1. Introduction
As an industry generating billions of dollars annually, microalgal biotechnology is opening up increasing new areas of research. Microalgal biomass is considered a promising source of raw materials for the sustainable production of a wide range of compounds and high value-added products, including functional foods, feed ingredients, animal feed, medicine, cosmetics, bio-stimulants, and bio-actives, as well as for use in agricultural algae extraction, bio-oil production, and biofuel production [
1]. Microalgal extracts are ideal sources of biotechnological compounds with high levels of bioactivity, e.g., potent antiviral, anti-microbial, anti-oxidant, anti-inflammatory, anti-cancer, and immune-enhancing properties; neuroprotective activity; and various therapeutic and restorative properties. Furthermore, compounds or metabolites derived from microalgae can be used as antibodies and raw materials for vaccine production against SARS-CoV-2 [
2,
3].
In recent decades, omega-3 and omega-6 polyunsaturated fatty acids (ω-3 and ω-6 PUFAs, respectively) have received special attention in nutrition and pharmaceutical research and application because of their important roles in human health, including the regulation of biological functions, the prevention and treatment of a variety of diseases related to neurological development [
4], improvements in cognition [
5], reductions in cardiovascular system stress [
6], and anti-inflammatory and anti-cancer actions in humans [
1]. ω-3 PUFAs include ALA (linolenic acid; C18:3 ω-3), EPA (eicosapentaenoic acid, C20:5ω-3), and DHA (docosahexaenoic acid, C22:6ω-3), and ω-6 PUFAs include LA (linoleic acid; C18:2 ω-6). All these PUFAs, especially DHA, are produced by the Thraustochytriidae (Thraustochytrids) family, which includes the genera
Schizochytrium, Thraustochytrium, Aurantiochytrium, Labyrinthuloides, and
Ulkenia. Studies have been published on the optimal growth conditions and the production of highly effective ω-3 and ω-6 PUFA strains belonging to these genera [
7,
8,
9]. In 2001, the US FDA affirmed that DHA and ARA oils derived from microalgae are generally recognized as safe (GRAS) for use in infant formula. Omega-3 products derived from heterotrophic marine microalgae and prepared for human consumption include Life’sDHA
®, Life’s™OMEGA, DHASCO (from
Schizochytrium sp. microalgae), and DHASCO-T (from
Crypthecodinium cohnii) marketed by DSM and the Martek Biosciences Corporation of Columbia, USA. Moreover, mixtures of PUFAs extracted from algae have been added to commercial products such as yogurt, confectionery, and beverages [
10]. Falk et al. [
11] evaluated the toxicity of DHA-rich oils from
Schizochytrium sp. (DHA accounted for 41.37% the total fatty acids—TFAs) and showed using a rat model that this oil was safe for the embryo or fetus and during growth. Omega-3 fatty acids can help improve factors that cause nerve cell damage, are able to protect cerebral blood vessels, and can increase metabolism and brain activity in mice with Alzheimer’s disease [
12]. Several companies including Algae, Parry Nutraceuticals, Fermentalg, Algaetech International, AlgaeBiotech, Algae to Omega Holdings, and Alltech Algae are performing nutraceutical research and conducting product development based on microalgal oils that are rich in EPA and DHA. PureOne
TM is an EPA- and DHA-rich algal oil supplement sold on the market as capsules [
13].
Various methods can be used for the extraction and purification of PUFAs, such as the use of urea complexes, low-temperature solvent crystallization, fractional distillation, hydrolysis, enzymatic techniques, and high-pressure liquid chromatography (HPLC) [
14,
15]. Of the above methods, the use of urea complexes is the most suitable for attaining PUFA-rich samples [
15].
Today, along with increases in life expectancy and changes of population demographics, neurodegenerative diseases such as Alzheimer’s and Parkinson’s disease (PD) are becoming seriously prevalent [
16,
17,
18]. The marine environment is known to be a rich source of chemical structures that have many beneficial biological effects on human health. It is widely accepted that natural marine products have many unusual and unique chemical properties. Around the world, based on molecular models and chemosynthetic methods, new drugs with greater efficacy and specificity for the treatment of human diseases have been developed [
19,
20]. Among marine organisms, heterotrophic marine microalgae have been the specific targets of numerous studies that have shown their potential as rich sources of structurally diverse and biologically active compounds with great pharmaceutical and biomedical potential, including PUFAs [
1,
2,
16,
17,
21,
22,
23]. Correlative epidemiological studies suggest that the high consumption of food rich in ω-3 PUFA is associated with better health and possibly the prevention of age-related cognitive impairment or Alzheimer’s disease (AD). Many longitudinal or case–control studies have shown an association between ω-3 PUFA consumption or blood levels and lower risks of dementia or AD [
24,
25,
26]. Recently, a cross-sectional study in a Finnish cohort reported that higher serum concentrations of long-chain ω-3 PUFA were associated with better performance in neuropsychological tests [
27]. Preclinical studies involving controlled diets have consistently shown that increasing DHA concentrations in the brain improves rodent performance in a wealth of different memory tests [
28]. This has been confirmed in various animal models of AD-like neuropathology. DHA-induced decreases have been reported in amyloid levels, neurofibrillary tangles (Tau), or synaptic neuropathologies in animal models of AD [
28,
29,
30]. More specifically, lower amyloid beta protein (Aβ) levels in the brains of amyloid protein precursor (APP) transgenic mice after high DHA intake have been reported by at least four groups [
31,
32,
33,
34]. Other evidence suggests that DHA may also act more directly on neuronal function by progressively integrating cell membranes without necessarily targeting AD neuropathology [
29]. A reduction in markers of neuroinflammation has also been observed following n-3 PUFA intake, which could contribute to a therapeutic effect in neurodegenerative diseases [
35].
Aurantiochytrium is an oleaginous microorganism genus in the Thraustochytriaceae family that has attracted attention because of its ability to produce high levels of PUFAs and other compounds such as squalene, astaxanthin, and exopolysaccharide. A few reports have demonstrated that ethanol extracts obtained from
Aurantiochytrium sp. have anti-inflammatory properties [
36,
37]. However, knowledge is limited on the effects of
Aurantiochytrium sp. and its bioactive compounds on neuroprotection. Thus, the main aims of the present study were to isolate
Aurantiochytrium sp. SC145 from decomposing leaves and seagrass collected from Sand Cay (Son Ca) Island, Vietnam, and to optimize its culture conditions. Then, PUFAs in the forms of alkyl esters (FAAE) and free fatty acids (FFA) were extracted, assessed regarding their acute and subchronic toxicity in rats, and evaluated in terms of their antioxidation and neuroprotective activities.
3. Materials and Methods
3.1. Strain and Culture
In this study, we isolated the heterotrophic marine microalga
Aurantiochytrium sp. SC145 (SC145 strain) from leaf and seagrass specimens taken from Sand Cay (Son Ca) Island, Vietnam (at 10°22′42″ N latitude–114°28′33″ E longitude), from May to June and from September to November 2021; these specimens were deposited at the Department of Algal Biotechnology, Institute of Biotechnology, Vietnam Academy of Science and Technology, Vietnam under accession number ASC145 (
Figure 1).
