

Stimulation of Neurite Outgrowth in Cerebrocortical Neurons by Sodium Channel Activator Brevetoxin-2 Requires Both N-Methyl-D-aspartate Receptor 2B (GluN2B) and p21 Protein (Cdc42/Rac)-Activated Kinase 1 (PAK1)



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
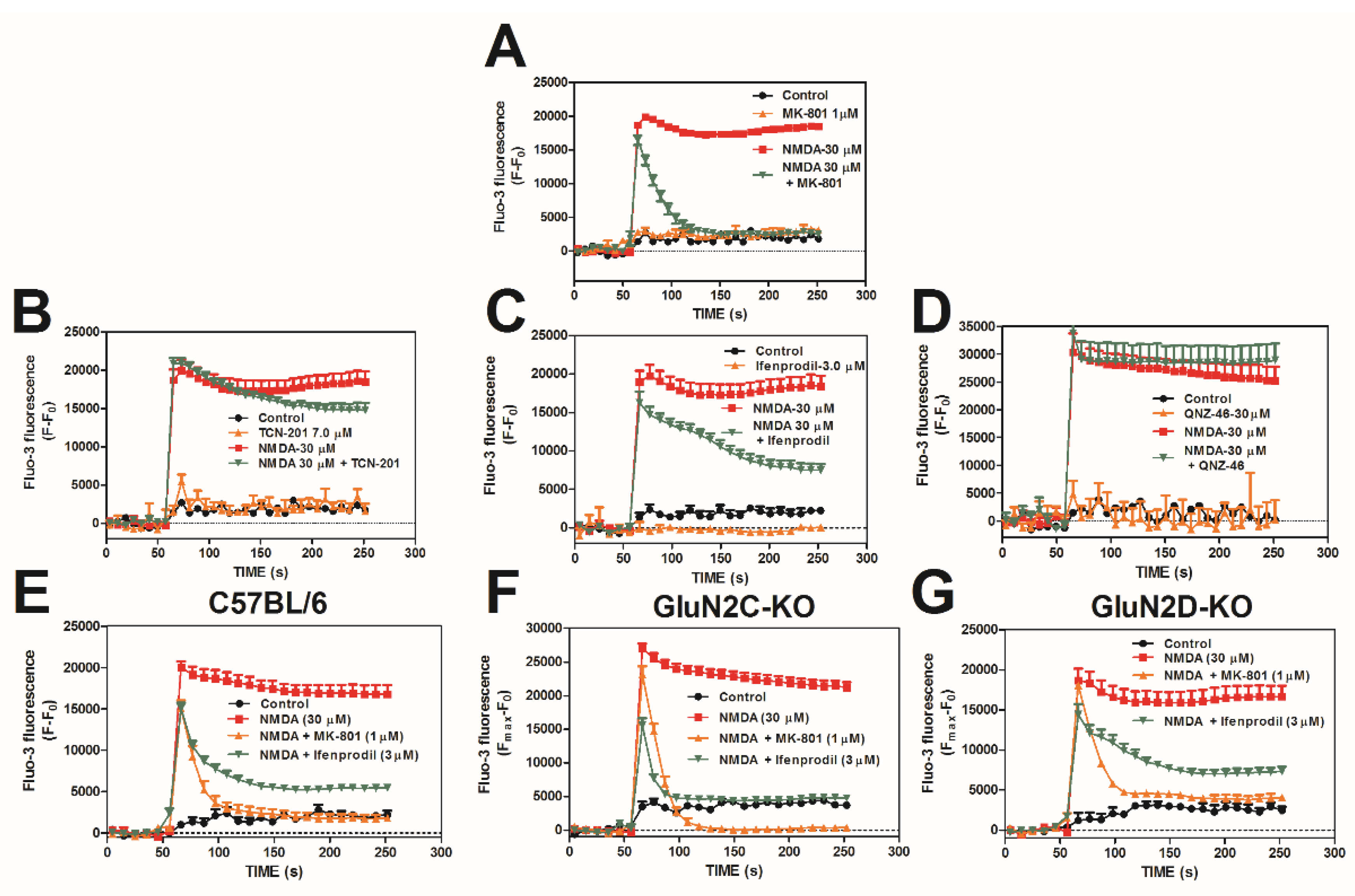
2.1. NMDA-Induced Ca2+ Influx Involves GluN2B but Not GluN2A, –2C, or –2D Complexes in Cerebrocortical Neurons
2.2. PbTx-2 and NMDA Exposure Produced Distinct Ca2+ Influx Pathways
2.3. PbTx-2-Induced Ca2+ Influx Involves the GluN2B but Not GluN2A Subtype of NMDAR
2.4. PbTx-2-Induced Neurite Outgrowth in Cerebrocortical Neurons Is Antagonized by Ifenprodil
2.5. PbTx-2-Induced Neurite Outgrowth in Cerebrocortical Neurons Is Antagonized by a CaMKII Inhibitor
2.6. PbTx-2-Induced Neurite Outgrowth Involves Rac1 but Not Rho A
2.7. PbTx-2-Induced Neurite Outgrowth Involves PAK1
2.8. PbTx-2 Induces Release of Glutamate into Extracellular Medium
2.9. PbTx-2-Induced Elevation of Miniature Excitatory Post-Synaptic Current (mEPSCs) Is Dependent on PAK1
2.10. PbTx-2 Enhancement of Dendritic Complexity Requires PAK1
2.11. PbTx-2 Treatment Produces Phosphorylation of Cofilin and Increases F-Actin Density
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cerebrocortical Neuron Culture
4.3. Intracellular Ca 2+ Monitoring
4.4. Determination of Total Neurite Length and Diolistic Labeling
4.5. Plasmids and Nucleofection
4.6. Measurement of Dendritic Complexity
4.7. Western Blot
4.8. Glutamate Release Assay
4.9. mEPSCs Recording
4.10. F-Actin Staining
4.11. Statistical Analyses and Graphical Illustration
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murray, T.F. Neurotoxic: Ciguatoxin and brevetoxin—From excitotoxicity to neurotherapeutics. Adv. Neurotoxicol. 2021, 6, 89–104. [Google Scholar] [CrossRef]
- Cao, Z.; George, J.; Gerwick, W.H.; Baden, D.G.; Rainier, J.D.; Murray, T.F. Influence of lipid-soluble gating modifier toxins on sodium influx in neocortical neurons. J. Pharmacol. Exp. Ther. 2008, 326, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Dravid, S.M.; Baden, D.G.; Murray, T.F. Brevetoxin augments NMDA receptor signaling in murine neocortical neurons. Brain Res. 2005, 1031, 30–38. [Google Scholar] [CrossRef] [PubMed]
- LePage, K.T.; Baden, D.G.; Murray, T.F. Brevetoxin derivatives act as partial agonists at neurotoxin site 5 on the voltage-gated Na+ channel. Brain Res. 2003, 959, 120–127. [Google Scholar] [CrossRef]
- Dravid, S.M.; Baden, D.G.; Murray, T.F. Brevetoxin activation of voltage-gated sodium channels regulates Ca dynamics and ERK1/2 phosphorylation in murine neocortical neurons. J. Neurochem. 2004, 89, 739–749. [Google Scholar] [CrossRef]
