1. Introduction
An unknown series of pneumonia cases were identified in December 2019 and emerged in Wuhan, Hubei province, China. According to the World Health Organization (WHO) office in China, the infected clusters were initially reported on the 31 December 2019. A novel type of coronavirus was identified by Chinese authorities on 7 January 2020, which caused a new, infectious, respiratory disease called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The identified coronavirus expressed significant differences compared to other respiratory pathogens, such as severe acute respiratory coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV), influenza, adenovirus, and avian influenza. The origin of SARS-CoV-2 remains unclear, but RaTG13, the coronavirus isolated from bats, expressed a genetic similarity close to SARS-CoV-2. Therefore, bats are considered the origin of this disease [
1]. However, the transmission mechanism of the virus from bats to human beings remains unclear.
Coronaviruses that belong to the
Coronaviridae family are enveloped, non-segmented, positive-sense, single-stranded RNA viruses in the
Nidovirales order [
2]. The abovementioned viruses infect humans and other mammals to a considerable extent. Furthermore, Chinese horseshoe bats were identified as natural reservoir hosts for SARS-CoV [
3]. SARS-CoV was therefore controlled using conventional methods such as travel restrictions and the isolation of patients. The infection mechanism of SARS-CoV-2 is not yet fully understood, including the reasons why human beings are the principal hosts of the virus and how it escapes their innate immune systems. Moreover, the interaction between the human Toll-like receptor (TLR), viral antigens, the mechanism of pro-inflammatory cytokine production, and its effect on important human organs, are not yet fully understood. However, the viral entry mechanism has been identified in the literature, and it invades the human body through the respiratory system using respiratory droplets via sneezing and coughing [
4]. SARS-CoV-2 consists of a protein capsid covered by glycoprotein with anchored spike proteins. These spike proteins initiate viral entry into the target cells. The entry of SARS-CoV-2 into the host cell is an important factor required to determine the infectivity and pathogenesis of the virus [
5]. Therefore, it is determined to be a key target for host immune monitoring and human intervention strategies [
6]. The SARS-CoV-2 spike protein initially binds to the cell surface receptor called angiotensin-converting enzyme 2 (ACE-2), and this is known as viral attachment; subsequently, it enters the endosome and, finally, the viral membrane fuses with the lysosomal membrane [
7]. However, the continuous mutation of the SARS-COV-2 spike protein has made the development of an antiviral drug for viral infections using a spike protein inhibition strategy challenging. Therefore, in the present study, we identified that viral replication occurring in the host cell is the most successful way to control the viral load in the host.
Coronaviruses contain RNA viral genomes that are 26 to 32 kb in length. The newly sequenced SARS-CoV-2 genome was submitted to the NCBI genome database under accession number NC_045512.2, and the size was ~29.9 kb [
8]. SARS-CoV-2 consists of 13 to 15 open reading frames (ORFs), including 12 functioning ORFs. The ORFs are arranged as nucleocapsid proteins. When considering the whole genome of SARS-CoV-2, it encodes for polyproteins that consist of ~7096 residues. It contains many structural and non-structural proteins (NSPs) as well as ORF1a and ORF1b, which encode for non-structural proteins and are mainly responsible for the nucleotide content of the genome. ORFs 1a and 1b encode the polyproteins pp1a and pp1b, respectively, and gene 1b employs the ribosomal frameshift mechanism to encode pp1ab. The virally encoded proteases cleave these polyproteins and produce 16 NSPs and the rest of the genomes responsible for the creation of structural proteins. These proteins play a pivotal role in viral-entry fusion, replication, and survival in host cells. Thus, these gene products are considered the main drug or vaccine targets [
9]. Polyprotein processing is mainly conducted by the 3-chymotrypsin-like protease (3CL
pro) and papain-like protease (PL
pro). The polyprotein is cleaved at 11 distinct sites by 3CL
pro. This leads to the production of NSPs that are important in the process of viral replication [
10]. A key role is played by 3CL
pro in SARS-CoV-2 replication in the host cell. According to the previous studies, high-throughput studies and structure-based activity analysis confirmed the value of the potential inhibitors of the activity of 3CL
pro against SARS-CoV and MERS-CoV, which successfully inhibited virus replication activity [
11,
12,
13]. Therefore, the 3CL
pro of SARS-CoV-2 is considered a potential drug candidate. The PL
pro of SARS-CoV and SARS-CoV-2 expresses an 83% sequence identity and diverges from MERS-CoV. However, the host substrate preference of PL
pro is the difference between these two strains. Furthermore, the PL
pro of SARS-CoV-2 cleaves the ubiquitin-like interferon-stimulated gene 15 protein (ISG15), and PL
pro of SARS-CoV predominantly targets the ubiquitin chain [
14]. ISG15 regulates various cellular signaling pathways and host immune responses. Therefore, 3CL
pro and PL
pro are identified as potential drug targets for the inhibition of SARS-CoV-2.
