
Discovery and Photoisomerization of New Pyrrolosesquiterpenoids Glaciapyrroles D and E, from Deep-Sea Sediment Streptomyces sp.

 , , ,
, , ,  , ,
, ,  , , , and
, , , and
Abstract
:1. Introduction
2. Results and Discussion
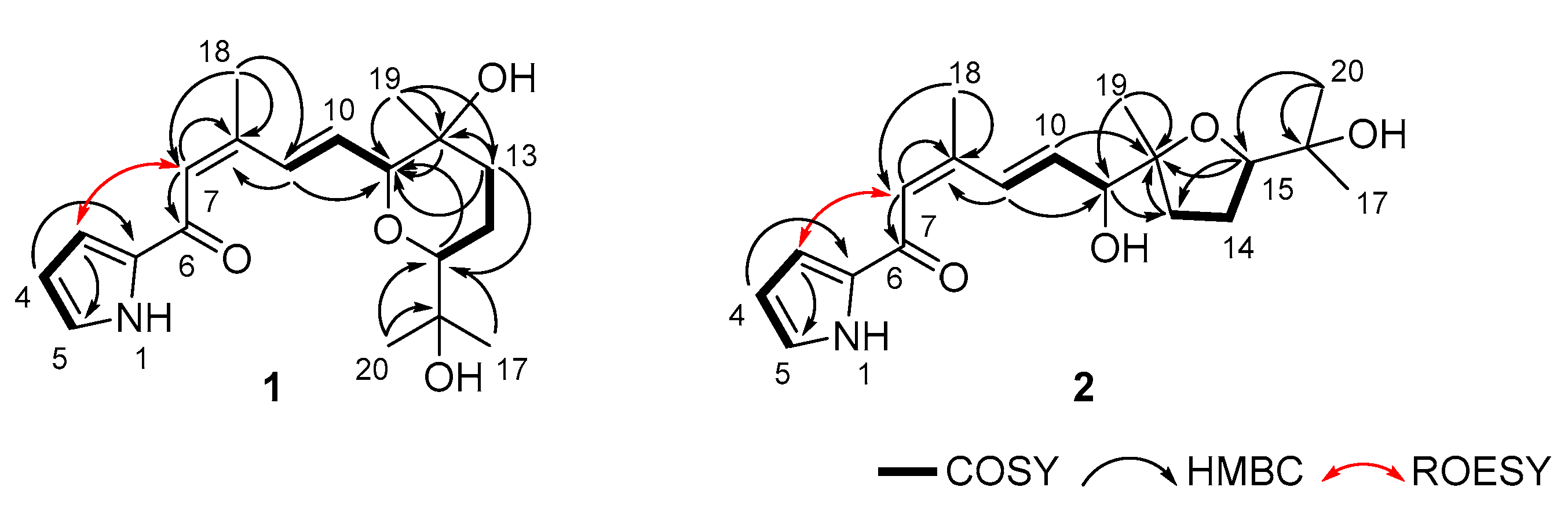
2.1. Structure Determination of Glaciapyrroles
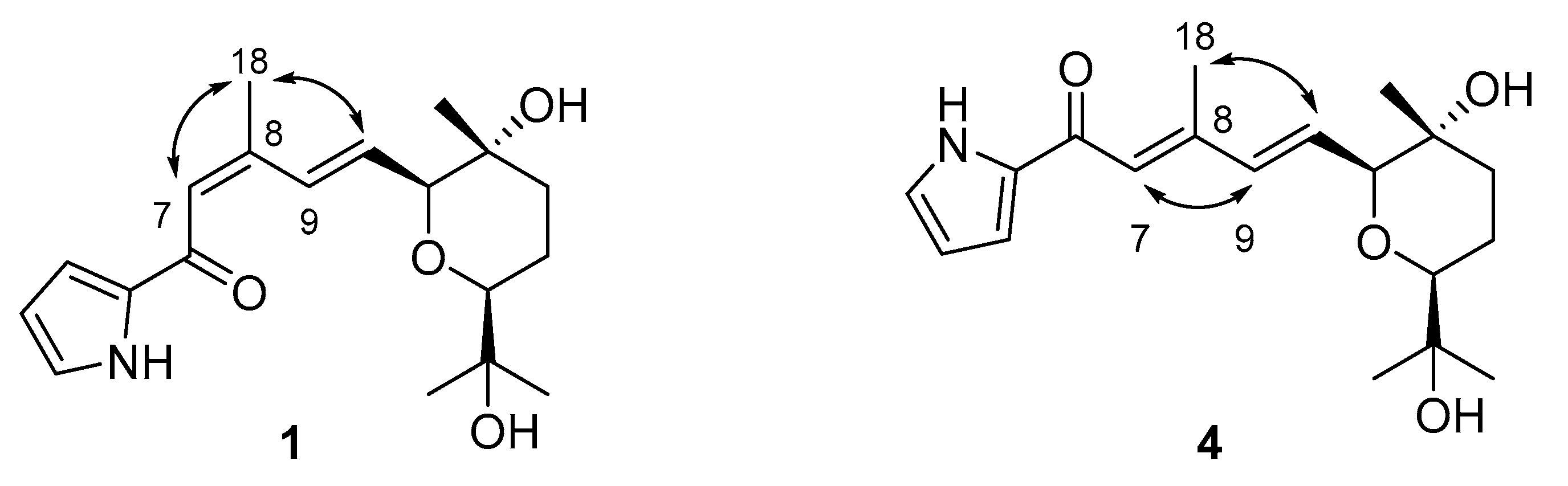
2.2. Photoirradiation of Glaciapyrroles
2.3. Structure Determination of Photoglaciapyrroles
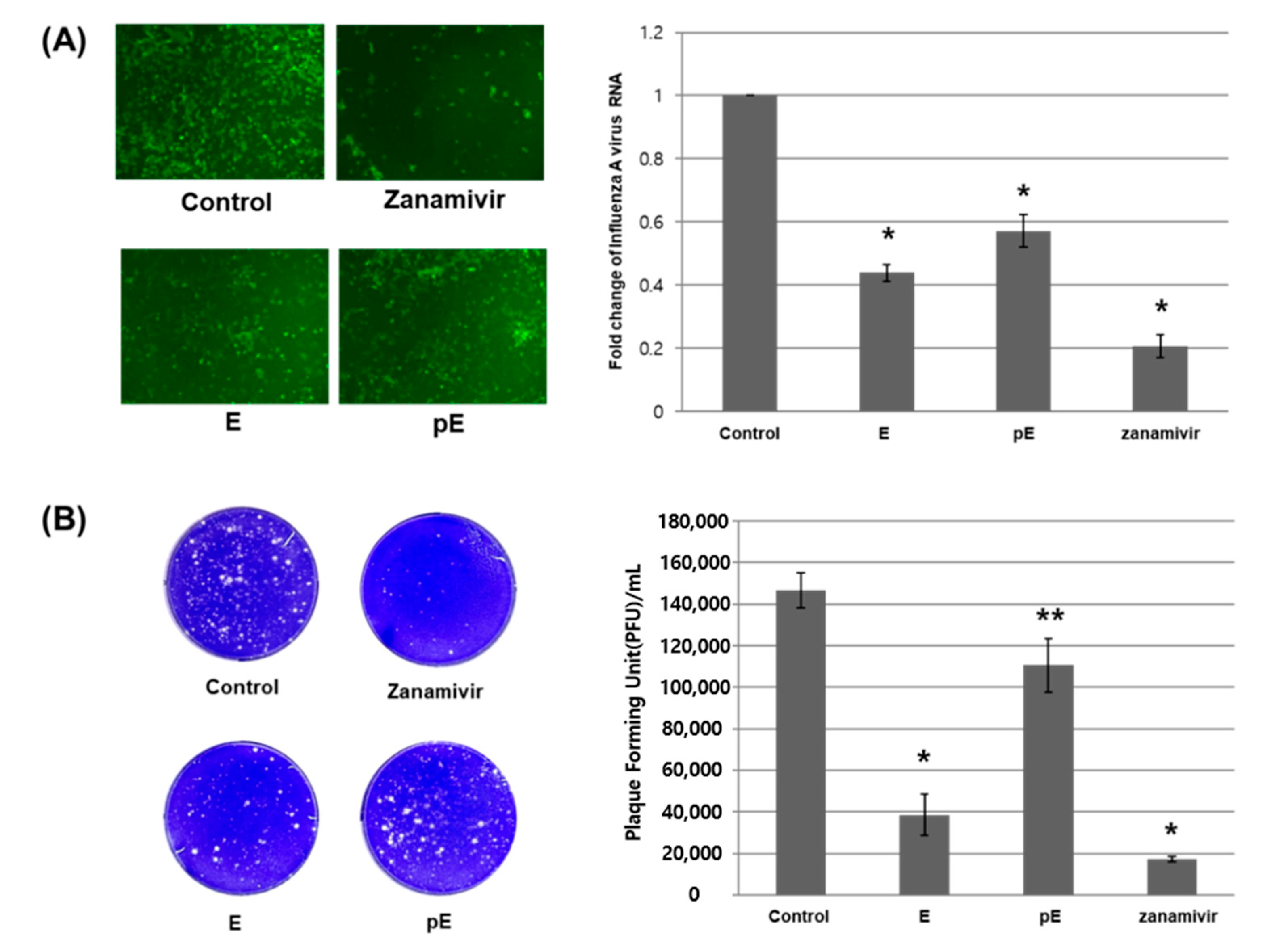
2.4. Evaluation of Antiviral Activity with Encapsulated Poly(lactic-co-glycolic acid) Nanoparticles
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Bacterial Isolation
3.3. Cultivation and Extraction
3.4. Isolation of Glaciapyrrole D, E, and A (1–3)
3.5. X-ray Crystallographic Data of glaciapyrrole E (2)
3.6. Photoirradiation
3.7. MTPA Esterification of Glaciapyrrole E
3.8. ECD Calculation
3.9. Synthesis and Characterization of Gla-PLGA NPs
3.10. Cells and Viruses
3.11. Quantitative RT-PCR (qRT-PCR)
3.12. Plaque Assay
3.13. Cell Viability Assay
3.14. In-Situ LED-Illuminated NMR Spectroscopy
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Molinski, T.F.; Dalisay, D.S.; Lievens, S.L.; Saludes, J.P. Drug Development from Marine Natural Products. Nat. Rev. Drug Discov. 2009, 8, 69–85. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, C. Marine Natural Products in Medicinal Chemistry. ACS Med. Chem. Lett. 2018, 9, 959–961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manivasagan, P.; Venkatesan, J.; Sivakumar, K.; Kim, S.-K. Pharmaceutically Active Secondary Metabolites of Marine Actinobacteria. Microbiol. Res. 2014, 169, 262–278. [Google Scholar] [CrossRef] [PubMed]