- George, J.; Dravid, S.M.; Prakash, A.; Xie, J.; Peterson, J.; Jabba, S.V.; Baden, D.G.; Murray, T.F. Sodium channel activation augments NMDA receptor function and promotes neurite outgrowth in immature cerebrocortical neurons. J. Neurosci. 2009, 29, 3288–3301. [Google Scholar] [CrossRef] [PubMed]
- Sequeira, E.; Pierce, M.L.; Akasheh, D.; Sellers, S.; Gerwick, W.H.; Baden, D.G.; Murray, T.F. Epicortical Brevetoxin Treatment Promotes Neural Repair and Functional Recovery after Ischemic Stroke. Mar. Drugs 2020, 18, 374. [Google Scholar] [CrossRef]
- Ghosh, A.; Greenberg, M.E. Calcium signaling in neurons: Molecular mechanisms and cellular consequences. Science 1995, 268, 239–247. [Google Scholar] [CrossRef]
- West, A.E.; Chen, W.G.; Dalva, M.B.; Dolmetsch, R.E.; Kornhauser, J.M.; Shaywitz, A.J.; Takasu, M.A.; Tao, X.; Greenberg, M.E. Calcium regulation of neuronal gene expression. Proc. Natl. Acad. Sci. USA 2001, 98, 11024–11031. [Google Scholar] [CrossRef]
- West, A.E.; Griffith, E.C.; Greenberg, M.E. Regulation of transcription factors by neuronal activity. Nat. Rev. Neurosci. 2002, 3, 921–931. [Google Scholar] [CrossRef]
- Wayman, G.A.; Impey, S.; Marks, D.; Saneyoshi, T.; Grant, W.F.; Derkach, V.; Soderling, T.R. Activity-dependent dendritic arborization mediated by CaM-kinase I activation and enhanced CREB-dependent transcription of Wnt-2. Neuron 2006, 50, 897–909. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.O.; Ghosh, A. Activity-dependent regulation of dendritic growth and patterning. Nat. Rev. Neurosci. 2002, 3, 803–812. [Google Scholar] [CrossRef] [PubMed]
- Ultanir, S.K.; Kim, J.E.; Hall, B.J.; Deerinck, T.; Ellisman, M.; Ghosh, A. Regulation of spine morphology and spine density by NMDA receptor signaling in vivo. Proc. Natl. Acad. Sci. USA 2007, 104, 19553–19558. [Google Scholar] [CrossRef]
- Saneyoshi, T.; Wayman, G.; Fortin, D.; Davare, M.; Hoshi, N.; Nozaki, N.; Natsume, T.; Soderling, T.R. Activity-dependent synaptogenesis: Regulation by a CaM-kinase kinase/CaM-kinase I/betaPIX signaling complex. Neuron 2008, 57, 94–107. [Google Scholar] [CrossRef] [PubMed]
- Behar, T.N.; Scott, C.A.; Greene, C.L.; Wen, X.; Smith, S.V.; Maric, D.; Liu, Q.Y.; Colton, C.A.; Barker, J.L. Glutamate acting at NMDA receptors stimulates embryonic cortical neuronal migration. J. Neurosci. 1999, 19, 4449–4461. [Google Scholar] [CrossRef]
- Scheetz, A.J.; Constantine-Paton, M. Modulation of NMDA receptor function: Implications for vertebrate neural development. FASEB J. 1994, 8, 745–752. [Google Scholar] [CrossRef]