Marine algae confront extreme environmental conditions, and their metabolism consists of a biochemical process that absorbs nutrients and converts them into materials that are important for survival in these specific environmental conditions [
15]. These accumulated defense metabolites express an elevated potential to develop novel therapeutic agents [
16]. Among these secondary metabolites, phlorotannins, such as polyphenolic compounds, have been identified in the research as potential antiviral agents for various types of viruses, including enveloped [
17,
18,
19] and non-enveloped viruses [
20,
21,
22], which exert their antiviral activity through inhibiting vital viral proteins.
Thus, the present study aims to inhibit SARS-CoV-2 through 3CLpro and PLpro using marine natural products isolated from marine algae. Molecular docking was utilized for the initial screening of selected natural products (MNPs) based on the 3CLpro and PLpro protein structures. Moreover, the resulting compounds were isolated and used for biological assays for further confirmation of the inhibition activity. In the present study, we utilize an in vitro assay kit and introduce a simplified method to determine the inhibitory activity of the compounds using a cell-based assay. To the best of our knowledge, this is the first study that assesses the function of marine natural products in relation to 3CLpro and PLpro of SARS-CoV-2 as a multi-target approach.
3. Discussion
The emergence of COVID-19 in December 2019 resulted in a pandemic that was responsible for millions of deaths. The health sector developed several vaccines and attempted to repurpose US Food and Drug Administration (FDA)-approved drugs. However, these attempts were