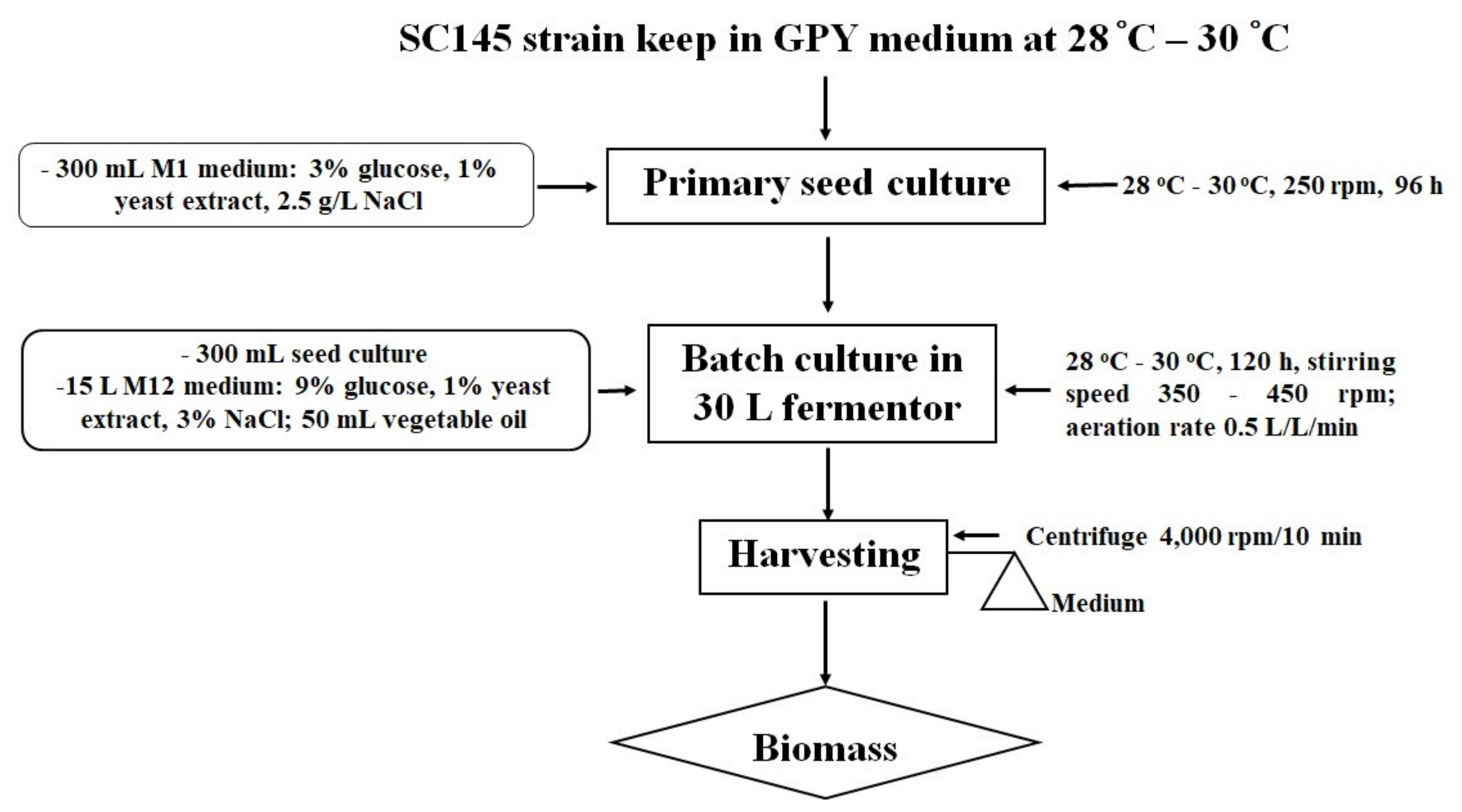
This strain was kept on a GPY medium (0.2% glucose, 0.1% polypeptide, 0.05% yeast extract, 1.5% agar, and 17.5 g/L artificial sea water—ASW). The primary seed culture was prepared by placing colonies of the SC145 strain in a Petri dish in a 1000 mL Erlenmeyer flask with 350 mL of an M1 liquid medium containing 30 g/L glucose, 10 g/L yeast extract, and 2.5% NaCl.
The primary seed culture flasks were incubated for 96 h at 28–30 °C with shaking at 250 rpm. The highest levels of the dry biomass and lipid content were 12.39 ± 1.62 g/L and 27.82 ± 1.67% dry cell weight (DCW), respectively.
The size of the inoculum was 2% of the total liquid volume in the fermentor. In the 30 L fermentor, the SC145 strain grew well in a medium containing 9% glucose and 1% industrial yeast extract. The temperature was kept at 28–30 °C, and the dissolved oxygen level was maintained above 10% by manually increasing the stirring speed from 350 rpm to a maximum of 450 rpm. The aeration rate was maintained at 0.5 volume air/(vol. medium)/min using a 0.2 mm filter. Instead of antifoam, 50 mL Neptune® Gold vegetable oil (made by Cai Lan Vegetable Oil Limited Company, Ha Long City, Quang Ninh Province, Vietnam) was added to the 30 L fermentor.