- Bérdy, J. Bioactive Micobial Metabolites. J. Antibiot. 2005, 58, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kormarov, I.V.; Afonin, S.; Babii, O.; Schober, T.; Ulrich, A.S. Efficiently Photocontrollable of Not? Biological Activity of Photoisomerizable Diarylethenes. Chem. Eur. J. 2018, 24, 11245–11254. [Google Scholar]
- Montsko, G.; Nikfardjam, M.S.P.; Szabo, Z.; Boddi, K.; Lorand, T.; Ohmacht, R.; Mark, L. Determination of Products Derived from Trans-Resveratrol UV Photoisomerisation by means of HPLC-APCI-MS. J. Photochem. Photobiol. 2008, 196, 44–50. [Google Scholar] [CrossRef]
- Simmler, C.; Lankin, D.C.; Nikolicé, D.; Breemen, R.B.V.; Pauli, G.F. Isolation and Structural Characterization of Dihydrobenzofuran Congeners of Licochalcone A. Fitoterapia 2017, 121, 6–15. [Google Scholar] [CrossRef]
- Zaki, M.A.; Balachandran, P.; Khan, S.; Wang, M.; Mohammed, R.; Hetta, M.H.; Pasco, D.S.; Muhamma, I. Cytotoxicity and Modulation of Cancer-Related Signaling by (Z)- and (E)-3,4,3′,5′-Tetramethoxystilbene Isolated from Eugenia Rigida. J. Nat. Prod. 2013, 76, 679–684. [Google Scholar] [CrossRef]
- Park, J.S.; Ko, K.; Kim, S.-H.; Lee, J.K.; Park, J.-S.; Park, K.; Kim, M.R.; Kang, K.; Oh, D.-C.; Kim, S.Y.; et al. Tropolone-Bearing Sesquiterpenes from Juniperus chinensis: Structures, Photochemistry and Bioactivity. J. Nat. Prod. 2021, 84, 2020–2027. [Google Scholar] [CrossRef]
- Seco, J.M.; Quiñoá, E.; Riguera, R. A Practical Guide for the Assignment of the Absolute Configuration of Alcohols, Amines and Carboxylic acids by NMR. Tetrahedron Asymmetry 2001, 12, 2915–2925. [Google Scholar] [CrossRef]
- Macherla, V.R.; Liu, J.; Bellows, C.; Teisan, S.; Nicholson, B.; Lam, K.S.; Potts, B.C.M. Glaciapyrroles A, B, and C, Pyrrolesesquiterpenes from a Streptomyces sp. Isolated from an Alaskan Marine Sediment. J. Nat. Prod. 2005, 68, 780–783. [Google Scholar] [PubMed]