not sufficient to eliminate COVID-19, and most vaccines failed due to the occurrence of viral mutations. This emphasized the development of an antiviral agent to be used against SARS-CoV-2. In the various studies that provide insights into the development of an antiviral agent for SARS-CoV-2, such as computational studies, in vitro, in vivo, and clinical trials can be observed. However, the authors of the present study attempted to develop an antiviral agent for several drug targets, and this multi-target approach was more successful than inhibiting individual drug targets.
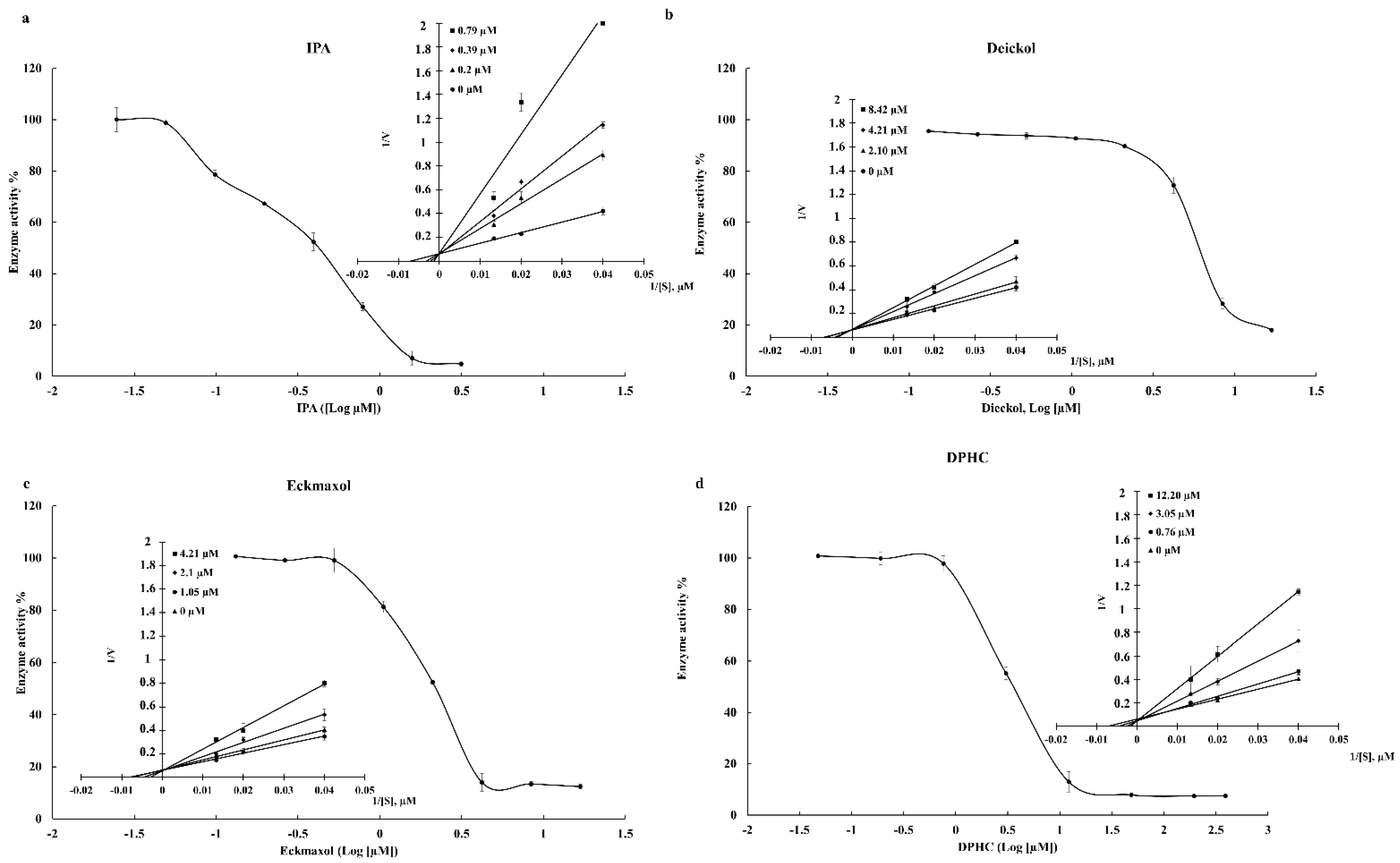
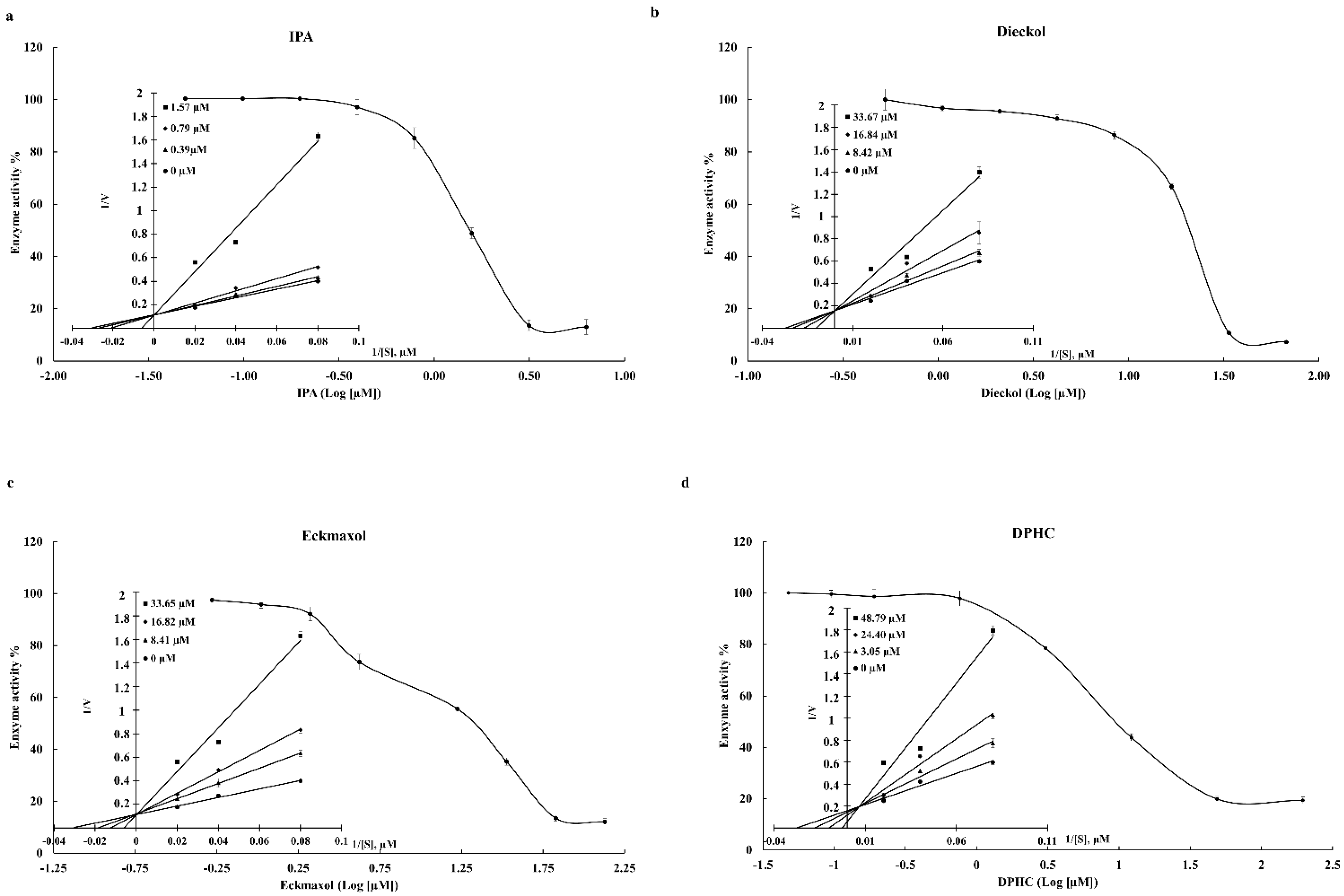
The present study used Discovery Studio software to perform the initial screening. The “Prepare protein” tool of the software successfully prepared the structures of 3CLpro and PLpro using 6LU7 and 7CMD structures of PDB. The “Prepare protein” tool was utilized to solve the most common problems, such as removing alternate conformations, removing heteroatoms, hydrogen additions, and correcting missing or incorrectly specified residues. The energy minimization of target receptor proteins was performed using the “Protein minimization” tool. DS provides three options to prepare the binding site of the receptor protein: “based on the PDB site records”, “from receptor cavities”, and “form current selection”. The present study used the final tool based on the available ligands of the crystal structures and previously published data. The results of the molecular docking reveal that compounds IPA, DPHC, Dieckol, and Eckmaxol showed the greatest binding affinity to the active sites of 3CLpro and PLpro. Therefore, these compounds were selected for further analysis.
