ilicicolin H and funiculosin. Biochim. Biophys. Acta (BBA) Bioenerg. 2008, 1777, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, K.; Koyama, N.; Kanamoto, A.; Tomoda, H. Discovery of nosiheptide, griseoviridin, and etamycin as potent anti-mycobacterial agents against Mycobacterium avium complex. Molecules 2019, 24, 1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Maloney, K.N.; Nam, S.J.; Haste, N.M.; Raju, R.; Aalbersberg, W.; Jensen, P.R.; Nizet, V.; Hensler, M.E.; Fenical, W. Fijimycins A-C, three antibacterial etamycin-class depsipeptides from a marine-derived Streptomyces sp. Bioorg. Med. Chem. 2011, 19, 6557–6562. [Google Scholar] [CrossRef] [Green Version]
- Stoye, A.; Nagalingam, G.; Britton, W.J.; Payne, R.J. Synthesis of Norfijimycin A with Activity against Mycobacterium tuberculosis. Aust. J. Chem. 2017, 70, 229–232. [Google Scholar] [CrossRef]
- Braña, A.F.; Sarmiento-Vizcaíno, A.; Pérez-Victoria, I.; Martín, J.; Otero, L.; Palacios-Gutiérrez, J.J.; Fernández, J.; Mohamedi, Y.; Fontanil, T.; Salmón, M.; et al. Desertomycin G, a New Antibiotic with Activity against Mycobacterium tuberculosis and Human Breast Tumor Cell Lines Produced by Streptomyces althioticus MSM3, Isolated from the Cantabrian Sea Intertidal Macroalgae Ulva sp. Mar. Drugs 2019, 17, 114. [Google Scholar] [CrossRef] [Green Version]