- Seegerer, A.; Nitschke, P.; Gschwind, R.M. Combined In Situ Illumination-NMR-UV/Vis Spectroscopy: A New Mechanistic Tool in Photochemistry. Angew. Chem. Int. Ed. 2018, 57, 7493–7497. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, S.; Goraya, M.U.; Maarouf, M.; Huang, S.; Chen, J.-L. Host Immune Response to Influenza A Virus Infection. Front. Immunol. 2018, 9, 320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, W.; Li, X.; Goraya, M.U.; Wang, S.; Chen, J.-L. Evolution of Influenza A Virus by Mutation and Re-Assortment. Int. J. Mol. Sci. 2017, 18, 1650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedford, T.; Suchard, M.A.; Lemey, P.; Dudas, G.; Gregory, V.; Hay, A.J.; McCauley, J.W.; Russell, C.A.; Smith, D.J.; Rambaut, A. Integrating Influenza Antigenic Dynamics with Molecular Evolution. eLife 2014, 3, e01914. [Google Scholar] [CrossRef] [PubMed]
- Morens, D.M.; Taubenberger, J.K.; Fauci, A.S. The Persistent Legacy of the 1918 Influenza Virus. N. Eng. J. Med. 2009, 361, 225–229. [Google Scholar] [CrossRef] [Green Version]
- Julkunen, I.; Melén, K.; Nyqvist, M.; Pirhonen, J.; Sareneva, T.; Matikainen, S. Inflammatory Responses in Influenza A Virus Infection. Vaccine 2001, 19, S32–S37. [Google Scholar] [CrossRef]
- Lamb, R.A.; Krug, R.M. Orthomyxoviridae: The Viruses and Their Replication. Fields Virol. Ed. 2001, 54, 1353–1395. [Google Scholar]
- Shaw, M.L. The Next Wave of Influenza Drugs. ACS Infect. Dis. 2017, 3, 691–694. [Google Scholar] [CrossRef]
- Danhier, F.; Ansorena, E.; Silva, J.M.; Coco, R.; Breton, A.L.; Préat, V. PLGA-based Nanoparticles: An Overview of Biomedical Applications. J. Control Release 2012, 161, 505–522. [Google Scholar] [CrossRef]
- Han, H.S.; Koo, S.Y.; Choi, K.Y. Emerging Nanoformulation Strategies for Phytocompounds and Applications from Drug Delivery to Phototherapy to Imaging. Bioact. Mater. 2022, 14, 182–205. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Parmar, A.; Kori, S.; Sandhir, R. PLGA-based nanoparticles: A New Paradigm in Biomedical Applications. Trends Anal. Chem. 2016, 80, 30–40. [Google Scholar] [CrossRef]