The current study used an in vitro assay kit that was specially designed to determine 3CL
pro and PL
pro inhibitions. The isolated polyphenolic compounds were utilized to determine inhibitory activity against 3CL
pro and PL
pro. All of the tested compounds exhibited dose-dependent inhibitory activity against 3CL
pro and PL
pro. GC376 (IC
50 0.4231 µM) and GRL0617 (IC
50 1.5 µM) were used as positive controls against 3CL
pro and PL
pro, respectively [
14,
25,
26]. Among these compounds, IPA presented remarkable inhibitory activity against both proteases 3CL
pro and PL
pro with IC
50 values of 0.4814 and 1.4048 µM, respectively. Furthermore, DPHC, Dieckol, and Eckmaxol also exhibited significant inhibitory activity. These results strengthen the in silico outcomes. The binding affinity of these compounds was evaluated based on the active site of the protease enzymes. Thus, a Lineweaver–Burk plot was created to determine the inhibitory patterns, according to the enzyme kinetic results, by increasing K
m and constant V
max with the increasing compound concentration. This confirmed that all compounds were competitive inhibitors. These results also confirm the in silico results that all the compounds are bound to the active site of the protease enzymes.
3CL
pro and PL
pro inhibitions were evaluated further using a cell-based inhibitory assay. This method did not require protein purification and was close to the natural physiological state. Thus, this method can be successfully used to strengthen the previous results. The in-frame construction of 3CL
pro or PL
pro with the substrate (the peptide sequence that contained a cleavage site) and firefly luciferase gene was designed as the plasmid transfected into the Vero E-9 cells. Normally, this method should be performed as a co-transfection process using the indicated vector. However, it can affect the accuracy of the final results. Therefore, we used a vector that contained both renilla and firefly luciferases. Furthermore, renilla luciferase was expressed independently from the protease gene or firefly luciferase. Thus, the luminescence from the firefly luciferase can be normalized using renilla luciferase expression. The luciferase protein that is bound to another protein with a value greater than 33 kDa remarkably decreased the luciferase activity [
27]. Thus, a peptide sequence that contains a cleavage site for 3CL
pro or PL
pro was used for binding the protease enzyme with the firefly luciferase. Thus, the inhibitory activity of MPNs downregulated the luciferase activity [
28] and did not interfere with renilla luciferase activity. These results also exhibit the significant inhibitory activity of MNPs against 3CL
pro and PL
pro. However, the IC
50 values of the results present a significant difference compared to the in vitro assay kit. The permeability of the compounds and cell membrane could be the reason for the results we obtained [
29]. Furthermore, the results confirm that IPA is the most efficient inhibitor of both proteases. SARS-CoV-2 can affect cells in numerous organs and systems present in the human body and mostly infected the upper and lower respiratory tracts. Among them, the lungs are the most infected organ due to the presence of ACE-2 receptors [
30]. Therefore, the CPE of the virus on these tissues and the reduction in CPE are important factors. The CPE reduction evaluation is a commonly used assay format for screening antiviral agents. The cell death caused by viral infection was measured using this assay [
31]. The CPE reduction effect of all compounds against the virus was evaluated, and among them, IPA, DPHC, and Eckmaxol exhibited remarkable CPE reduction activity. However, further studies are required to reveal and confirm the exact behavior of these compounds in an in vivo model.