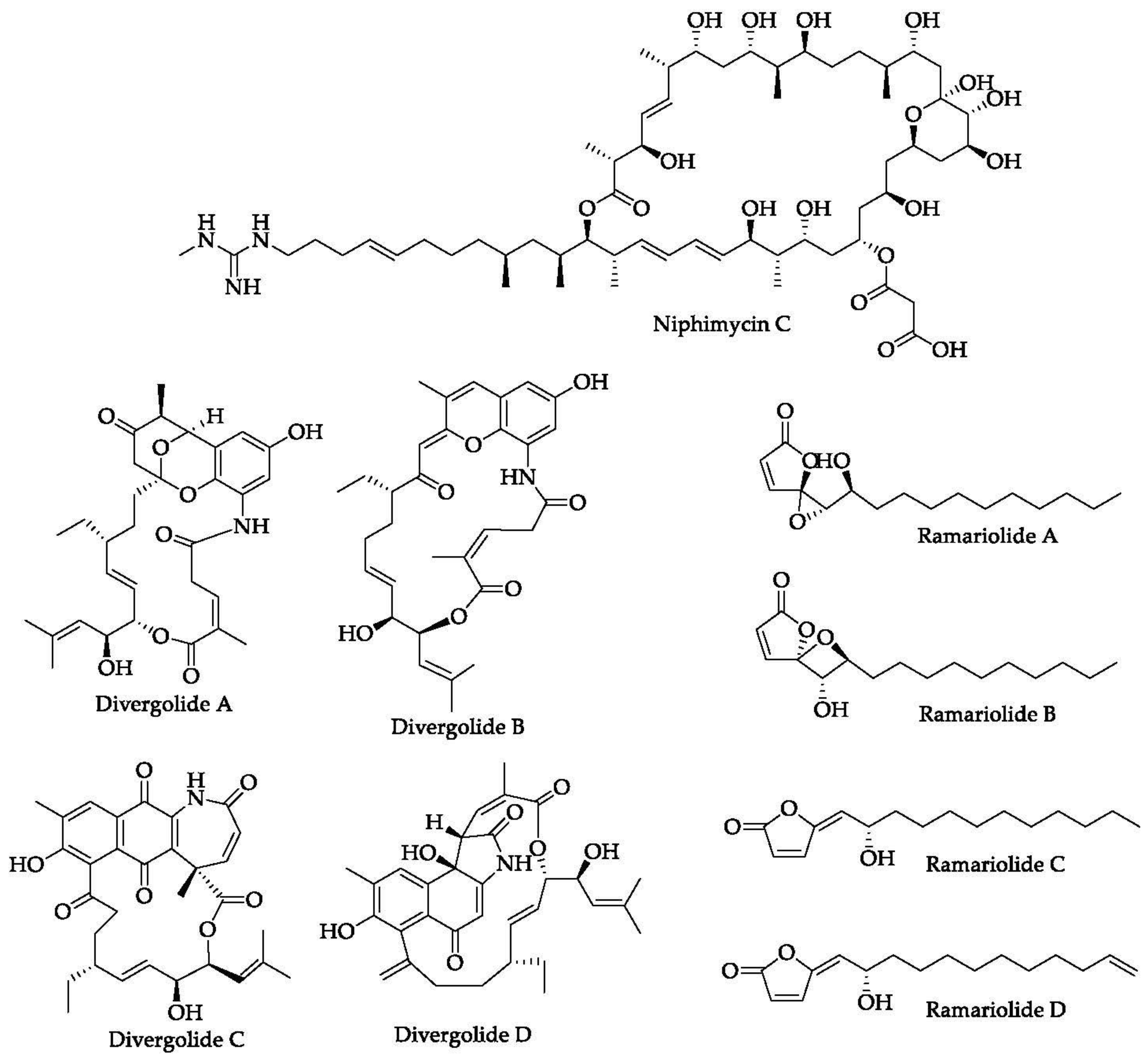
- Hu, Y.; Wang, M.; Wu, C.; Tan, Y.; Li, J.; Hao, X.; Duan, Y.; Guan, Y.; Shang, X.; Wang, Y.; et al. Identification and Proposed Relative and Absolute Configurations of Niphimycins C-E from the Marine-Derived Streptomyces sp. IMB7-145 by Genomic Analysis. J. Nat. Prod. 2018, 81, 178–187. [Google Scholar] [CrossRef]
- Gutierrez-Lugo, M.T.; Bewley, C.A. Susceptibility and mode of binding of the Mycobacterium tuberculosis cysteinyl transferase mycothiol ligase to tRNA synthetase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 2480–2483. [Google Scholar] [CrossRef] [Green Version]
- Sun, J.; Shao, J.; Sun, C.; Song, Y.; Li, Q.; Lu, L.; Hu, Y.; Gui, C.; Zhang, H.; Ju, J. Borrelidins F-I, cytotoxic and cell migration inhibiting agents from mangrove-derived Streptomyces rochei SCSIO ZJ89. Bioorg. Med. Chem. 2018, 26, 1488–1494. [Google Scholar] [CrossRef]
- Yu, M.; Li, Y.; Banakar, S.P.; Liu, L.; Shao, C.; Li, Z.; Wang, C. New Metabolites from the Co-culture of Marine-Derived Actinomycete Streptomyces rochei MB037 and Fungus Rhinocladiella similis 35. Front. Microbiol. 