- Wang, W.; Wu, J.; Zhang, X.; Hao, C.; Zhao, X.; Jiao, G.; Shan, X.; Tai, W.; Yu, G. Inhibition of Influenza A Virus Infection by Fucoidan Targeting Viral Neuraminidase and Cellular EGFR Pathway. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Feldmeier, C.; Bartling, H.; Riedle, E.; Gschwind, R.M. LED Based NMR Illumination Device for Mechanistic Studies on Photochemical Reactions—Versatile and Simple, yet Surprisingly Powerful. J. Magn. Reson. 2013, 232, 39–44. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 a | 2 a | ||||
|---|---|---|---|---|---|---|
| δC | Type | δH, mult (J in Hz) | δC | Type | δH, mult (J in Hz) | |
| 1 | NH | NH | ||||
| 2 | 135.5 | C | 135.3 | C | ||
| 3 | 117.6 | CH | 7.00, dd (3.9, 1.4) | 117.4 | CH | 6.98, dd (3.9, 1.3) |
| 4 | 111.2 | CH | 6.24, dd (3.9, 2.5) | 111.2 | CH | 6.23, dd (3.9, 2.5) |
| 5 | 126.7 | CH | 7.06, dd (2.5, 1.4) | 126.6 | CH | 7.06, dd (2.5, 1.3) |
| 6 | 182.4 | C | 182.1 | C | ||
| 7 | 123.1 | CH | 6.67, s | 123.6 | CH | 6.68, s |
| 8 | 150.1 | C | 149.6 | C | ||
| 9 | 130.0 | CH | 7.88, d (16.1, 1.3) | 131.5 | CH | 7.86, d (16.0) |
| 10 | 135.4 | CH | 6.37, dd (16.1, 4.3) | 137.5 | CH | 6.21, dd (16.0, 6.8) |
| 11 | 85.0 | CH | 3.87, dd (4.3, 1.3) | 79.2 | CH | 4.16, dd (6.8, 1.0) |
| 12 | 70.8 | C | 86.8 | C | ||
| 13a | 40.0 | CH2 | 1.90–1.86, m | 33.6 | CH2 | 2.17, ddd (12.4, 8.6, 6.0) |
| 13b | 1.71–1.67, m | 1.58, dt (12.4, 8.0) | ||||
| 14a | 25.0 | CH2 | 1.75–1.71, m | 27.5 | CH2 | 1.96–1.89, m |
| 14b | 1.61–1.56, m | |||||
| 15 | 85.5 | CH | 3.25, dd (11.4, 2.2) | 86.4 | CH | 3.84, t (7.3) |
| 16 | 72.8 | C | 72.6 | C | ||
