
Structural and Molecular Characterization of Squalene Synthase Belonging to the Marine Thraustochytrid Species Aurantiochytrium limacinum Using Bioinformatics Approach

 ,
,  , ,
, , 
 and
and 
Abstract
:
1. Introduction
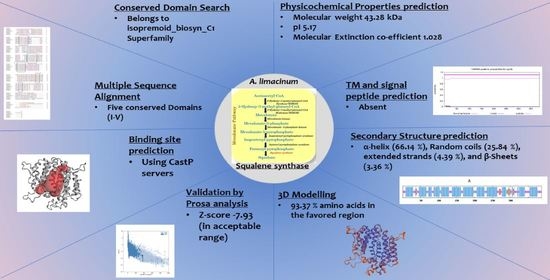
2. Results and Discussions
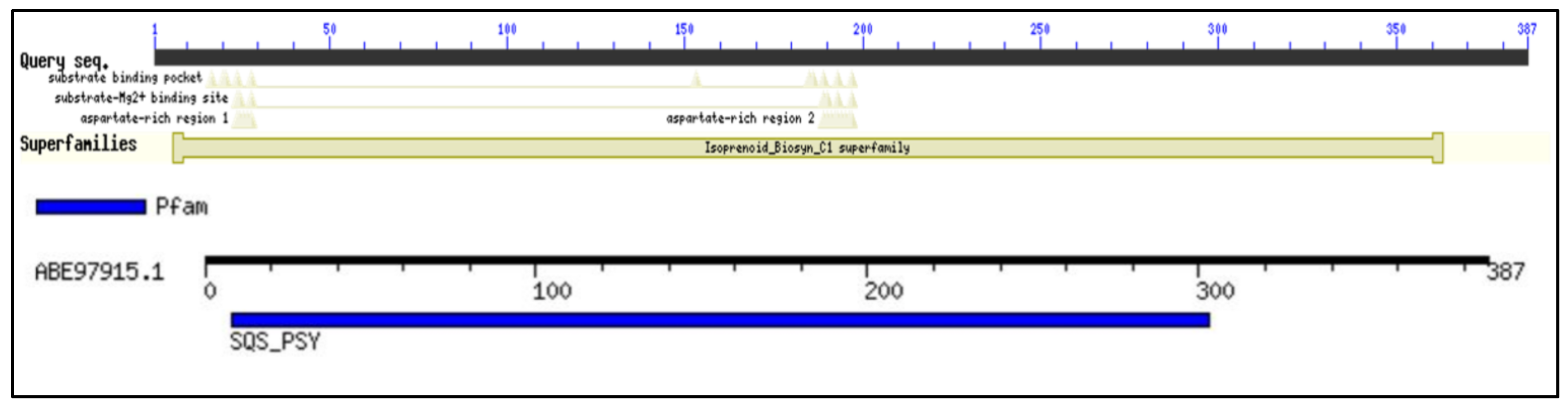
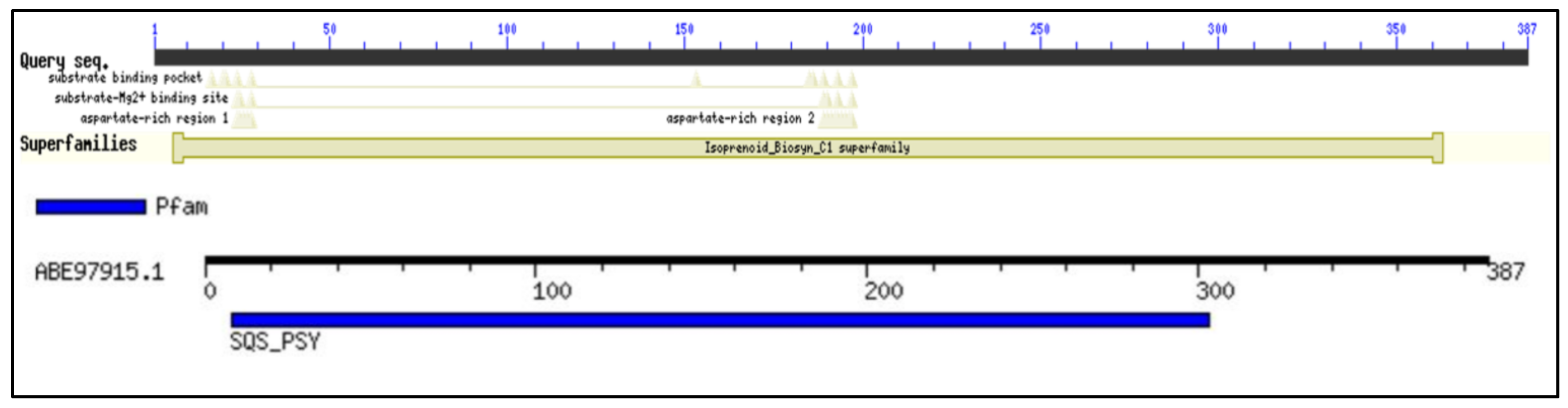
2.1. Conserved Domain Search and Multiple Sequence Analysis
2.2. Physicochemical Characteristics In-Silico Analysis
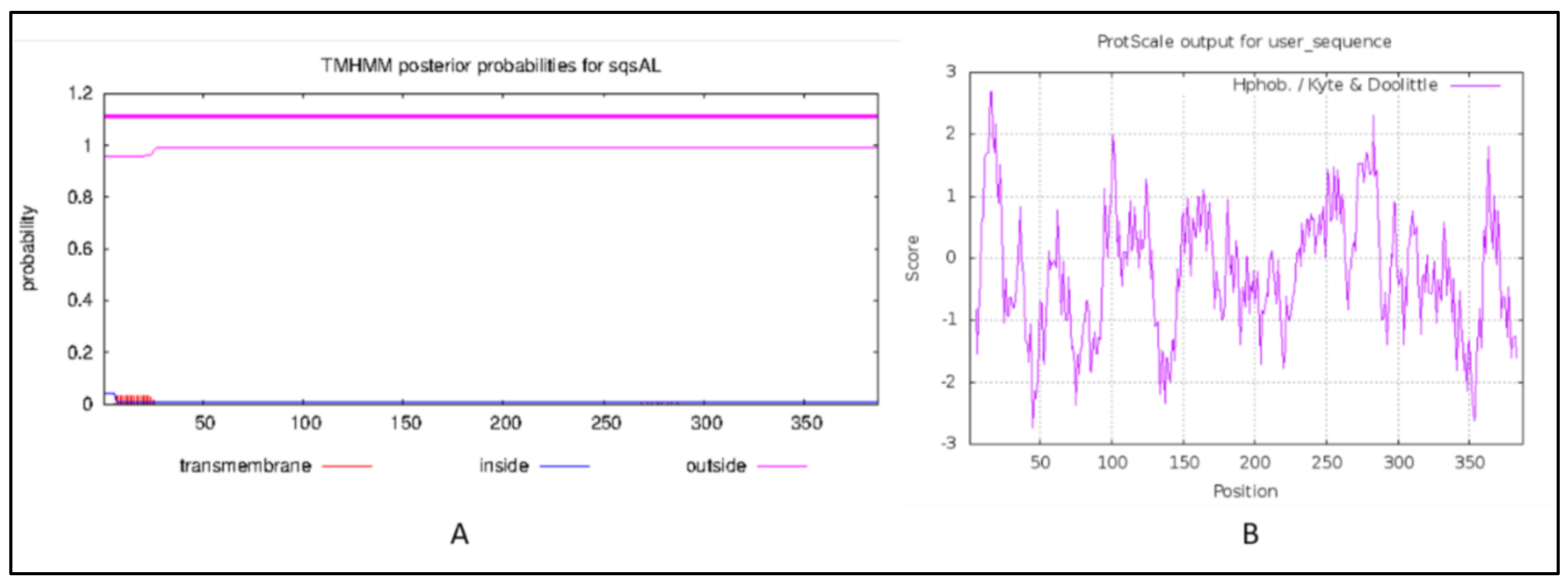
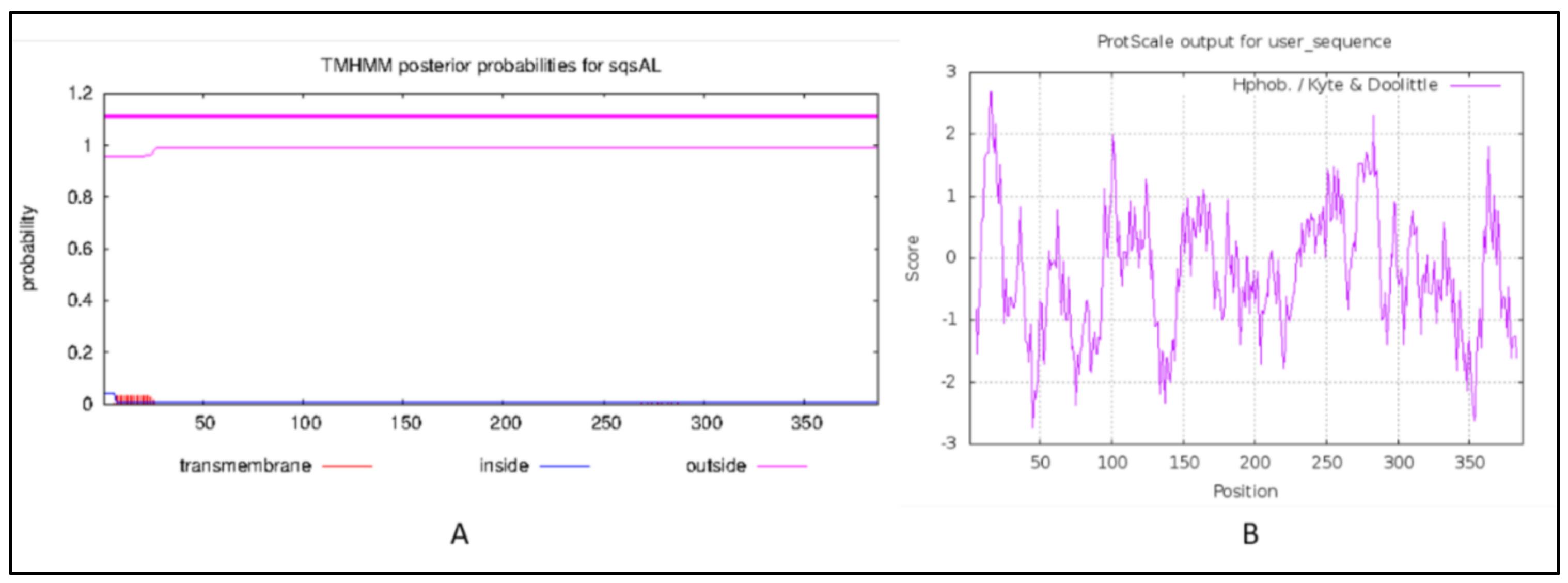
2.3. Transmembrane Domain and Hydropathy Analysis
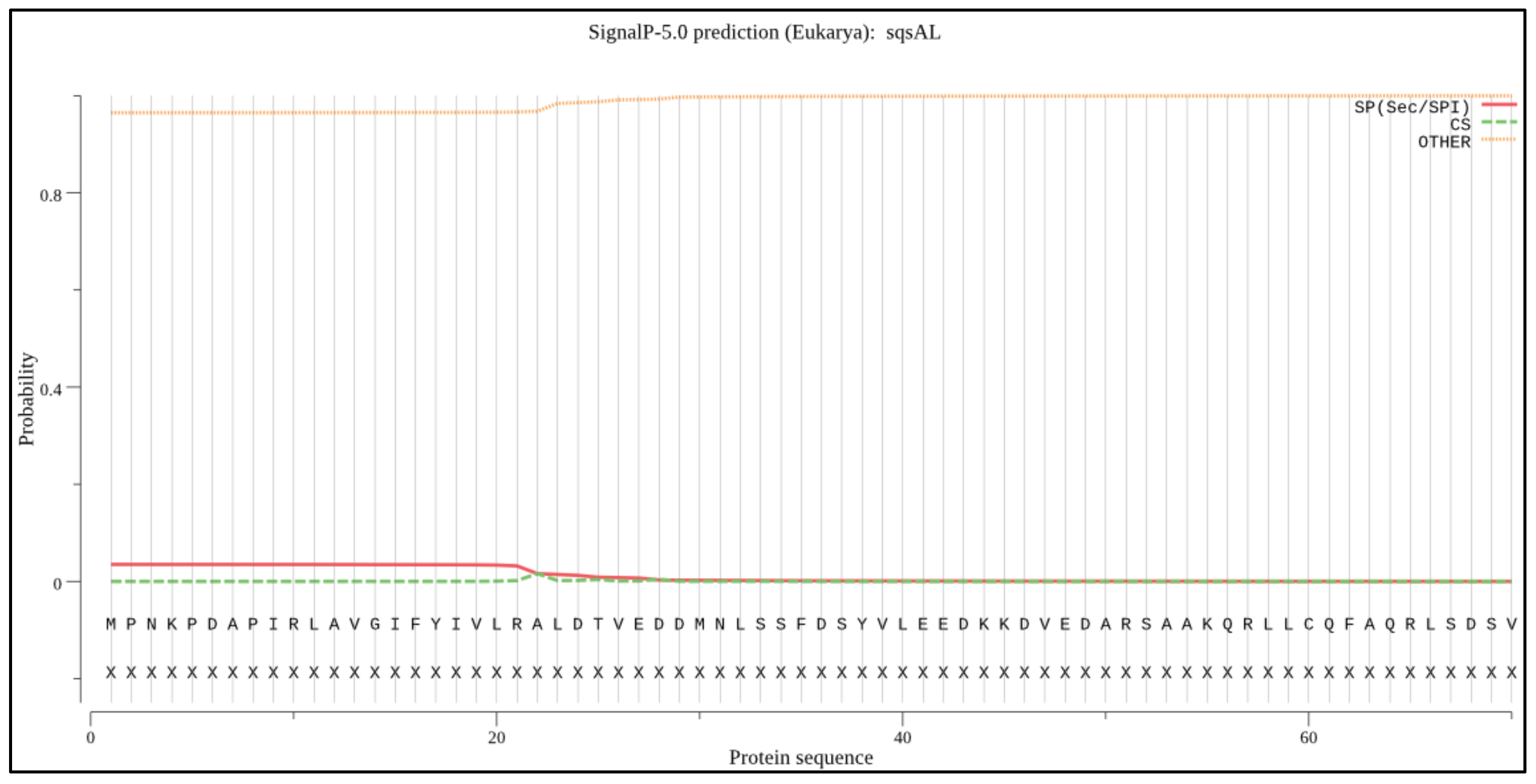
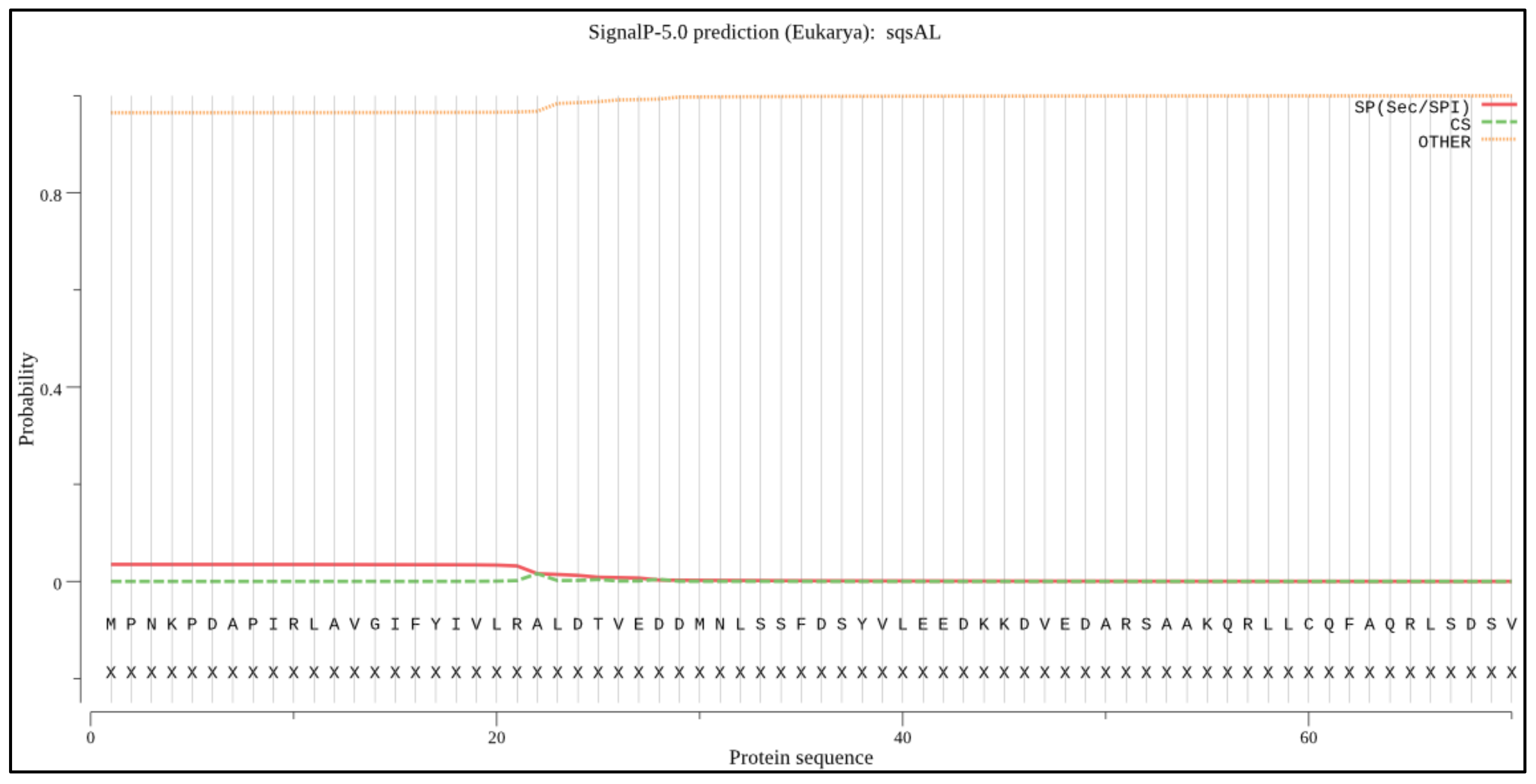
2.4. Signal Peptide and Peroxisome Targeting Signal Prediction
2.5. Secondary Structure Prediction and Analysis
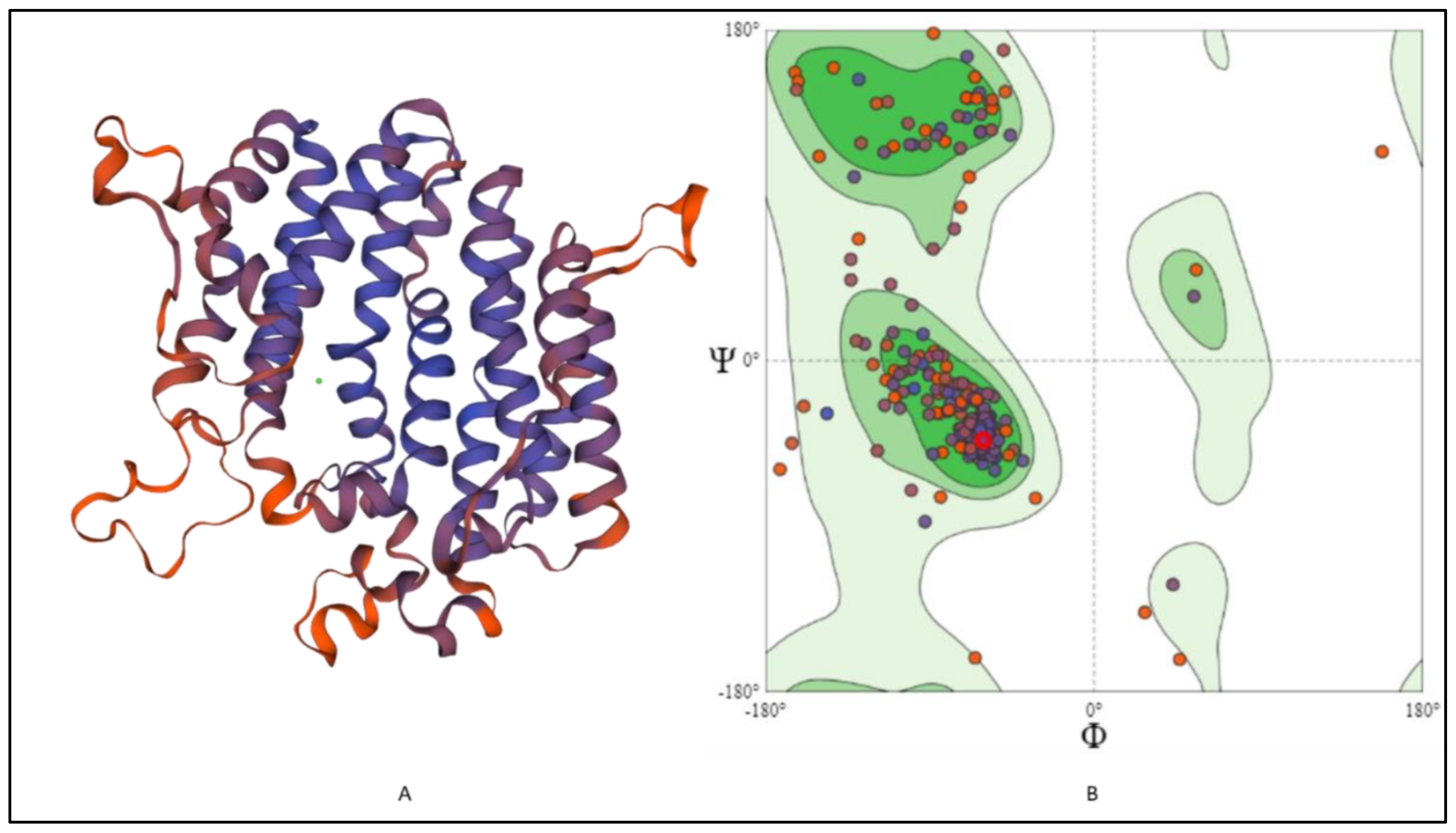
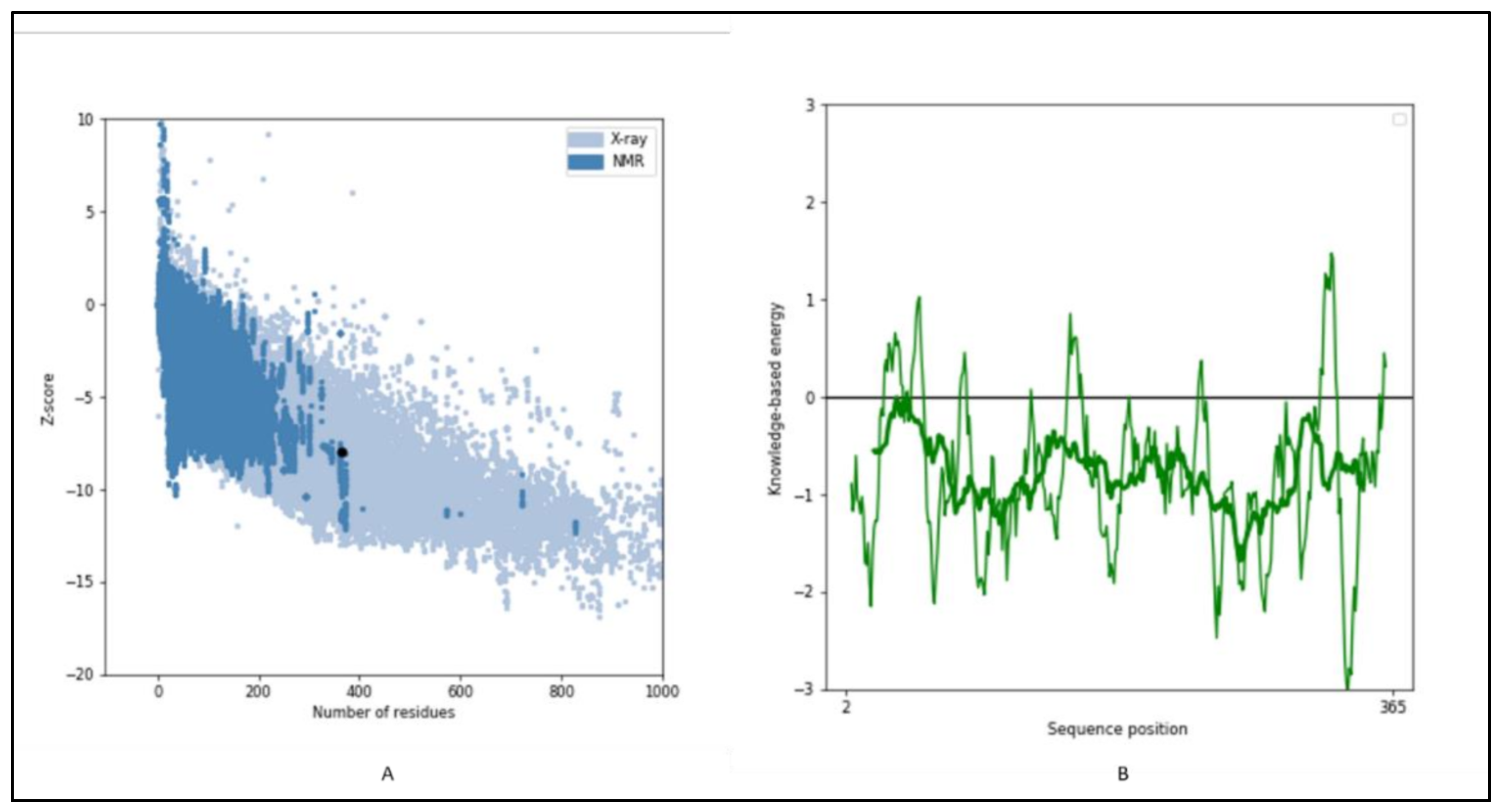
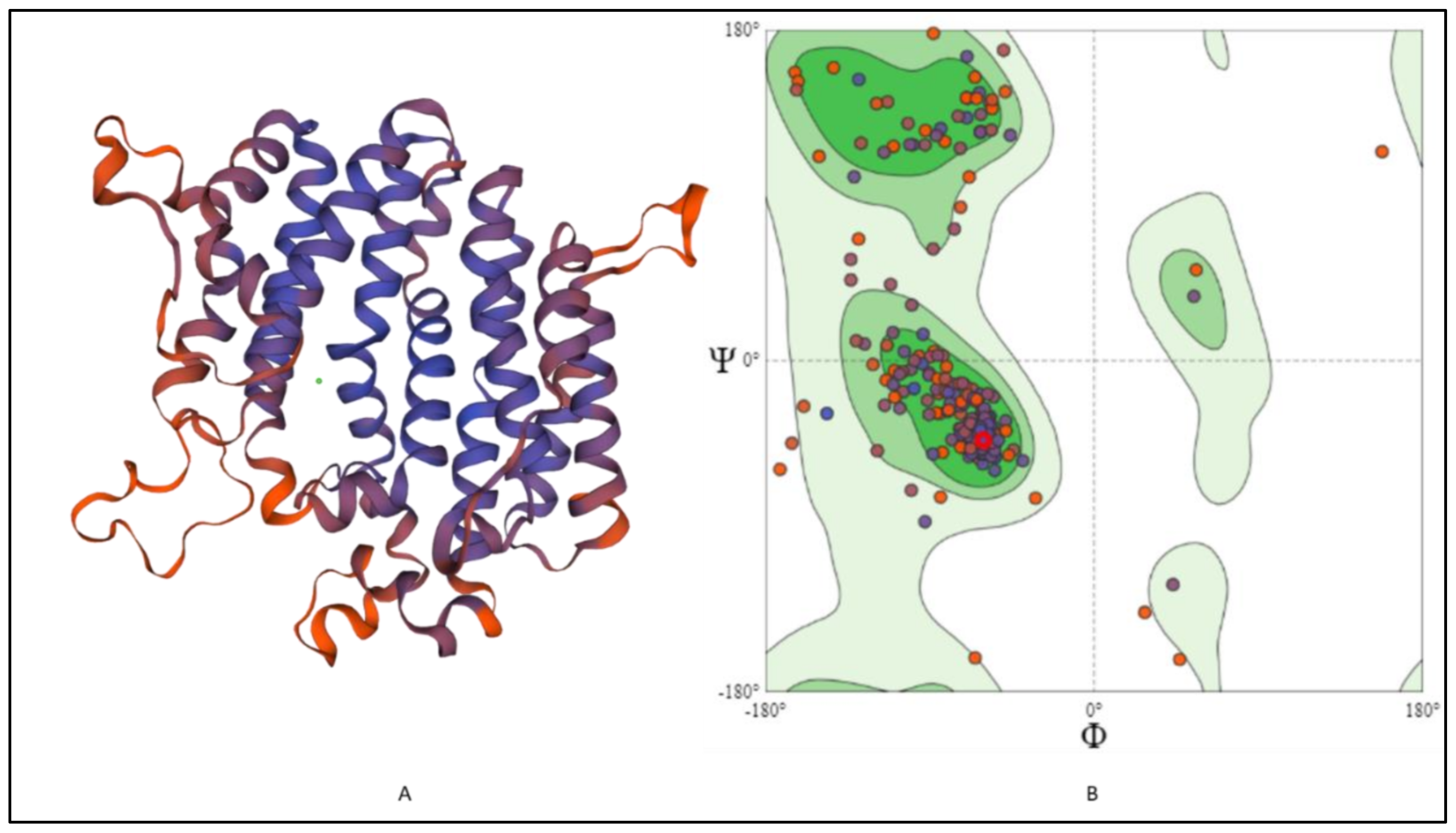
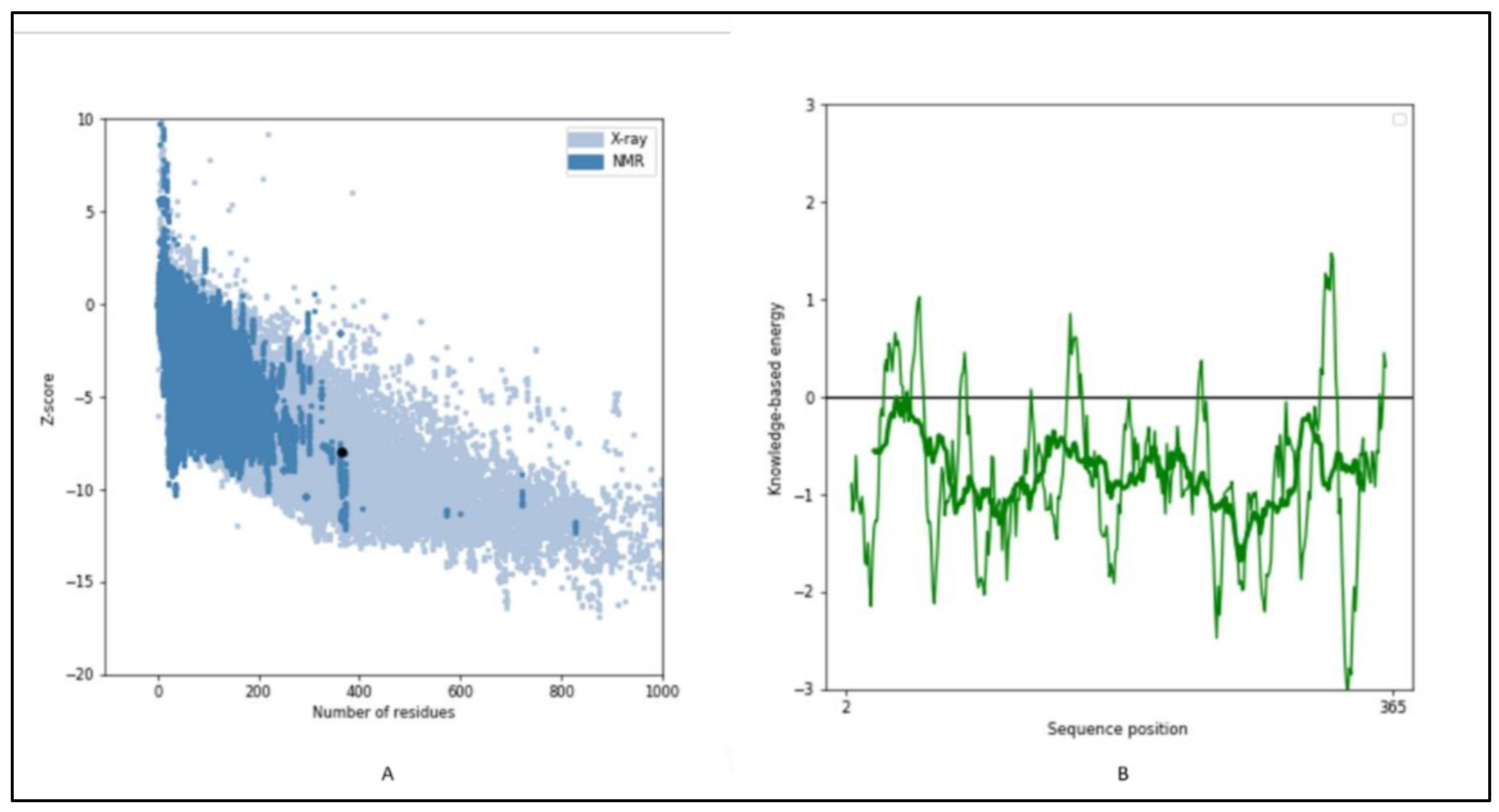
2.6. 3-D Structure Modeling and Analysis
2.7. Prediction of Anticipated Binding Sites in SQS
3. Materials and Methods
3.1. Retrieval of the Sequence Data:
3.2. Physicochemical Characteristics Prediction
3.3. Secondary and 3-D Structure Modelling
3.4. Binding Site and Ligand Binding Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Patel, A.; Karageorgou, D.; Katapodis, P.; Sharma, A.; Rova, U.; Christakopoulos, P.; Matsakas, L. Bioprospecting of Thraustochytrids for Omega-3 Fatty Acids: A Sustainable Approach to Reduce Dependency on Animal Sources. Trends Food Sci. Technol. 2021, 115, 433–444. [Google Scholar] [CrossRef]