2019, 10, 915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, L.; Maier, A.; Fiebig, H.H.; Görls, H.; Lin, W.H.; Peschel, G.; Hertweck, C. Divergolides A–D from a Mangrove Endophyte Reveal an Unparalleled Plasticity in ansa-Macrolide Biosynthesis. Angew. Chem. 2011, 123, 1668–1672. [Google Scholar] [CrossRef]
- Centko, R.M.; Ramón-García, S.; Taylor, T.; Patrick, B.O.; Thompson, C.J.; Miao, V.P.; Andersen, R.J. Ramariolides A–D, Antimycobacterial Butenolides Isolated from the Mushroom Ramaria cystidiophora. J. Nat. Prod. 2012, 75, 2178–2182. [Google Scholar] [CrossRef] [PubMed]
- Bahbah, E.I.; Ghozy, S.; Attia, M.S.; Negida, A.; Emran, T.B.; Mitra, S.; Albadrani, G.M.; Abdel-Daim, M.M.; Uddin, M.S.; Simal-Gandara, J. Molecular Mechanisms of Astaxanthin as a Potential Neurotherapeutic Agent. Mar. Drugs 2021, 19, 201. [Google Scholar] [CrossRef]
- Hanh, B.T.B.; Kim, T.H.; Park, J.W.; Lee, D.G.; Kim, J.S.; Du, Y.E.; Yang, C.S.; Oh, D.C.; Jang, J. Etamycin as a Novel Mycobacterium abscessus Inhibitor. Int. J. Mol. Sci. 2020, 21, 6908. [Google Scholar] [CrossRef]
- Lukarska, M.; Palencia, A. Chapter Eleven—Aminoacyl-tRNA synthetases as drug targets. In The Enzymes; Ribas de Pouplana, L., Kaguni, L.S., Eds.; Academic Press: Cambridge, MA, USA, 2020; Volume 48, pp. 321–350. [Google Scholar]