- Furukawa, H.; Singh, S.K.; Mancusso, R.; Gouaux, E. Subunit arrangement and function in NMDA receptors. Nature 2005, 438, 185–192. [Google Scholar] [CrossRef]
- Cull-Candy, S.G.; Leszkiewicz, D.N. Role of distinct NMDA receptor subtypes at central synapses. Sci. STKE 2004, 2004, re16. [Google Scholar] [CrossRef]
- Erreger, K.; Chen, P.E.; Wyllie, D.J.; Traynelis, S.F. Glutamate receptor gating. Crit. Rev. Neurobiol. 2004, 16, 187–224. [Google Scholar] [CrossRef]
- Dammerman, R.S.; Kriegstein, A.R. Transient actions of neurotransmitters during neocortical development. Epilepsia 2000, 41, 1080–1081. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Feldmeyer, D.; Suzuki, N.; Onodera, K.; Cull-Candy, S.G.; Sakimura, K.; Mishina, M. Functional correlation of NMDA receptor epsilon subunits expression with the properties of single-channel and synaptic currents in the developing cerebellum. J. Neurosci. 1996, 16, 4376–4382. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Mishina, M.; Inoue, Y. Distinct spatiotemporal expressions of five NMDA receptor channel subunit mRNAs in the cerebellum. J. Comp. Neurol. 1994, 343, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Akazawa, C.; Shigemoto, R.; Bessho, Y.; Nakanishi, S.; Mizuno, N. Differential expression of five N-methyl-D-aspartate receptor subunit mRNAs in the cerebellum of developing and adult rats. J. Comp. Neurol. 1994, 347, 150–160. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, M.D. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat. Neurosci. 2003, 6, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Fujisawa, S.; Aoki, C. In vivo blockade of N-methyl-D-aspartate receptors induces rapid trafficking of NR2B subunits away from synapses and out of spines and terminals in adult cortex. Neuroscience 2003, 121, 51–63. [Google Scholar] [CrossRef]
- Li, B.; Chen, N.; Luo, T.; Otsu, Y.; Murphy, T.H.; Raymond, L.A. Differential regulation of synaptic and extra-synaptic NMDA receptors. Nat. Neurosci. 2002, 5, 833–834. [Google Scholar] [CrossRef]
- Rumbaugh, G.; Vicini, S. Distinct synaptic and extrasynaptic NMDA receptors in developing cerebellar granule neurons. J. Neurosci. 1999, 19, 10603–10610. [Google Scholar] [CrossRef]
- Tovar, K.R.; Westbrook, G.L. The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J. Neurosci. 1999, 19, 4180–4188. [Google Scholar] [CrossRef]
- van Zundert, B.; Yoshii, A.; Constantine-Paton, M. Receptor compartmentalization and trafficking at glutamate synapses: A developmental proposal. Trends Neurosci. 2004, 27, 428–437. [Google Scholar] [CrossRef]