- Xie, Y.; Sen, B.; Wang, G. Mining Terpenoids Production and Biosynthetic Pathway in Thraustochytrids. Bioresour. Technol. 2017, 244, 1269–1280. [Google Scholar] [CrossRef] [PubMed]
- Aasen, I.M.; Ertesvåg, H.; Heggeset, T.M.B.; Liu, B.; Brautaset, T.; Vadstein, O.; Ellingsen, T.E. Thraustochytrids as Production Organisms for Docosahexaenoic Acid (DHA), Squalene, and Carotenoids. Appl. Microbiol. Biotechnol. 2016, 100, 4309–4321. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Mu, L.; Shi, Y.; Rova, U.; Christakopoulos, P.; Matsakas, L. Novel Biorefinery Approach Aimed at Vegetarians Reduces the Dependency on Marine Fish Stocks for Obtaining Squalene and Docosahexaenoic Acid. ACS Sustain. Chem. Eng. 2020, 8, 8803–8813. [Google Scholar] [CrossRef]
- Gohil, N.; Bhattacharjee, G.; Khambhati, K.; Braddick, D.; Singh, V. Engineering Strategies in Microorganisms for the Enhanced Production of Squalene: Advances, Challenges and Opportunities. Front. Bioeng. Biotechnol. 2019, 7, 50. [Google Scholar] [CrossRef] [PubMed]
- Pyne, M.E.; Narcross, L.; Martin, V.J.J. Engineering Plant Secondary Metabolism in Microbial Systems. Plant Physiology 2019, 179, 844–861. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, L.J.; Kwak, S.; Liu, J.J.; Lane, S.; Hua, Q.; Kweon, D.H.; Jin, Y.S. Improved Squalene Production through Increasing Lipid Contents in Saccharomyces Cerevisiae. Biotechnol. Bioeng. 2018, 115, 1793–1800. [Google Scholar] [CrossRef]
- Potijun, S.; Jaingam, S.; Sanevas, N.; Vajrodaya, S.; Sirikhachornkit, A. Green Microalgae Strain Improvement for the Production of Sterols and Squalene. Plants 2021, 10, 1673. [Google Scholar] [CrossRef]