5. Methods and Materials
5.1. Chemicals and Reagents
Dimethylsulfoxide (DMSO) and all the organic solvents (HPLC grade) used in the experiments were purchased from Sigma-Aldrich (St Louis, MO, USA). The in vitro inhibition assay kits for 3CLpro and PLpro were purchased from the AMSBIO company (Madrid, Spain). Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Gibco/BRL (Burlington, ON, Canada)), 10% fetal bovine serum (FBS) and 5% penicillin/streptomycin were purchased from WELGENE (Gyeongsan, Korea), and 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma Aldrich (St. Lois, MO, USA). The Nano-Glo® Dual-Luciferase® Reporter Assay System was purchased from Promega (Madison, Wisconsin, USA), the genes were purchased from Sino Biological (Beijing, China), and the pcDNA3 RLUC POLIRES FLUC vector was purchased from addgene (Watertown, Massachusetts, USA). The Vero E6 cell line was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA).
5.2. Preparation of Receptors
The protein Data Bank (PDB) (
http://www.pdb.org, accessed on 20 October 2020) was used to obtain the crystal structures of 3CL
pro and PL
pro under PDB ID: 6LU7 and 7CMD, respectively. The molecular docking studies were conducted using Discovery Studio (DS-Client v18.1.100.18065). Briefly, the crystal structure of each protein was downloaded from PDB. The water molecules and heteroatoms were removed and the “Clean protein” tool was used to correct any minor problems, such as missing-side chain atoms, which were added in an extended confirmation. Then, the “Prepare protein” tool was utilized for further preparations of the receptor protein, and the energy was minimized using the “Protein minimization” tool. The crystal structure of 3CL
pro was available in the PDB as a complex with an inhibitor called the Michael-acceptor or N3 inhibitor. The binding site of 3CL
pro was determined based on the abovementioned inhibitor and previous studies [
32]. PL
pro was available in the PDB as a complex with a GRL0617 inhibitor. Therefore, the binding site of PL
pro was determined using GRL0617 and previous studies [
24]. Briefly, the binding site was prepared as a sphere in the ligand-binding site of the crystal structure, and the prepared binding sites of the target proteins were identified by specifying a sphere of a given radius located in the active site. The geometric center of the ligand in the crystal structure was used as the center of the sphere. A Python-enhanced molecular graphics tool (PyMOL, version 2.4.1) was used to calculate the root-mean-square deviation of the atomic position (RMSD) value between the prepared 3CL
pro, PL
pro, and raw 3CL
pro and PL
pro to determine any significant differences in the structures.
5.3. Preparation of Ligands
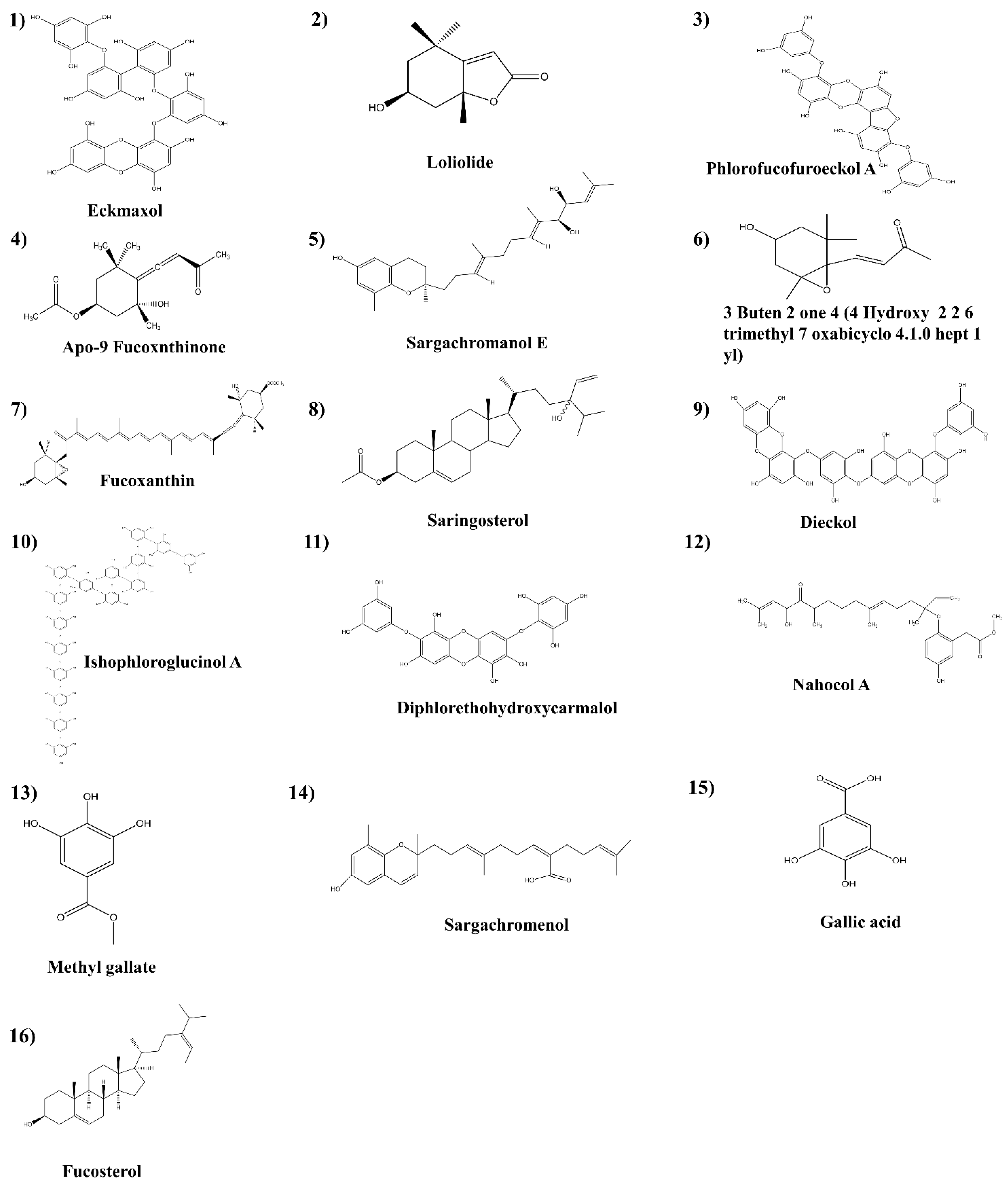
The 3D structure of each compound was generated and the hydrogen atoms were added. The energy of the ligand was minimized using the “Clean geometry” tool and by applying the CHARMm force field. The final ligand structure generated using the “Prepare ligand” tool was optimized using the DS ligand optimization tool, and the energy of the ligands was minimized using the DS minimization tool. The summary of the ligands presented in this study is shown in