- Song, X.; Yuan, G.; Li, P.; Cao, S. Guanidine-Containing Polyhydroxyl Macrolides: Chemistry, Biology, and Structure-Activity Relationship. Molecules 2019, 24, 3913. [Google Scholar] [CrossRef]

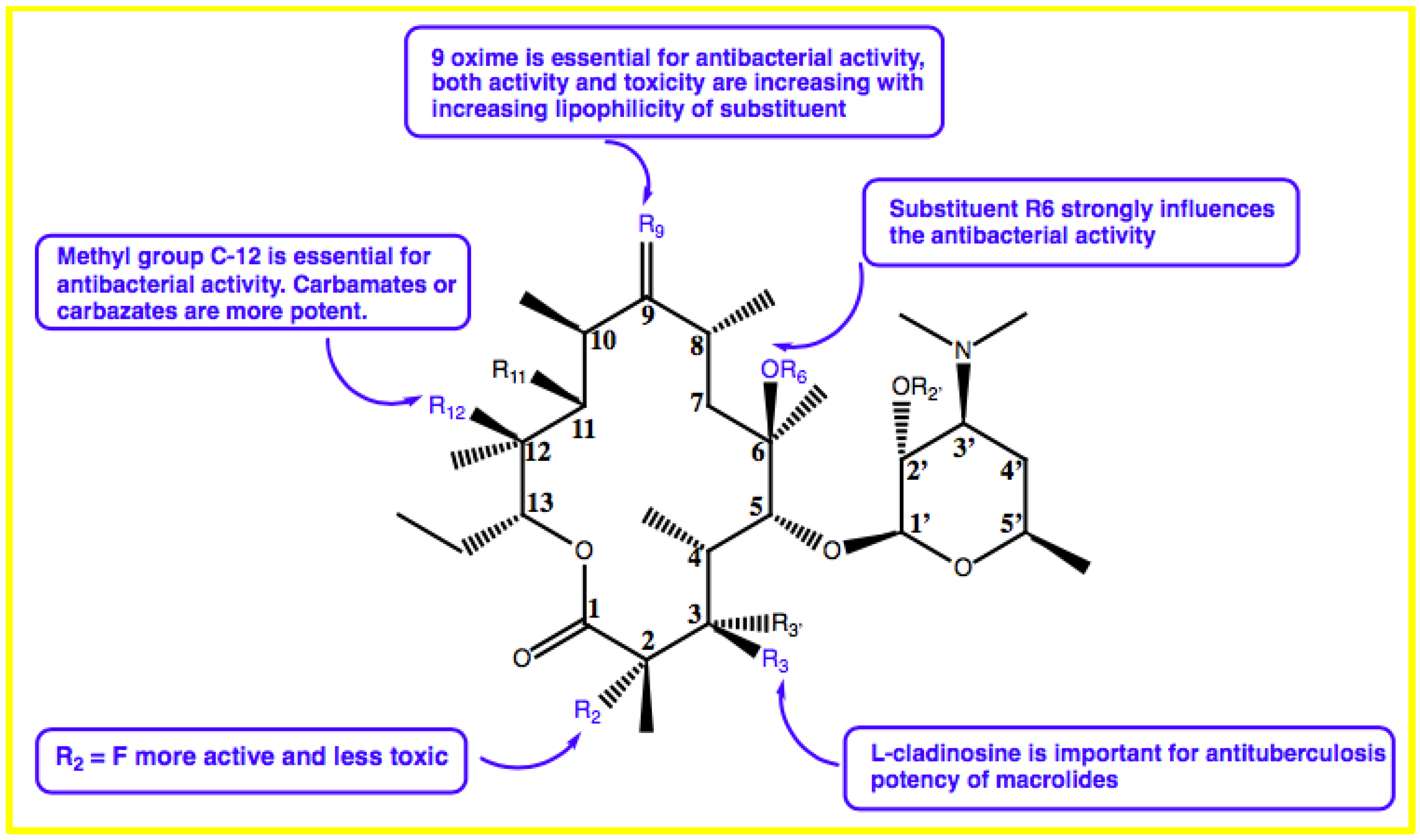






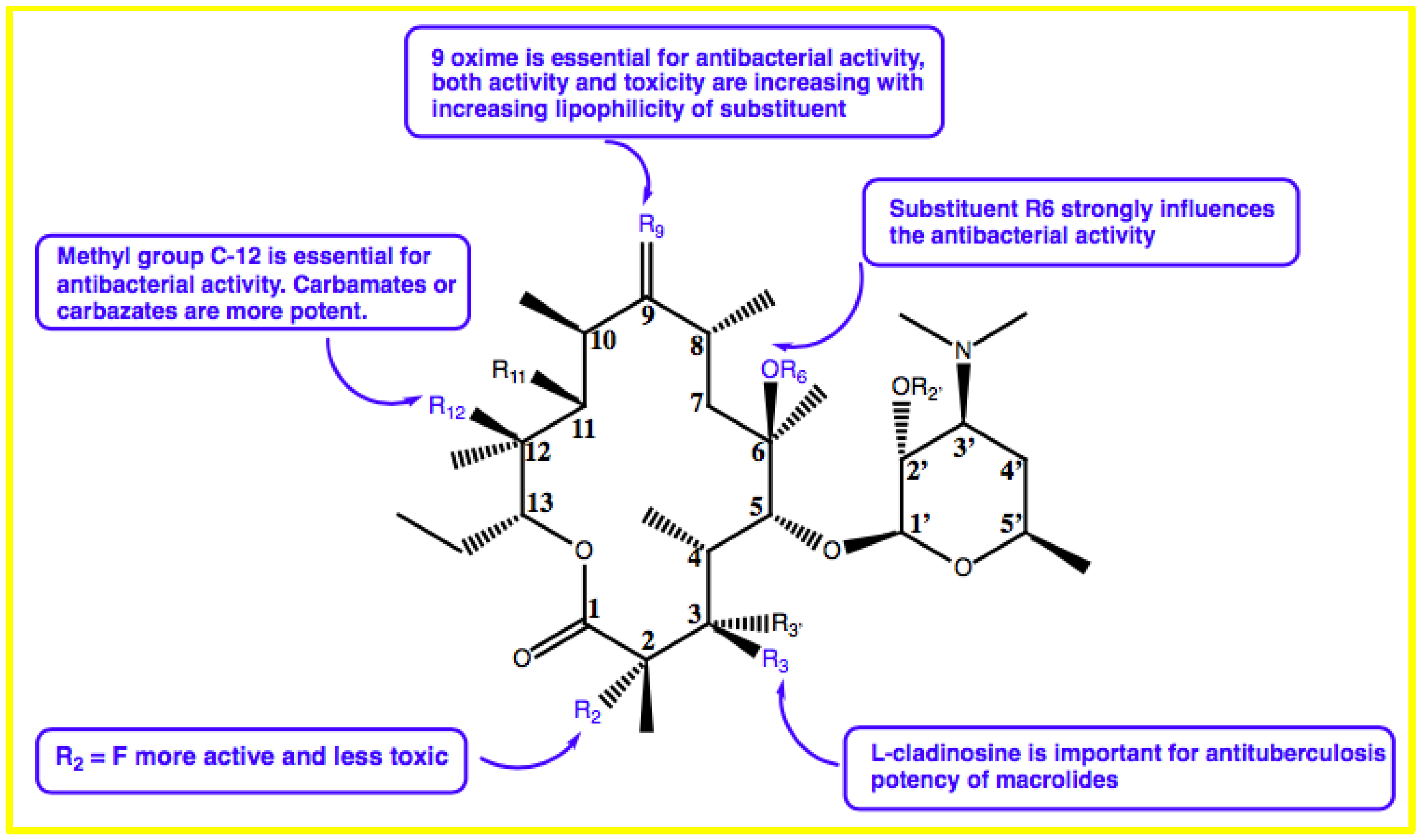
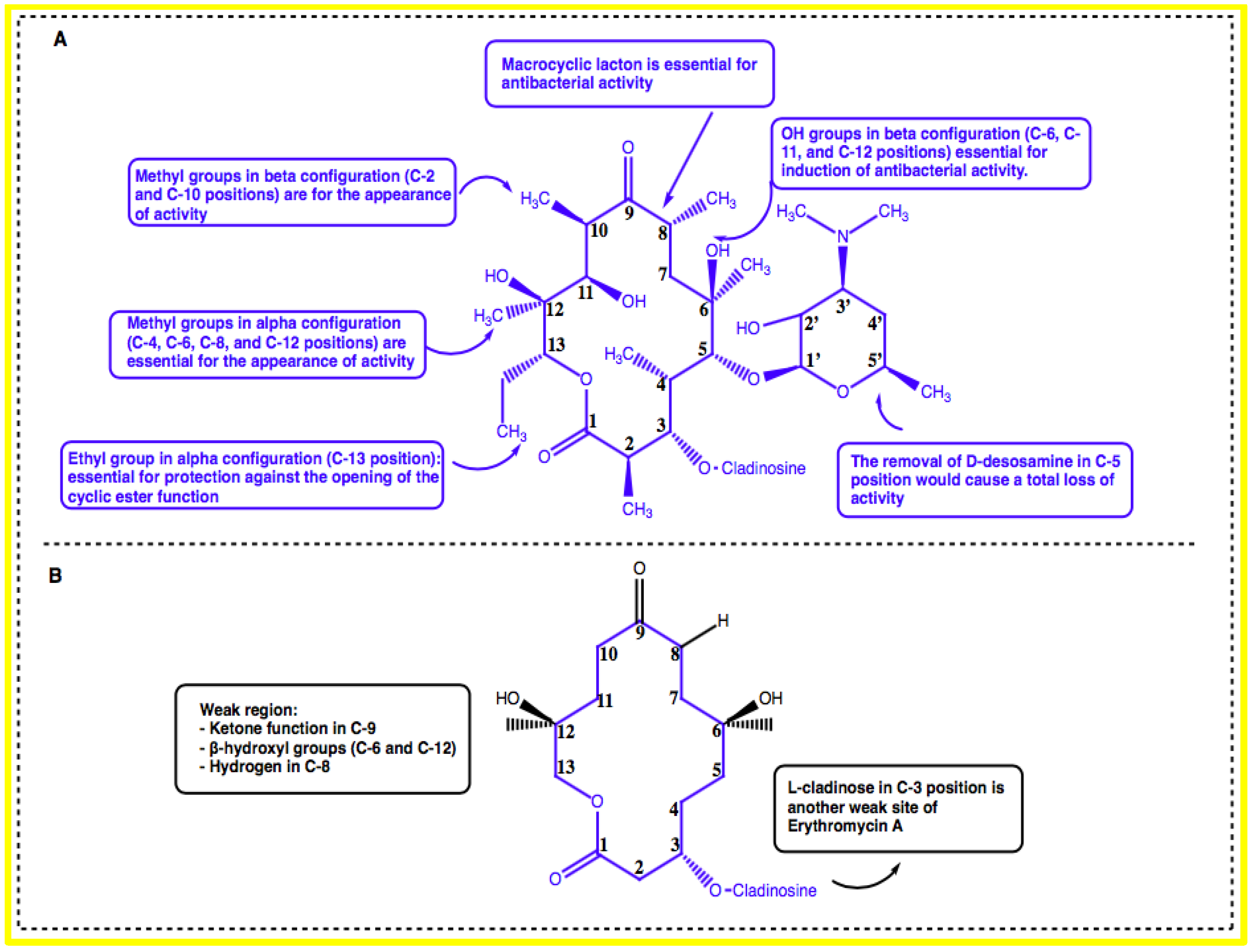{kind=link}
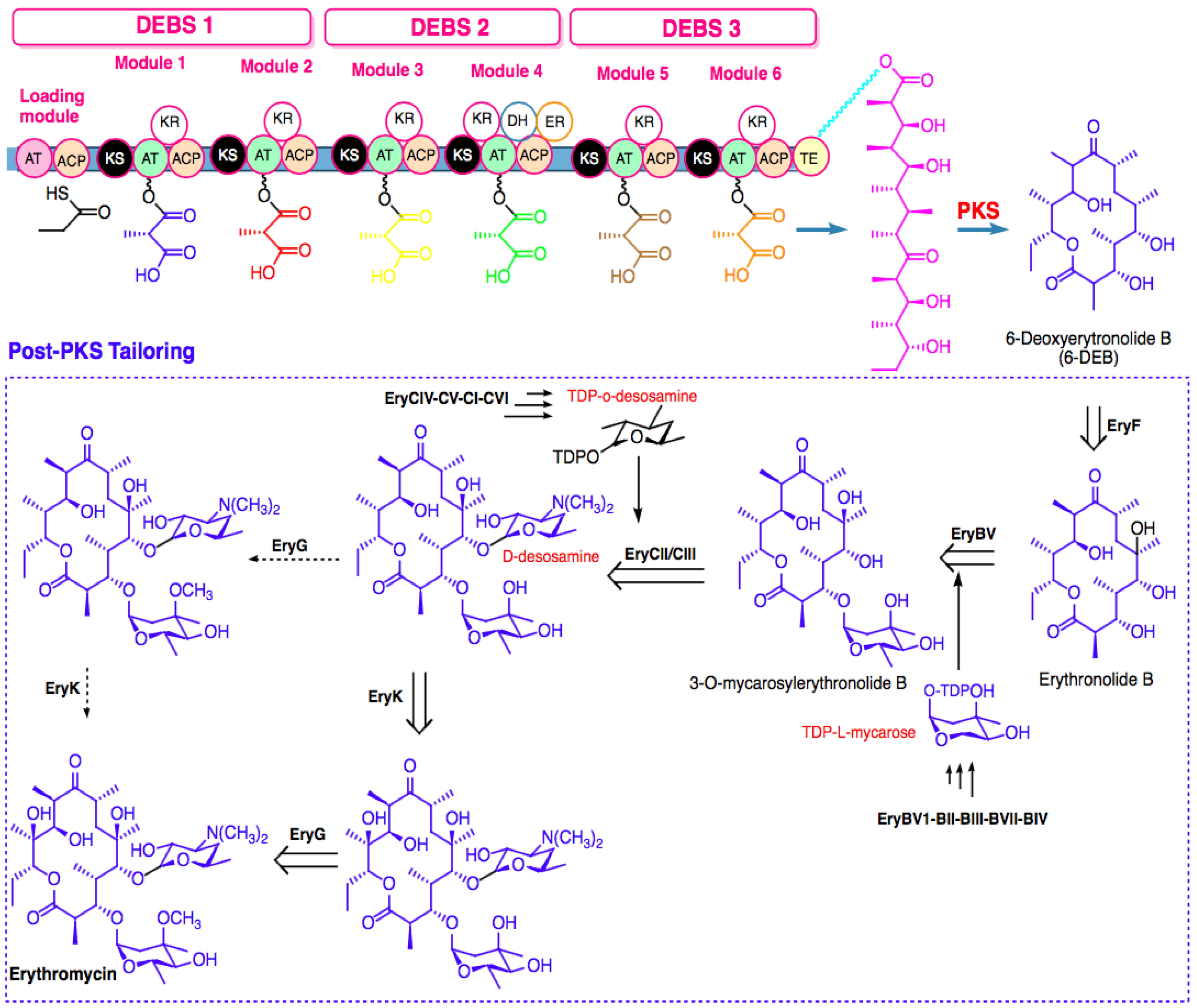
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Strain | Strategy | Key Findings | Ref. |
|---|---|---|---|---|
| Erythromycin | Saccharopolyspora erythraea | Inactivate a number of TetR-family transcriptional regulator (TFR) candidates. | Overexpression of SACE_7301 in wild-type and industrial S. erythraea strains enhanced erythromycin yields. | [51] |
| Pikromycin | Streptomyces venezuelae | Manipulate three key enzymes of branched-chain amino-acid (BCAA) catabolism, branched-chain α-keto acid dehydrogenase (BCDH), acyl-CoA dehydrogenase, and 3-ketoacyl acyl carrier-protein synthase III. | Overexpression of BCAA resulted in the highest titer of total macrolide production of 43 mg/L, which was about a 2.2-fold increase compared to that of the wild type. | [52] |