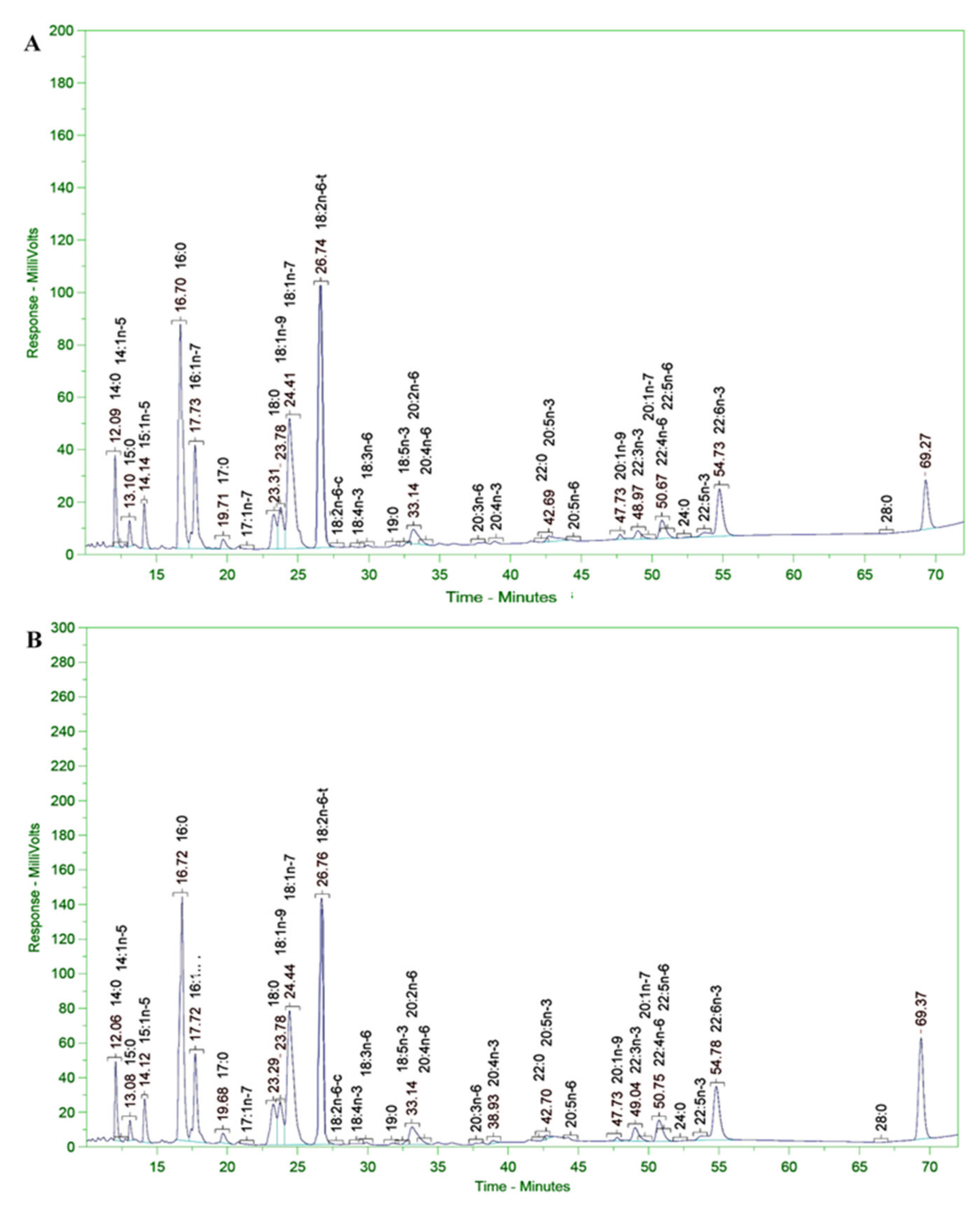
The SC145 strain biomass was harvested after 120 h of fermentation. The culture was conducted in a nutrient medium containing 9% glucose and 1% industrial yeast extract of 3% salinity at room temperature (from 28 to 30 °C) in a 30 L fermentor (handmade). The growth of the Aurantiochytrium sp. SC145 species reached its maximum after 120 h of culture, with dry biomass and lipid content reaching 31.183 ± 2.630 g/L and 25.292 ± 1.430% of DCW, respectively. The fatty acid profile was mainly dominated by C16:0 (palmitic acid; 1.288% of DCW), C18:0 (stearic acid; 0.738% of DCW), C18:1 ω-9 (oleic acid; 2.134% of DCW), C18:2ω-6 (linoleic acid; 0.016% of DCW), C18:3 ω-3 (linolenic acid; 1.076% of DCW), C20:0 (arachidic acid; 0.178% of DCW), C20:5ω-3 (EPA; 0.051% of DCW), C22:6 ω-3 (DHA; 0.129% of DCW), total fatty acid (TFA; 6. 655% of DCW), saturated fatty acid (SFA; 2.566% of DCW), monounsaturated fatty acid (MUFA; 2.193% of DCW), and polyunsaturated fatty acids (PUFAs; 1.318% of DCW).
Algal biomass was harvested at the early stationary phase in the growth curve after centrifugation at 4000 rpm for 10 min. The algal paste was washed three times with sterile distilled water and then dried to a constant weight in a drying oven at 50 °C and stored in desiccators. The procedure followed for cultivation of
Aurantiochytrium sp. SC145 for the exploitation of secondary metabolites is shown in
Figure 5.
3.2. Determination of Microalgal Growth
For the determination of algal growth in terms of DCW, a 10 mL culture broth after 120 h of culture (in the early stationary phase of the growth curve) was collected and centrifuged to obtain the biomass. After centrifugation, the cell biomass was transferred to a cup with an identified weight mass and dried at 105 °C to a constant weight, three consecutive times, as described in a report by Dang et al. [
62]. The DCW of each sample was determined according to the formula:
3.3. Morphological Observation
The
Aurantiochytrium sp. SC145 strain was maintained in a slant culture with a GPY⁄agar medium at 28 °C. Living cells in the colony on the agar plate and the M1 liquid medium were observed with Olympus CX21 and BX51 light microscopes and a JSM-5410L scanning electron microscope (JEOL Company, Tokyo, Japan) [
62].
3.4. Transmission Electron Microscopy (TEM)
After cultivation, the cell suspension was centrifuged at 3000×
g for 5 min. The cells were fixed in 2.5% glutaraldehyde in a 0.1 M cacodylate buffer (pH 7.2) and kept overnight at 4 °C. The sample was then carefully rinsed with a 0.1 M cacodylate buffer (pH 7.2) several times. The final cells were fixed with 1% OsO
4 (osmium tetroxide) in the same buffer for 60 min, rinsed carefully with a 0.1 M cacodylate buffer (pH 7.2) several times, dehydrated in graded ethanol (from 50 to 100%), washed in propylene oxide, and infiltrated for 6 h in a 1:1 mixture of propylene oxide and epoxydic resin. The cells were then embedded in the resin. Thin sections were obtained with an ultramicrotome (Ultracut UC6; Leica Microsystems GmbH, Wetzlar, Germany) and stained with uranyl acetate and lead citrate. The ultrathin sections were then observed with a transmission electron microscope operating at 80 kV [
63].
3.5. Scanning Electron Microscopy (SEM)
The specimens were fixed with 2.5% glutaraldehyde in 0.1 M cacodylate at pH 7.2 for 1 h or overnight. Then, they were washed three times in 0.1 M cacodylate, followed by post-fixing treatment in 0.1 M cacodylate with 1% OsO
4 for 20 min. The samples were then washed three times in 0.1 M cacodylate and dehydrated with 50%, 70%, 90%, and 100% ethanol. After dehydration, the samples underwent critical point drying, and were mounted onto metal stubs then sputter-coated with gold and an ion coater. The prepared samples were examined with an FE-SEM S4800 scanning electron microscope [
64].