| 17 | 25.6 | CH3 | 1.23, s | 27.3 | CH3 | 1.12, s |
| 18 | 21.3 | CH3 | 2.13, s | 21.3 | CH3 | 2.10, s |
| 19 | 20.7 | CH3 | 1.10, s | 24.1 | CH3 | 1.17, s |
| 20 | 26.2 | CH3 | 1.23, s | 25.8 | CH3 | 1.24, s |
| Position | 4 a | 5 a | 6 a | ||||||
|---|---|---|---|---|---|---|---|---|---|
| δC | Type | δH, mult (J in Hz) | δC | Type | δH, mult (J in Hz) | δC | Type | δH, mult (J in Hz) | |
| 1 | NH | NH | NH | ||||||
| 2 | 135.7 | C | 135.6 | C | 135.6 | C | |||
| 3 | 117.2 | CH | 7.01, d (3.8) | 117.3 | CH | 7.01, d (3.8) | 117.2 | CH | 6.99, d (3.7) |
| 4 | 111.2 | CH | 6.24, dd (3.8, 2.5) | 111.2 | CH | 6.24, dd (3.8, 2.5) | 111.2 | CH | 6.24, dd (3.7, 2.5) |
| 5 | 126.5 | CH | 7.06, d (2.5) | 126.6 | CH | 7.07, d (2.5) | 126.5 | CH | 7.06, d (2.5) |
| 6 | 183.0 | C | 182.8 | C | 182.9 | C | |||
| 7 | 124.9 | CH | 6.79, s | 125.3 | CH | 6.80, s | 125.0 | CH | 6.78, s |
| 8 | 151.5 | C | 151.1 | C | 151.2 | C | |||
| 9 | 135.6 | CH | 6.61, d (15.7) | 136.8 | CH | 6.57, d (15.8) | 136.3 | CH | 6.54, d (16.0) |
| 10 | 134.3 | CH | 6.38, dd (15.7, 4.3) | 136.6 | CH | 6.27, dd (15.8, 6.2) | 136.9 | CH | 6.29, dd (16.0, 6.0) |
| 11 | 85.0 | CH | 3.87, d (4.3) | 78.8 | CH | 4.21, d (6.2) | 78.4 | CH | 4.16, d (6.0) |
| 12 | 70.8 | C | 86.9 | C | 86.7 | C | |||
| 13a | 40.1 | CH2 | 1.90–1.87, m | 33.6 | CH2 | 2.18–2.14, m | 34.7 | CH2 | 2.15–2.10, m |
| 13b | 1.71–1.67, m | 1.61–1.57, m | 1.65–1.62, m | ||||||
| 14a | 25.1 | CH2 | 1.75–1.72, m | 27.6 | CH2 | 1.97–1.91, m (2H) | 27.7 | CH2 | 1.91–1.87, m |
| 14b | 1.61–1.55, m | 1.85–1.80, m | |||||||
| 15 | 85.6 | CH | 3.26, dd (11.3, 1.9) | 86.5 | CH | 3.85, dd (7.3) | 88.5 | CH | 3.85, dd (9.7, 5.9) |
| 16 | 72.8 | C | 72.7 | C | 72.3 | C | |||
| 17 | 25.7 | CH3 | 1.24, s | 25.9 | CH3 | 1.13, s | 26.3 | CH3 | 1.18, s |
| 18 | 14.5 | CH3 | 2.35, s | 14.6 | CH3 | 2.34, s | 14.6 | CH3 | 2.33, s |