- Morabito, C.; Bournaud, C.; Maës, C.; Schuler, M.; Aiese Cigliano, R.; Dellero, Y.; Maréchal, E.; Amato, A.; Rébeillé, F. The Lipid Metabolism in Thraustochytrids. Prog. Lipid Res. 2019, 76, 101007. [Google Scholar] [CrossRef]
- Li, Q.; Chen, G.Q.; Fan, K.W.; Lu, F.U.P.; Aki, T.; Jiang, Y. Screening and Characterization of Squalene-Producing Thraustochytrids from Hong Kong Mangroves. J. Agric. Food Chem. 2009, 57, 4267–4272. [Google Scholar] [CrossRef]
- Patel, A.; Rova, U.; Christakopoulos, P.; Matsakas, L. Simultaneous Production of DHA and Squalene from Aurantiochytrium sp. Grown on Forest Biomass Hydrolysates. Biotechnol. Biofuels 2019, 12, 255. [Google Scholar] [CrossRef] [PubMed]
- Varela, J.C.; Pereira, H.; Vila, M.; León, R. Production of Carotenoids by Microalgae: Achievements and Challenges. Photosynth. Res. 2015, 125, 423–436. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Ma, X.; Wang, Y. Production of Squalene by Microbes: An Update. World J. Microbiol. Biotechnol. 2016, 32, 195. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Wang, Y.Z.; Xu, Y.S.; Shi, T.Q.; Liu, W.Z.; Sun, X.M.; Huang, H. Biotechnological Production of Lipid and Terpenoid from Thraustochytrids. Biotechnol. Adv. 2021, 48, 107725. [Google Scholar] [CrossRef]
- Hong, W.K.; Heo, S.Y.; Park, H.M.; Kim, C.H.; Sohn, J.H.; Kondo, A.; Seo, J.W. Characterization of a Squalene Synthase from the Thraustochytrid Microalga Aurantiochytrium Sp. KRS101. J. Microbiol. Biotechnol. 