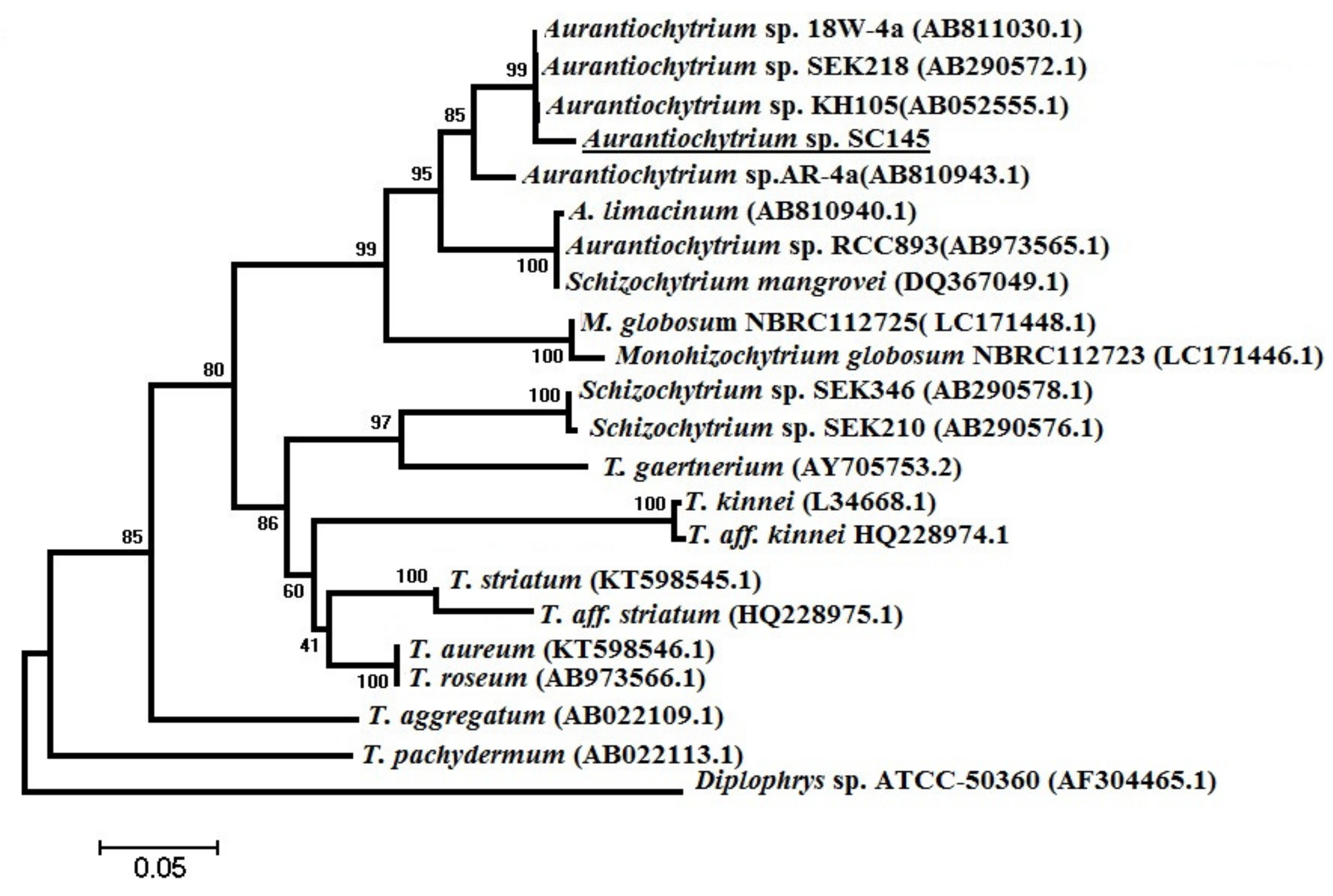
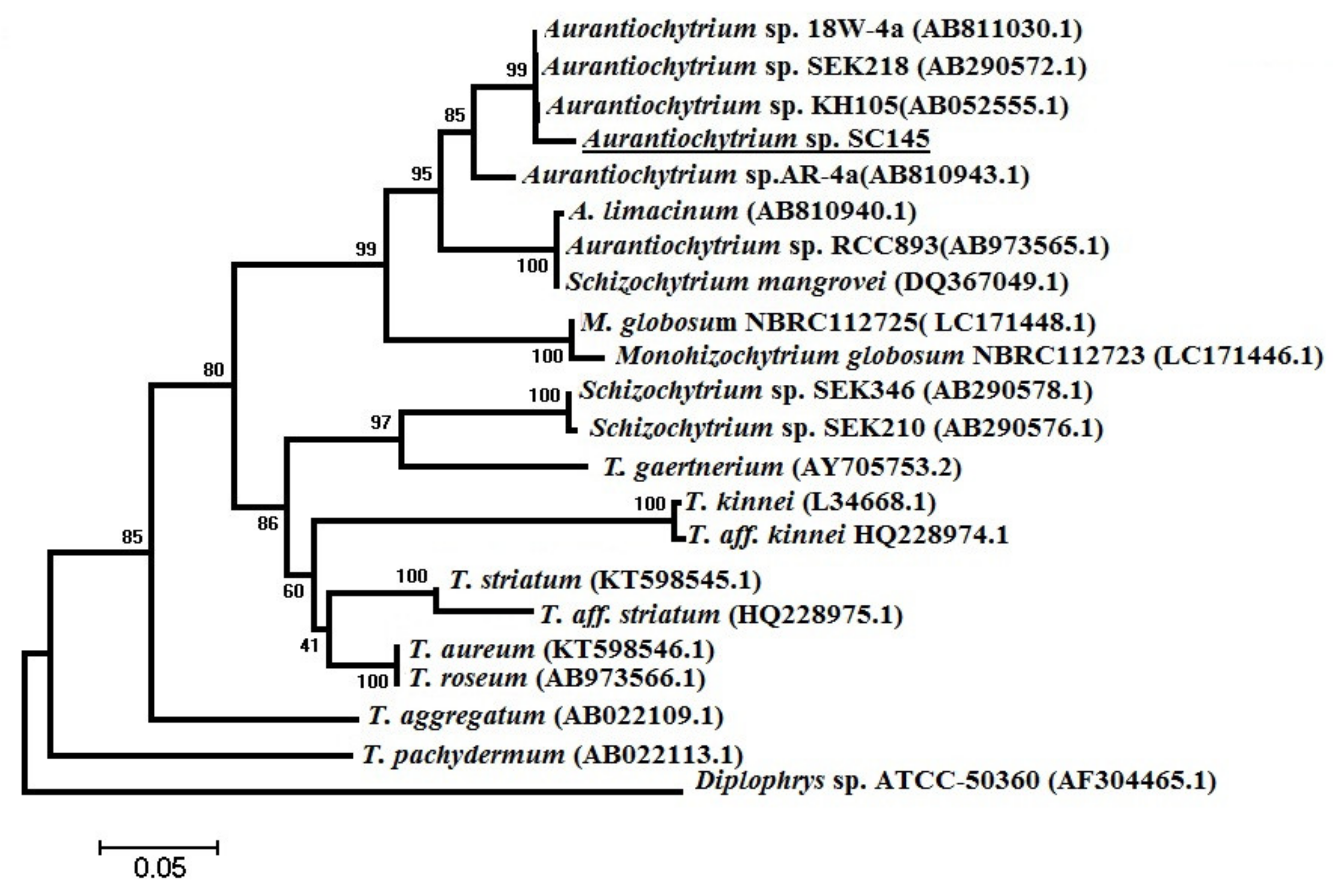
3.6. Phylogenetic Analysis
The scientific name of the marine microalgae strain of
Aurantiochytrium sp. SC145 was identified by comparing the nucleotide sequences of a partial 18S rRNA gene. The specific FA1/RA1 primer pair was utilized to amplify the partial 18S rRNA gene of the marine microalgae genera of
Aurantiochytrium, Schizochytrium, and
Thraustochytrium with a predicted size of 650 bps and sequences of FA1: 5′-AAAGATTAAGCCATGCATGT-3′ and RA1: 5′-AGCTTTTTAACTGCAACAAC-3′, as published by Mo et al. [
65]. The PCR reaction mixture had a total volume of 20 μL, consisting of 2 μL of a green buffer (2X, Thermo Fisher Scientific, Waltham, MA, USA), 1 μL of each primer (10 pmol/μL), 2 μL of a DNA template (50–100 ng/μL), 0.5 μL Taq polymerase (2 u/μL), 1.5 μL dNTP, 0.5 μL MgCl
2 (50 mM), and 11.5 μL deionized water. PCR amplification was performed with a Veriti
® 96-well thermal cycler (Applied Biosystems
®, Life Technology, Thermo Fisher Scientific, Waltham, MA, USA) in a cycling program of denaturation for 3 min at 95 °C and 35 amplification cycles of 1 min at 94 °C, 1 min at 50 °C, and 1 min at 72 °C, with a 10 min extended elongation step. PCR production was visualized on an ethidium bromide-stained 1.2% agarose gel. The 18S rRNA gene sequences of species belonging to the genera
Aurantiochytrium, Schizochytrium, Thraustochytrium, and
Monohizochytrium, and species belonging to the genus
Diplophylus registered in the GeneBank database were used as outgroups to construct the phylogenetic tree [
66]. PCR products were precipitated with ethanol and amplified with a BigDye Terminator v3.1 cycle sequencing kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s recommendations. Sequence analysis was conducted using standard nucleotide BLAST, blastn (
https://blast.ncbi.nlm.nih.gov/ (accessed on 21 July 2022)), a nucleotide collection (nr/nt) database, and ClustalX (1.81) and MEGA X software [
67].