Figure 5. (
1. Eckmaxol,
2. Loliolide,
3. Phlorofucofuroeckol A,
4. Apo-9 Fucoxnthinone,
5. Sargachromanol E,
6. 3 Buten 2 one 4 (4 Hydroxy 2 2 6 trimethyl 7 oxabicyclo 4.1.0 hept 1 yl),
7. Fucoxanthin,
8. Saringosterol,
9. Dieckol,
10. Ishophloroglucinol A,
11. Diphlorethohydroxycarmalol.
12. Nahocol A,
13. Methyl gallate,
14. Sargachromenol,
15. Gallic acid,
16. Fucosterol). The ligands that were selected for validation were 16 compounds obtained from marine algae.
5.4. Molecular Docking Analysis
The docking of the selected ligands with prepared proteins was performed using DS. The crystal structure of 3CLpro bound to the N3 inhibitor and PLpro bound to GRl0617 were reproduced, and the RMSD values of the raw crystal structure and docking results were calculated to confirm the accuracy of the process. Initially, flexible docking experiments were performed using the 3D crystal structures of 3CLpro and PLpro. Flexible docking is a fully automated workflow process. The flexible docking protocol permits receptor flexibility during the docking of flexible ligands. The side chains of specified amino acids in the target receptor protein are permitted to move during the docking process. Moreover, the receptor was adapted to different ligands in an induced-fit model. Therefore, flexible docking was performed to determine the suitable orientation of the ligand in the active site of each receptor protein. The results obtained from the flexible docking process were used to calculate the binding energy between each ligand and receptor protein using CHARMm-based energy. The free energy of the complex, ligand, and receptor was used to calculate the free energy of the binding.
Energy binding = Energy Complex − (Energy Ligand + Energy Receptor).
The best four ligands were selected for the biological assays based on the results of the molecular docking process.
5.5. Sample Collection and Extraction
Brown algae Ishige okamurae (IO) and Ecklonia cava (EC) were collected from the coastal area of Seongsan, Jeju, South Korea in February 2019. Ecklonia maxima (EM) was collected from the coastal area of Cape Town, South Africa in 2019 February. The samples were washed 4 times, immediately after being harvested, with running water to remove any salt, attached sand, and epiphytes. The washed seaweeds were stored at temperatures below −70 °C. The frozen seaweeds were lyophilized using a freeze dryer and the dried seaweeds were ground into powder. Sample extraction was performed using 70% ethanol 3 times at room temperature. The subsequent solution was evaporated using a rotary evaporator and the resulting ethanol extract powders of IO (IOE), EC (ECE), and EM (EME) were obtained. IOE, ECE, and EME were dissolved in deionized water and successfully fractionated using n-hexane, chloroform, ethyl acetate, and butanol, respectively. Each resulting fraction was evaporated and the ethyl acetate fractions of IO (IOEA), EC (ECEA), and EM (EMEA) were utilized to isolate the desired compounds. The centrifugal partition chromatography (CPC 240, Tokyo, Japan) and ODS cartridge in the FlashPrep system (C-850 FlashPrep, BUCHI, Switzerland) were utilized to further separate the IOEA, ECEA, and EMEA.