2013, 23, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Rong, Q.; Chen, Y.; Yuan, Q.; Shen, Y.; Guo, J.; Yang, Y.; Zha, L.; Wu, H.; Huang, L.; et al. Molecular Cloning and Functional Analysis of Squalene Synthase (SS) in Panax Notoginseng. Int. J. Biol. Macromol. 2017, 95, 759–765. [Google Scholar] [CrossRef]
- Kalra, S.; Kumar, S.; Lakhanpal, N.; Kaur, J.; Singh, K. Characterization of Squalene Synthase Gene from Chlorophytum Borivilianum (Sant. and Fernand.). Mol. Biotechnol. 2013, 54, 944–953. [Google Scholar] [CrossRef]
- Pandit, J.; Danley, D.E.; Schulte, G.K.; Mazzalupo, S.; Pauly, T.A.; Hayward, C.M.; Hamanaka, E.S.; Thompson, J.F.; Harwood, H.J. Crystal Structure of Human Squalene Synthase. J. Biol. Chem. 2000, 275, 30610–30617. [Google Scholar] [CrossRef] [Green Version]
- Zhan, D.; Zhang, Y.; Song, Y.; Sun, H.; Li, Z.; Han, W.; Liu, J. Computational Studies of Squalene Synthase from Panax Ginseng: Homology Modeling, Docking Study and Virtual Screening for a New Inhibitor. J. Theor. Comput. Chem. 2012, 11, 1101–1120. [Google Scholar] [CrossRef]
- Gu, P.; Ishii, Y.; Spencer, T.A.; Shechter, I. Function-Structure Studies and Identification of Three Enzyme Domains Involved in the Catalytic Activity in Rat Hepatic Squalene Synthase. J. Biol. Chem. 1998, 273, 12515–12525. [Google Scholar] [CrossRef] [Green Version]
- Robinson, G.W.; Tsay, Y.H.; Kienzle, B.K.; Smith-Monroy, C.A.; Bishop, R.W. Conservation between Human and Fungal Squalene Synthetases: Similarities in Structure, Function, and Regulation. Mol. Cell. Biol. 1993, 13, 2706–2717. [Google Scholar] [CrossRef] [Green Version]