3.7. Methods for Determining Optimal Culture Conditions in Erlenmeyer Flask
In order to determine the optimal culture conditions for the SC145 strain in a 250 mL Erlenmeyer flask, the effects on algal growth of Bajpai, M1, and GPY nutritional media; glucose, maltose, maltodextrin, and starch carbon sources; 1, 3, 5, 7, and 9% glucose levels; yeast extract, (NH4)2SO4, CH3COONH4, and NaNO3 nitrogen sources; 0.5, 1, 1.5, and 2% concentrations of yeast extract; and temperatures of 15, 20, 25, 28, 30, and 35 °C were studied. After 120 h of cultivation, samples were taken to evaluate the parameters of cell density, DCW, and lipid content.
3.8. Methods for Determining Optimal Culture Conditions in a 30 L Fermentor
To determine the optimal culture conditions of the SC145 strain in a 30 L fermentor, the effects on algal growth of glucose concentrations (3, 6, 9, and 12%) and nitrogen sources (yeast extract and industrial yeast extract) were studied. After 120 h of cultivation, samples were taken for evaluation of the parameters of cell density, DCW, and lipid content.
3.9. Extraction and Determination of Total Carotenoid and Astaxanthin
The total carotenoid content was determined as described by Furlan et al. [
68]. First, 1 g of fresh biomass was ground using 10 mL 90% acetone with a mortar and pestle. This mixture was transferred to tubes enveloped by silver foil. After washing the porcelain mortar and pestle, the second step was repeated. The samples were kept in the cold for at least 2 h, and after extraction or centrifugation at 10,000 rpm at 4 °C for 10 min the mixture was then filtered using filter paper to obtain the supernatant. Then, 10 mL of this solution was poured into a 90% acetone solution and placed in a bottle of penicillin. The final solution was light yellow. Note that the extraction was carried out in the dark and cold. The solution was studied using a spectrophotometer to measure the absorbance (OD) at wavelengths of 477 nm and 480 nm.
The total carotenoid content (μg/g of DCW) was calculated with the formula:
where A477 nm is the optical density absorbed at a wavelength of 477 nm, V
extract is the extract volume (mL), DF is the dilution factor (final volume divided by the initial volume), 0.2 is the value of the 1 μg/mL carotenoid solution measured at A477 nm, and Wsample is the sample weight (g).
The astaxanthin concentration (mg/L) was calculated with the formula [
69]:
where A480 nm is the optical density absorbed at a wavelength of 480 nm, Va (mL) is the volume of the solvent, Vb (mL) is the volume of the algal sample, and f is the dilution ratio.
3.10. Extraction and Determination of Total Protein and Carbohydrate Contents
The total protein content was determined according to the method described by Cakmak et al. [
70] and Bradford [
71]. The carbohydrate content was determined according to the method described by Sun et al. [
72].
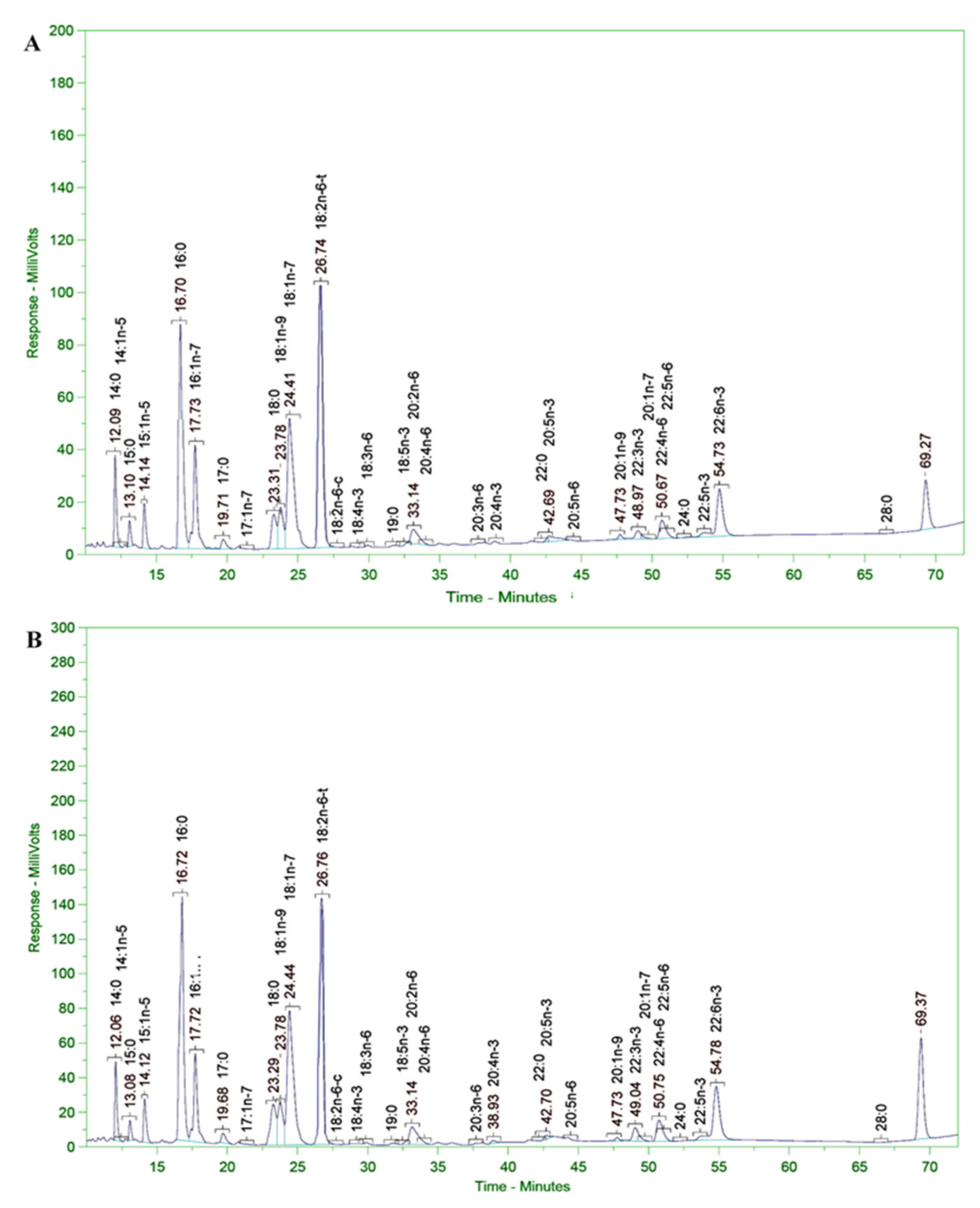
3.11. Analysis of Fatty Acid Composition of Microalgal Biomass
The fatty acid composition of the microalgal biomass (in terms of PUFAs in the alkyl ester and free forms) was determined according to the LFOD-TST-8444 (GC–FID) method conducted at SGS Vietnam Ltd. The fatty acid composition and content were analyzed using an HP-6890 gas chromatograph coupled with an HP-5MS Agilent 5973 mass selective detector column (0.25 m × 30 m × 0.25 mm); the carrier gas was He and the temperature program was 80 °C for 1 min, increased to 150 °C at a rate of 4 °C/min, then increased to 260 °C and held for 10 min at a rate of 10 °C/min. The WILEY275. L and NIST 98. L mass spectrum libraries were used according to the German ISO/FDIS 5590:1998 standard [
73].