5.6. Isolation of Ishophloroglucin A (IPA) and Diphlorethohydroxycarmalol (DPHC)
Centrifugal partition chromatography (CPC 240, Tokyo, Japan) was utilized to isolate the IPA. The rotor volume was 1 L. The method was continued in a two-phase solvent system, which consisted of n-hexane:ethyl acetate:methanol:water at a 1:9:4.5:6.5
v/v ratio. These solvents were thoroughly mixed and equilibrated in a separatory funnel. The upper organic phase acted as a stationary phase and the lower aqueous phase acted as a mobile phase. The CPC instrument was conditioned until it attained hydrostatic equilibrium, and 500 mg of IOEA was dissolved in 6 mL of a 1:1
v/v water:methanol ration of the CPC solvent system and injected using an isocratic pump (Kromaton). The effluent was monitored at 230 nm and the fractions were collected into test tubes using a fraction collector (3 min for each tube). All the fractions collected from the same compounds were pooled to continue further purification processes. The high-performance liquid chromatography (HPLC) system (Milford, Massachusetts, USA) equipped with a PDA detector was used for further purification. The semi-preparative column HPLC column YMC-Pack ODS-A 10 mm × 250 mm, 5 µm (YMC Co., Ltd., Kyoto, Japan) was used in an isocratic solvent mode (32% acetonitrile with 1% formic acid), and the flow rate was 2 mL/min [
33,
34] (
Supplementary Figure S11)
5.7. Isolation of Dieckol
Dieckol was isolated from ECEA using an ODS cartridge in the Flashprep system (C-850 FlashPrep, BUCHI, Switzerland) equipped with PDA and ELSD detectors. A packing PREP C18, 55–105 µm, 125 Å (Waters, Milford, CT, USA) column was used with a 20 mL/min flow rate. The mobile phase consisted of water and acetonitrile with a gradient method (0 min 90:10
v/v, 0–12 min 90:10
v/v, 12–36 min 85:15
v/v, 36–68 min 80:20
v/v, 68–80 min 0:100
v/v). The fractions were collected based on the results obtained at 230 nm (
Supplementary Figure S12).
5.8. Isolation of Eckmaxol
Eckmaxol was isolated from EMEA using the centrifugal partition chromatography (CPC 240, Tokyo, Japan) method composed of n-hexane:ethyl acetate:methanol:water at a ratio of 3:7:4:6:
v/v. The abovementioned solvents were vigorously mixed and equilibrated to separate two phases at room temperature. The upper organic phase was used as a stationary phase and the lower aqueous phase was used as a mobile phase. The organic stationary phase was filled into the CPC column and rotated at 1000 rpm, and the aqueous mobile phase was pumped into the column in a descending mode at a flow rate of 2 mL/min. The hydrodynamic equilibrium was maintained before injecting the sample, and 500 mg of EMEA dissolved in 6 mL of a 1:1
v/v ratio of water:methanol was injected through the injection valve. The automatic fraction collector was utilized to collect the fractions (6 mL for each tube) under the 230 nm UV detection range. The HPLC system equipped with a PDA detector was used for further purification processes. A YMC-Pack ODS-A 10 mm × 250 mm, 5 µm column with acetonitrile +0.1% formic acid and deionized water + 0.1% formic acid was used as a mobile phase, and the flow rate was 2 mL/min [
35] (
Supplementary Figure S13).
5.9. In Vitro Cleavage Inhibition Assay
The inhibitory activity of the compounds of interest was measured using the “3CLpro and PLpro (SARS-CoV-2)” assay kit (AMSBIO, Madrid, Spain). The compounds were dissolved in dimethyl sulfoxide (DMSO) and diluted into the assay buffer available with the kit. The final DMSO concentration of the highest concentration of each compound used in the assay was lower than 1%. IPA, Dieckol, and Eckmaxol were incubated with 120 ng of the 3CLpro enzyme for 30 min at room temperature with slow shaking. Broad-spectrum antiviral medication GC376 was used as a test inhibitor in the assay, and 50 µM of the fluorogenic substrate was added to each well to determine the 3CLpro inhibitory activity. The diluted concentrations of IPA, Dieckol, and Eckmaxol were treated into each well that contained 1.2 ng of PLpro enzyme and incubated for 30 min with slow shaking at room temperature. The blank well was treated with only 25 µM of fluorogenic substrate and the positive control well contained only PLpro enzyme and fluorogenic substrate to evaluate the PLpro inhibitory activity. The experiment was performed on a 96-well plate. The negative control was the well that only contained 3CLpro or PLpro enzymes, and the fluorogenic substrate used to measure the enzyme activity and blank well contained only substrate. The enhanced fluorescence emission as a result of substrate cleavage was monitored at an excitation of 360 nm and emission of 460 nm using a Synergy HTX multi-mode microplate reader (Winooski, VT, USA).