- Zhang, B.; Liu, Y.; Chen, M.; Feng, J.; Ma, Z.; Zhang, X.; Zhu, C. Cloning, Expression Analysis and Functional Characterization of Squalene Synthase (SQS) from Tripterygium Wilfordii. Molecules 2018, 23, 269. [Google Scholar] [CrossRef] [Green Version]
- Zha, L.; Liu, S.; Su, P.; Yuan, Y.; Huang, L. Cloning, Prokaryotic Expression and Functional Analysis of Squalene Synthase (SQS) in Magnolia Officinalis. Protein Expr. Purif. 2016, 120, 28–34. [Google Scholar] [CrossRef]
- Gao, R.; Yu, D.; Chen, L.; Wang, W.; Sun, L.; Chang, Y. Cloning and Functional Analysis of Squalene Synthase Gene from Dryopteris Fragrans (L.) Schott. Protein Expr. Purif. 2019, 155, 95–103. [Google Scholar] [CrossRef]
- Boyd, D.; Beckwith, J. Positively Charged Amino Acid Residues Can Act as Topogenic Determinants in Membrane Proteins. Proc. Natl. Acad. Sci. USA 1989, 86, 9446–9450. [Google Scholar] [CrossRef] [Green Version]
- Aminfar, Z.; Tohidfar, M. In Silico Analysis of Squalene Synthase in Fabaceae Family Using Bioinformatics Tools. J. Genet. Eng. Biotechnol. 2018, 16, 739–747. [Google Scholar] [CrossRef]
- Zhao, H.; Tang, Q.; Mo, C.; Bai, L.; Tu, D.; Ma, X. Cloning and Characterization of Squalene Synthase and Cycloartenol Synthase from Siraitia Grosvenorii. Acta Pharm. Sin. B 2017, 7, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Krebs, E.G.; Beavo, J.A. Phosphorylation-Dephosphorylation of Enzymes. Annu. Rev. Biochem. 1979, 48, 923–959. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [Green Version]