3.12. PUFAs Rich in Omega 3–6 Fatty Acids in the Free Form (FFA)
The total lipid extraction was carried out as described by Bligh and Dyer [
74] with some modifications described by Dang et al. [
2]. Twenty grams of dried biomass was ground using a porcelain mortar and pestle with distilled water (at 10% of the total volume or available water in fresh biomass) for 45 min. Then, the mixture was placed into a Pyrex bottle and n-hexane was added at a 1:10 (
w:
v) ratio of biomass–n-hexane. The mixture was continuously stirred at 70 °C for 3 h. After the reaction, the mixture was allowed to cool to room temperature, then centrifuged at 3000 rpm for 5 min in order to collect the total lipids in the upper layer. Oil was obtained following the removal of the n-hexane.
Ten grams of total lipids were added to 1.8 g of NaOH, 4.4 mL of distilled water, and 21 mL of ethanol. The mixture was heated to 70 °C and continuously stirred for 3 h. After finishing this step, the mixture was added to 20 mL of 3% NaCl and centrifuged at 6000 rpm for 40 min at room temperature to remove the unsaponifiable fraction. The saponification fraction was added to a solution of HCl until the pH reached 2, and then centrifuged at 6000 rpm for 40 min. Then, n-hexane was added to the solution in order to obtain free fatty acids (FFAs). FFAs were collected after the removal of n-hexane using a rotary evaporator at 70 °C.
The obtained FFAs were then added to a mixture of urea and ethanol at the FFA–urea–ethanol ratio of 1:4:36 (w:w:v). The mixture was completely dissolved in a magnetic stirrer at 60 °C for 10 min and then kept at 4 °C for 15 h. After the complexing process, the mixture was divided into two phases—urea and non-urea complexing—and separated with filter paper. The non-urea complexing phase was completely removed using ethanol and washed with warm water (at 60 °C) to eliminate redundant urea. Then, the mixture was added to a mixture of n-hexane and omega 3–6 PUFAs; FFAs were collected after removing n-hexane with a vacuum rotary evaporator at 70 °C.
3.13. PUFAs Rich in Omega 3–6 Fatty Acids in the Alkyl Ester Form (FAAE)
Twenty grams of dried biomass was methylated in a mixture of 10% HCl in methanol and dichlomethane at a biomass–methanol–dichlomethane ratio of 1:10:5 (
w:
v:
v). The mixture was heated at 70 °C for 3 h and then mixed well. After the reaction, the mixture was allowed to cool to room temperature. Then, the mixture was added to a 40 mL solution of saturated NaCl and n-hexane. The mixture was shaken and the n-hexane phase containing the TFA was collected. The TFA content was obtained after removing the n-hexane with a rotary evaporator. The obtained TFA sample was added to a mixture of urea and methanol at a TFA–urea–methanol ratio of 1:4:20 (
w:
w:
v). The mixture was completely dissolved in a magnetic stirrer, heated at 60 °C for 10 min, and complexed at 4 °C for 15 h. After the complexing process, the mixture was divided into two phases consisting of urea and non-urea complexing, before being separated using filter paper. The non-urea complexing phase was completely removed with methanol, and washed with warm water (at 60 °C) to eliminate the redundant urea. Then, n-hexane was added to the mixture to dissolve the omega 3–6 fatty acids. Finally, n-hexane was evaporated with a vacuum rotary evaporator at 70 °C to obtain the omega 3–6 PUFA mixture in the alkyl ester form (FAAEs) [
75].
3.14. Extraction and Purification of ALA, LA, EPA and DHA
The ALA, LA, EPA, and DHA polyunsaturated fatty acids were extracted and purified as described by Hang et al. [
76]. The structures of purified compounds were confirmed with nuclear magnetic resonance (NMR) spectroscopy. NMR experiments were performed using a Bruker Avance e 500 MHz spectrometer (Bruker, Karlsruhe, Germany) at operating frequencies of 500 MHz (
1H-NMR), at the Center for Apply Spectroscopy, Institute of Chemistry, Vietnam Academy of Science and Technology, Hanoi, Vietnam.
Docosahexaenoic acid (DHA): Yellow oil.
1H NMR (500 MHz, CDCl
3, δ, ppm, J/Hz): 5.41–5.36 (12H, m, H-4,5, 7, 8, 10, 11, 13, 14 16, 17, 19, 20), 2.84–2.80 (10H, m, H-6, 9, 12, 15, 18), 2.41 (4H, m, H-2, 3), 2.08 (2H, m, H-21), 0.97(3H, t, J = 7.5, H-22) [
77].
Eicosapentaenoic acid (EPA): Colorless oil.
1H-NMR (500 MHz, CDCl
3, δ, ppm, J/Hz): 0.97 (3H, t, J = 7.7; H-20), 1.66 (2H, m, H-3), 2.08 (2H, m, H-19), 2.13 (2H, m, H-4), 2.29 (2H, t, J = 7.3; H-2), 2.86–2.81 (8H, m, H-7, 10, 13, 16), 5.40–5.30 (10H, m, H-5, 6, 8, 9, 11, 12, 14, 15, 17, 18) [
78].