| Nargenicin A1 | Nocardia sp. CS682 | Increase the pool of precursors for glycosylation. | Over-expression of the ACCase complex enhanced the titer. | [53] |
| Epothilone | Sorangium cellulosum and Burkholderiales strain DSM 7029 | Introduce the epothilone biosynthetic gene cluster from the myxobacterium Sorangium cellulosum to the chromosome of Burkholderiales strain DSM 7029 by transposition. | Overexpression of rare tRNA genes and introduction of the exogenous methylmalonyl-CoA biosynthetic pathway elevated the total yields of epothilones to 307 μg/L. | [54] |
| Galbonolide B | Streptomyces sp. LZ35 and S. coelicolor | Introduce the galbonolide-expression constructs (gbnA–E) to the host strain S. coelicolor ZM12 by intergeneric conjugation and integrate it into the chromosome. | Galbonolides B was successfully produced by heterologous expression. | [55] |
| Quinolidomicin | Micromonospora sp. JY16 and Streptomyces lividans | Introduce LAL regulator genes (qnmRI and qnmRII) from Micromonospora sp. JY16 into S. lividans TK23ΔredDX::pKU518quiP9-L5 to activate the transcription level of the biosynthetic genes. | Heterologous expression of the biosynthetic gene cluster for quinolidomicin A1 as the 213.7 kb region. The yield of quinolidomicin A1 was approximately 0.1 mg/L in the culture broth. | [56] |