- Sanchita; Singh, G.; Sharma, A. In Silico Study of Binding Motifs in Squalene Synthase Enzyme of Secondary Metabolic Pathway Solanaceae Family. Mol. Biol. Rep. 2014, 41, 7201–7208. [Google Scholar] [CrossRef]
- Ding, C.; Zhao, C.L.; Chen, Z.J.; Wei, F.G.; Song, D.G.; Miao, K.R. Bioinformatics Analysis of the Squalene Synthase Gene and the Amino Acid Sequence in Ginseng Species. Int. J. Clin. Exp. Med. 2015, 8, 12818–12825. [Google Scholar] [PubMed]
- Kang, J.; Zhang, Q.; Jiang, X.; Zhang, T.; Long, R.; Yang, Q.; Wang, Z. Molecular Cloning and Functional Identification of a Squalene Synthase Encoding Gene from Alfalfa (Medicago Sativa L.). Int. J. Mol. Sci. 2019, 20, 4499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kharwar, S.; Bhattacharjee, S.; Mishra, A.K. Bioinformatics Analysis of Enzymes Involved in Cysteine Biosynthesis: First Evidence for the Formation of Cysteine Synthase Complex in Cyanobacteria. 3 Biotech 2021, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Wilkins, M.R.; Gasteiger, E.; Bairoch, A.; Sanchez, J.C.; Williams, K.L.; Appel, R.D.; Hochstrasser, D.F. Protein Identification and Analysis Tools in the ExPASy Server. Methods Mol. Biol. 1999, 112, 531–552. [Google Scholar]
- Stothard, P. The Sequence Manipulation Suite: JavaScript Programs for Analyzing and Formatting Protein and DNA Sequences. BioTechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef] [Green Version]
- Geourjon, C.; Deléage, G. Sopma: Significant Improvements in Protein Secondary Structure Prediction by Consensus Prediction from Multiple Alignments. Bioinformatics 1995, 11, 681–684. [Google Scholar] [CrossRef]