Alpha-linolenic acid (ALA): Colorless oil
1H NMR (500 MHz, CDCl
3, δ ppm, J/Hz): 11.4 (1H, br. s, -COOH), 5.3 ~ 5.4 (m, 6H, H-9, 10, 12, 13, 15, 16), 2.8 (4H, t, H-11, 14), 2.35 (2H, t, H-2), 2.06 (4H, m, H-8, 17), 1.63 (2H, m, H-3), 1.32 (8H, m, H-4, 5, 6, 7), 0.98 (3H, t, H-8) [
79].
Linoleic acid (LA): Colorless oil;
1H-NMR (500 MHz, CDCl3, δ ppm, J/Hz): 5.50–5.30 (4H, m, H-9, 10, 12, 13), 2.77 (2H, t, J = 6,5 Hz, H-11), 2.30 (2H, t, J = 7.5 Hz, H-2), 2.06–2.02 (4H, q, J = 7.0 Hz, H-8, 14), 1.62 (2H, m), 1.37–1.25 (14H, m), 0.89 (3H, t, J = 7,0 Hz, H-18) [
78].
3.15. Acute Oral Toxicity Study
Acute toxicity and LD
50 (50% lethal dose) values were evaluated as described by the guidelines of the World Health Organization [
80] and the Organization for Economic Co-operation and Development (OECD) [
81]. Ninety-six white Swiss mice (male and female, weights ranging from 18 to 22 g) were randomly divided into 12 groups with 8 mice per group and treated with PUFAs rich in omega 3–6 fatty acids in the form of FFA or FAAE at different dosages (3.82, 6.11, 8.40, 10.69, 12.98, and 15.35 g/kg body weight/day), as described previously [
15]. Mortality or signs of morbidity in mice in each group were recorded over 72 h. Then, animals were continuously monitored until the end of the 7th day after the first oral administration.
3.16. Subchronic Oral Toxicity Study
Subchronic toxicity was evaluated as described in the guidelines of the WHO [
56] and OECD [
81]. Forty white Wistar rats (male and female, weights ranging from 160 to 180 g) were randomly divided into 5 groups with 8 rats per group. Groups 1 and 2 were exposed to doses of 78.5 and 353.3 mg/kg/day of FAAEs, respectively. Groups 3 and 4 were exposed to doses of 78.5 and 353.3 mg/kg/day of FFA, respectively. An untreated group (control group) received normal saline at a dosage of 1.00 mL/kg/day. All experimental and control groups were evaluated over 90 days. The hematological parameters, biochemical parameters, and liver and kidney functions of the rats were analyzed before (D0) and after oral administration for 45 days (D45) and 90 days (D90), as described by Thom and Hong [
15].
3.17. DPPH Assay
The 2,2-diphenyl-1-picrylhydrazyl (DPPH) assay was used as described by Alam et al. [
82]. Firstly, PUFAs in the FAAE and FFA forms, LA, ALA, EPA, and DHA were prepared in dimethyl sulfoxide (DMSO). DMSO was also used as a control group. Ascorbic acid, a potent reducing and antioxidant agent, was used as a positive control. A DPPH solution was dissolved in methanol to a concentration of 0.5 mM. Then, 100 μL of each sample or DMSO was added to a 100 μL DPPH solution and left to stand for 30 min in the dark at room temperature. After incubation, the absorbance of the reaction mixtures was measured at an absorption wavelength of 517 nm using a microplate reader (Thermo Fisher Scientific, Inc., Waltham, MA, USA). The percentage of the scavenging effect on the DPPH inhibition was calculated with equation:
where A
0 is the absorbance of the control and A1 is the absorbance of the samples. All experiments were performed in triplicate. The results are given as IC
50 corresponding to the concentration achieving 50% inhibition.
3.18. AChE Inhibitory Activity Assay
The AChE inhibitory activities of PUFAs in the FAAE and FFA forms, LA, ALA, EPA and DHA were evaluated using an acetylcholinesterase inhibitor screening kit (Sigma, St. Louis, MO, USA) according to the provided instructions. Galantamine, which is used for treatment of Alzheimer’s disease, was used as a positive control. Absorbance was measured at 412 nm with a microplate reader (Thermo Fisher Scientific, Inc., Waltham, MA, USA). All experiments were performed in triplicate. The results are given as IC
50. The percentage inhibition of AChE activity was calculated as follows:
where ΔA
Test Cpd is the A
412 value of a sample well at 0 min subtracted from the A
412 value of the same well at 10 min, and ΔA
No Inhibitor is the A
412 value of the No Inhibitor control well at 0 min subtracted from the A
412 value of the No Inhibitor control well at 10 min.
3.19. Cell Culture and Treatment
C6 rat glial cells (ATCC, CCL-107™) were obtained from Dr. Duong Hoang Nguyen, Center for Soft Matter and Biological Physics, Center for High Technology Development, Vietnam Academy of Science and Technology. The cells were cultured in high-glucose Dulbecco’s Minimum Essential Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% (v/v) penicillin/streptomycin under 5% CO2 at 37 °C.