| Compound | Source | Key Findings | Ref. |
|---|---|---|---|
| Jasplakinolide | Marine sponge Jaspis johnstoni |
| [100,101] |
| Lobophorins | Marine-derived Streptomyces sp. MS100061 |
| [102] |
| Mollusk-associated Streptomyces sp. 1053U.I.1a.3b |
| [103] | |
| Antimycin A and its analogues | Marine-derived actinomycete Streptomyces lusitanus |
| [104,105,106,107] |
| Etamycin | Marine-derived actinomycetes Streptomyces spp. OPMA1730 |
| [108,109] |
| Norfijimycin A | Its parent compound, fijimycin A, was first isolated from a marine-derived Streptomyces spp. CNS-575Norfijimycin A is a simplified analogue of fijimycin A |
| [110] |
| Desertomycin G | Marine-derived actinomycete Streptomyces althioticus MSM3 |
| [111] |
| Niphimycins | Marine-derived Streptomyces sp. IMB7-145 |
| [112] |
| Borrelidin | Mangrove-derived Streptomyces rochei SCSIO ZJ89Co-culture of marine-derived Streptomyces rochei MB037 and fungus Rhinocladiella similis 35 |
| [113,114,115] |
| Divergolides A–D | Mangrove endophyte Bruguiera gymnorrhiza |
| [116] |
| Ramariolides | Coral mushroom Ramaria cystidiophora |
| [117] |