- Klausen, M.S.; Jespersen, M.C.; Nielsen, H.; Jensen, K.K.; Jurtz, V.I.; Sønderby, C.K.; Sommer, M.O.A.; Winther, O.; Nielsen, M.; Petersen, B.; et al. NetSurfP-2.0: Improved Prediction of Protein Structural Features by Integrated Deep Learning. Proteins Struct. Funct. Bioinform. 2019, 87, 520–527. [Google Scholar] [CrossRef] [Green Version]
- Wiederstein, M.; Sippl, M.J. ProSA-Web: Interactive Web Service for the Recognition of Errors in Three-Dimensional Structures of Proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef] [Green Version]
- Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. CASTp: Computed Atlas of Surface Topography of Proteins with Structural and Topographical Mapping of Functionally Annotated Residues. Nucleic Acids Res. 2006, 34, W116–W118. [Google Scholar] [CrossRef] [Green Version]
- Wass, M.N.; Kelley, L.A.; Sternberg, M.J.E. 3DLigandSite: Predicting Ligand-Binding Sites Using Similar Structures. Nucleic Acids Res. 2010, 38, W469–W473. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UniProtKB Number | Isoelectric Point (pI) | Total Number of Atoms | No. of Negatively Charged Residues | No. of Positively Charged Residues | Estimated Half-Life (h) | Aliphatic Index | Instability Index | Extinction Co-Efficient | Grand Average of Hydropathicity (GRAVY) |
|---|---|---|---|---|---|---|---|---|---|
| ABE97915.1 | 5.17 | 6029 | 56 | 43 | 30 | 86.72 | 41.82 | 44515 | −0.256 |
| Name of the Organism | UniProtKb Number | Length of AA Sequence | Molecular Mass (kDa) | Taxonomic Identifier (NCBI) |
|---|---|---|---|---|
| Schizochytrium limacinum SR21 | ABE97915.1 | 387 | 43.284 | 87102 |