The IC
50 value of each compound was calculated and the experimental data were fit to a logistic curve using the equation below:
“B” is the fluorescence of the blank (substrate and assay buffer), “P” is the fluorescence of the negative control (substrate and enzyme), and “S” is the fluorescence of the tested sample.
5.10. Enzyme Kinetic Evaluation
The enzyme kinetic mechanism of these isolated compounds was studied with a series of substrate concentrations with various concentrations of inhibitors. The data were plotted in the graph (y axis—1/V and x axis—1/[S]).
5.11. Cell-Based Inhibition Assay
The enzyme kinetic mechanism of the interaction between the isolated compounds and 3CLpro and PLpro was evaluated with various inhibitor concentrations and substrate concentration series.
5.11.1. Cell Culture
Vero-E6 cells were purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). Cells were cultured using Dulbecco’s modified Eagle’s medium (DMEM) (Gibco/BRL; Burlington, ON, Canada) containing 10% FBS and 1% antibiotics under 37 ºC and 5% CO2.
5.11.2. 3CLpro and PLpro Cell-Based Cleavage Inhibition Assay
The 3CLpro and PLpro genes were fused in-frame with a cleavage site and a firefly luciferase gene at the C-terminus. Both genes were purchased from SinoBiological (Taizhou, Zhejiang, China). The 3CLpro gene was amplified using PCR with designed forward and reverse primers 5′-GAGAGAGCGGCCGCATGGCATTCCCATCTGGTAAAGTTGAGG- 3′ and 5′-GAGAGAGGATCCCCTTCCTGAAGCCGCTCTGCAGCACGGCGCTTTGGAAAGTAACACCTGAGCATTGTCTAACAAC-3′, respectively. The PLpro gene was amplified using PCR with designed forward and reverse primers 5′-GAGAGAGCGGCCGCATGGAAGTGAGGACTATTAAGGTGTTTACAACAG-3′ and 5′-GAGAGAGGATCCCGCACATGGCGCCGCCCCTCAGCCTAACTGGTTTTATGGTTGTTGTGTAACTGTTTTCTTTG, respectively.
The forward primer consisted of a NotI restriction site and the reverse primer contained a BamHI restriction site and in-frame gene encoding for the 3CL
pro-cleavage site (AGCGCCGTGCTGCAGAGCGGCTTCAGGAAG) luciferase gene and PL
pro-cleavage site (AGGCTGAGGGGCGGCGCCATGTGC) luciferase gene. The plasmid that contained the 3CL
pro gene was amplified by PCR using the abovementioned primers and cloned into the NotI/BamHI restriction sites of the pcDNA RLUC POLIRES FLUC vector (addgene, Watertown, MA, USA). The recombinant plasmid was transfected into Vero-E6 cells using an X-tremeGENE HP DNA transfection reagent (Sigma-Aldrich, St. Louis, MO, USA). Vero-E6 cells at 80–90% confluence in 24-well plates were transfected with 0.25 µg of total plasmid per 1 well that contained DMEM with 0% FBS and 0% antibiotics. The media was replaced with DMEM that contained 10% FBS and 1% antibiotics following 23 h of incubation (37 °C and 5% CO
2) and the samples were treated. The dual-luciferase reporter assay system (Promega) was utilized to evaluate the expression of firefly luciferase activity after an 18–20 h incubation (37 °C and 5% CO
2) period. The expression level of firefly and renilla luciferases was measured by a Luminometer (
Figure 6).
5.12. Cytopathic Effect (CPE) Reduction Evaluation
The CPE reduction assay was performed according to the previously reported method [
36]. Briefly, the cells were seeded with 5 × 10
4 cells/well in a 96-well plate and incubated at 37 °C for 48 h. A total of 100 µL of the virus was treated in each well using DMEM media that was FBS free and 1% antibiotic to infect a 0.01 multiplicity of infection (MOI) and incubated for an additional 1 h at 37 °C. Subsequently, the infected cells were treated with samples after removing the virus and 72 h of incubation was performed, and the MTT solution was added and allowed to stand for a 4 h incubation period. The formazan crystals were dissolved and the absorbance was measured. The cytotoxicity ratio was calculated according to the following equation:
5.13. Statistical Analysis
All the compounds were examined in the set of triplicate experiments. IC
50 (50% inhibitory concentration) values of the compounds represent the concentration that caused 50% enzyme activity loss. Using a minimum of three samples, standard deviation was calculated in all the experiments. The inhibition mechanism of the compounds was determined by comparing the statistical results, including the Akaike’s information criterion values, of different inhibition models and selecting the one with the best fit [
37].





 ,
, 







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}