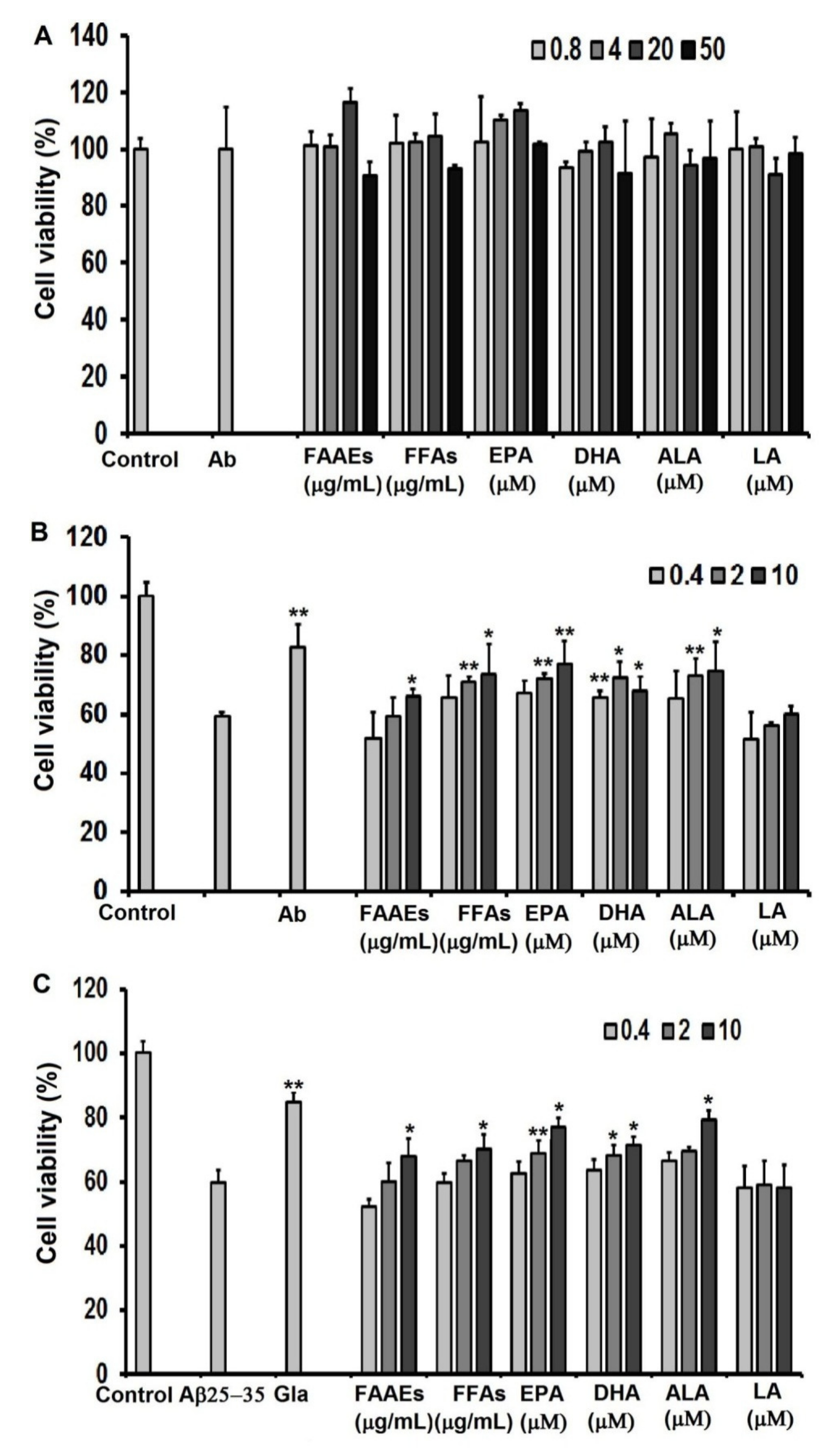
For assessment of the protective ability of PUFAs in the FAAE and FFA forms, EPA, DHA, LA, and ALA against oxidative stress caused by H2O2, C6 cells were cultured in high-glucose DMEM in 96-well culture plates at a density of 0.5 × 105 cells/well for 24 h. Then, the cells were incubated with PUFAs in the FAAE and FFA forms (0.4, 2 and 10 µg/mL), EPA, DHA, LA, ALA (0.4, 2, 10 μM), or ascorbic acid (20 µg/mL) as a positive control for another 24 h, followed by 1 h of incubation with a H2O2 solution (10 μM). The samples’ cytoprotective effects against oxidative stress induced by H2O2 on C6 cells were assessed in terms of the cell survival rate using a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay.
For the assessment of neuroprotection against Aβ
25-35-induced cytotoxicity in the C6 cell line provided by PUFAs in the FAAE and FFA forms, EPA, DHA, LA, and ALA, cells were cultured for 24 h in high-glucose DMEM in 96-well culture plates at a density of 0.5 × 10
5 cells/well. Then, the cells were incubated with PUFAs in the FAAE and FFA forms (0.4, 2 and 10 µg/mL), EPA, DHA, LA, ALA (0.4, 2 and 10 µM) or galantamine (1 µg/mL; Sopharma AD, Sofia, Bulgaria) as a positive control for another 24 h, followed by 24 h of incubation with an Aβ
25-35 protein (20 μM) [
56]. Cell viability was determined using the MTT assay method.
3.20. MTT Assay
After treatment, C6 cells were incubated with 5 μL MTT solution (5 mg/mL) at 37 °C and 5% CO
2 for 4 h. After 4 h of incubation, the media were removed and purple MTT formazan crystals were dissolved with the addition of 100 μL of DMSO to each well. The absorbance was measured at 570 nm with a microplate reader (Thermo Fisher Scientific, Inc., Waltham, MA, USA). In this experiment, each concentration was assessed in triplicate. The cell survival rate was calculated as follows:
where A
0 is the absorbance of control and A
1 is the absorbance of the samples. All experiments were performed in triplicate.
3.21. Statistics Analysis
All experiments were performed in triplicate. Data are expressed as the mean ± standard error of the mean (SEM). Statistically significant differences for control-group comparisons were evaluated using Student’s t test. Differences were considered statistically significant at p < 0.05. Experimental data were processed using Excel software and statistically analyzed via one-way ANOVA and Duncan’s post hoc test at a significance level of p < 0.05.
4. Conclusions
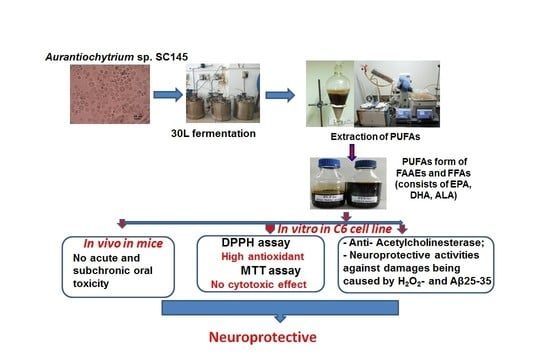
From samples collected in May–June 2021 in the coastal area of Sand Cay (Son Ca) Island, Vietnam, Aurantiochytrium sp. SC145 heterotrophic marine microalga (capable of producing high levels of PUFAs and other components) was isolated and stably cultured under laboratory conditions. The most suitable conditions for the biomass cultivation of the SC145 strain for use as a raw material for PUFA extraction were found to be the M1 medium containing 3% glucose and 1% yeast extract at a temperature of 28–30 °C on the flask scale, and the M12 medium containing 9% glucose and 1% industrial yeast extract on the 30 L fermentor scale. In the 30 L fermentor, the dry cell weight, lipid content, protein content, and carbohydrate content reached their highest values of 31.18 ± 2.63 g/L, 25.29 ± 1.43%, 7.93 ± 0.17%, and 15.21 ± 0.02% of DCW, respectively. In addition, carotenoid and astaxanthin contents reached 143.67 ± 0.35 and 8.07 ± 0.17 µg/L, respectively. The fatty acid composition of the SC145 strain biomass mainly comprised omega 3–6–9 fatty acids, which accounted for 51.60% of TFA. The PUFAs extracted from the dry biomass of the Aurantiochytrium sp. SC145 strain in the free and alkyl ester fatty acid forms rich in omega 3–6–9 fatty acids (with main constituents of EPA, DHA, and ALA) showed antioxidant and AChE inhibitory properties and neuroprotective activity against H2O2- and amyloid-ß protein fragment 25–35 (Aβ25-35)-induced damage in C6 cells. The mixtures of PUFAs in the FFA and FAAE forms were found to be safe according to acute and subchronic oral toxicity tests in the animal model. This study on PUFAs rich in omega-3–6–9 acids present in the Aurantiochytrium sp. SC145 strain contributes to the general understanding of the exploitation and development of drugs and functional foods based on marine microalgae. These PUFAs were also shown to be promising ingredients for the prevention and treatment of Alzheimer’s disease.
The neuroprotective mechanism of PUFA mixtures in the FFA or FAAE forms (including the main effects caused by the individual fatty acids ALA, EPA, and DHA) requires further elucidation in future studies.


 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}