| Aurantiochytrium limacinum | Q1KNJ1 | 387 | 43.284 | 87102 |
| Aurantiochytrium sp. TA4 | A0A0M4QHX0 | 387 | 43.285 | 1513508 |
| Aurantiochytrium sp. Qe4 | A0A0M5LMP0 | 387 | 43.285 | 1716546 |
| Aurantiochytrium sp. KRS101 | U3KZM8 | 387 | 43.285 | 797128 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vyas, S.; Bettiga, M.; Rova, U.; Christakopoulos, P.; Matsakas, L.; Patel, A. Structural and Molecular Characterization of Squalene Synthase Belonging to the Marine Thraustochytrid Species Aurantiochytrium limacinum Using Bioinformatics Approach. Mar. Drugs 2022, 20, 180. https://doi.org/10.3390/md20030180
Vyas S, Bettiga M, Rova U, Christakopoulos P, Matsakas L, Patel A. Structural and Molecular Characterization of Squalene Synthase Belonging to the Marine Thraustochytrid Species Aurantiochytrium limacinum Using Bioinformatics Approach. Marine Drugs. 2022; 20(3):180. https://doi.org/10.3390/md20030180
Chicago/Turabian StyleVyas, Sachin, Maurizio Bettiga, Ulrika Rova, Paul Christakopoulos, Leonidas Matsakas, and Alok Patel. 2022. "Structural and Molecular Characterization of Squalene Synthase Belonging to the Marine Thraustochytrid Species Aurantiochytrium limacinum Using Bioinformatics Approach" Marine Drugs 20, no. 3: 180. https://doi.org/10.3390/md20030180
APA StyleVyas, S., Bettiga, M., Rova, U., Christakopoulos, P., Matsakas, L., & Patel, A. (2022). Structural and Molecular Characterization of Squalene Synthase Belonging to the Marine Thraustochytrid Species Aurantiochytrium limacinum Using Bioinformatics Approach. Marine Drugs, 20(3), 180. https://doi.org/10.3390/md20030180