| [118] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mamada, S.S.; Nainu, F.; Masyita, A.; Frediansyah, A.; Utami, R.N.; Salampe, M.; Emran, T.B.; Lima, C.M.G.; Chopra, H.; Simal-Gandara, J. Marine Macrolides to Tackle Antimicrobial Resistance of Mycobacterium tuberculosis. Mar. Drugs 2022, 20, 691. https://doi.org/10.3390/md20110691
Mamada SS, Nainu F, Masyita A, Frediansyah A, Utami RN, Salampe M, Emran TB, Lima CMG, Chopra H, Simal-Gandara J. Marine Macrolides to Tackle Antimicrobial Resistance of Mycobacterium tuberculosis. Marine Drugs. 2022; 20(11):691. https://doi.org/10.3390/md20110691
Chicago/Turabian StyleMamada, Sukamto S., Firzan Nainu, Ayu Masyita, Andri Frediansyah, Rifka Nurul Utami, Mirnawati Salampe, Talha Bin Emran, Clara Mariana Gonçalves Lima, Hitesh Chopra, and Jesus Simal-Gandara. 2022. "Marine Macrolides to Tackle Antimicrobial Resistance of Mycobacterium tuberculosis" Marine Drugs 20, no. 11: 691. https://doi.org/10.3390/md20110691
APA StyleMamada, S. S., Nainu, F., Masyita, A., Frediansyah, A., Utami, R. N., Salampe, M., Emran, T. B., Lima, C. M. G., Chopra, H., & Simal-Gandara, J. (2022). Marine Macrolides to Tackle Antimicrobial Resistance of Mycobacterium tuberculosis. Marine Drugs, 20(11), 691. https://doi.org/10.3390/md20110691