| 19 | 20.7 | CH3 | 1.11, s | 24.1 | CH3 | 1.18, s | 23.7 | CH3 | 1.16, s |
| 20 | 26.0 | CH3 | 1.25, s | 27.3 | CH3 | 1.25, s | 25.1 | CH3 | 1.16, s |
| Sample | Size | Poly Disperse Index (PDI) | Encapsulation Efficiency (%) |
|---|---|---|---|
| GlaE-PLGA NPs | 96.3 ± 3.5 | 0.08 ± 0.03 | 14.4 ± 2.3 |
| pGlaE-PLGA NPs | 108.7 ± 2.5 | 0.12 ± 0.07 | 24.6 ± 1.5 |
| PLGA NPs | 72.4 ± 5.8 | 0.12 ± 0.07 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ko, K.; Kim, S.-H.; Park, S.; Han, H.S.; Lee, J.K.; Cha, J.W.; Hwang, S.; Choi, K.Y.; Song, Y.-J.; Nam, S.-J.; et al. Discovery and Photoisomerization of New Pyrrolosesquiterpenoids Glaciapyrroles D and E, from Deep-Sea Sediment Streptomyces sp. Mar. Drugs 2022, 20, 281. https://doi.org/10.3390/md20050281
Ko K, Kim S-H, Park S, Han HS, Lee JK, Cha JW, Hwang S, Choi KY, Song Y-J, Nam S-J, et al. Discovery and Photoisomerization of New Pyrrolosesquiterpenoids Glaciapyrroles D and E, from Deep-Sea Sediment Streptomyces sp. Marine Drugs. 2022; 20(5):281. https://doi.org/10.3390/md20050281
Chicago/Turabian StyleKo, Keebeom, Seong-Hwan Kim, Subin Park, Hwa Seung Han, Jae Kyun Lee, Jin Wook Cha, Sunghoon Hwang, Ki Young Choi, Yoon-Jae Song, Sang-Jip Nam, and et al. 2022. "Discovery and Photoisomerization of New Pyrrolosesquiterpenoids Glaciapyrroles D and E, from Deep-Sea Sediment Streptomyces sp." Marine Drugs 20, no. 5: 281. https://doi.org/10.3390/md20050281
APA StyleKo, K., Kim, S.-H., Park, S., Han, H. S., Lee, J. K., Cha, J. W., Hwang, S., Choi, K. Y., Song, Y.-J., Nam, S.-J., Shin, J., Nam, S.-I., Kwon, H. C., Park, J.-S., & Oh, D.-C. (2022). Discovery and Photoisomerization of New Pyrrolosesquiterpenoids Glaciapyrroles D and E, from Deep-Sea Sediment Streptomyces sp. Marine Drugs, 20(5), 281. https://doi.org/10.3390/md20050281