- Ethell, I.M.; Pasquale, E.B. Molecular mechanisms of dendritic spine development and remodeling. Prog. Neurobiol. 2005, 75, 161–205. [Google Scholar] [CrossRef]
- Papadia, S.; Stevenson, P.; Hardingham, N.R.; Bading, H.; Hardingham, G.E. Nuclear Ca2+ and the cAMP response element-binding protein family mediate a late phase of activity-dependent neuroprotection. J. Neurosci. 2005, 25, 4279–4287. [Google Scholar] [CrossRef] [PubMed]
- Leveille, F.; El Gaamouch, F.; Gouix, E.; Lecocq, M.; Lobner, D.; Nicole, O.; Buisson, A. Neuronal viability is controlled by a functional relation between synaptic and extrasynaptic NMDA receptors. FASEB J. 2008, 22, 4258–4271. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, M.; Queisser, G.; Eder, A.; Wiegert, J.S.; Bengtson, C.P.; Hellwig, A.; Wittum, G.; Bading, H. Synaptic activity induces dramatic changes in the geometry of the cell nucleus: Interplay between nuclear structure, histone H3 phosphorylation, and nuclear calcium signaling. J. Neurosci. 2009, 29, 14687–14700. [Google Scholar] [CrossRef] [PubMed]
- Leveille, F.; Papadia, S.; Fricker, M.; Bell, K.F.; Soriano, F.X.; Martel, M.A.; Puddifoot, C.; Habel, M.; Wyllie, D.J.; Ikonomidou, C.; et al. Suppression of the intrinsic apoptosis pathway by synaptic activity. J. Neurosci. 2010, 30, 2623–2635. [Google Scholar] [CrossRef] [PubMed]
- Hardingham, G.E.; Fukunaga, Y.; Bading, H. Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat. Neurosci. 2002, 5, 405–414. [Google Scholar] [CrossRef]
- Gouix, E.; Leveille, F.; Nicole, O.; Melon, C.; Had-Aissouni, L.; Buisson, A. Reverse glial glutamate uptake triggers neuronal cell death through extrasynaptic NMDA receptor activation. Mol. Cell. Neurosci. 2009, 40, 463–473. [Google Scholar] [CrossRef]
- Soderling, T.R.; Stull, J.T. Structure and regulation of calcium/calmodulin-dependent protein kinases. Chem. Rev. 2001, 101, 2341–2352. [Google Scholar] [CrossRef]
- Bennett, M.K.; Erondu, N.E.; Kennedy, M.B. Purification and characterization of a calmodulin-dependent protein kinase that is highly concentrated in brain. J. Biol. Chem. 1983, 258, 12735–12744. [Google Scholar] [CrossRef]
- Jourdain, P.; Fukunaga, K.; Muller, D. Calcium/calmodulin-dependent protein kinase II contributes to activity-dependent filopodia growth and spine formation. J. Neurosci. 2003, 23, 10645–10649. [Google Scholar] [CrossRef]
- Lisman, J.; Schulman, H.; Cline, H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat. Rev. Neurosci. 2002, 3, 175–190. [Google Scholar] [CrossRef]
- Newey, S.E.; Velamoor, V.; Govek, E.E.; Van Aelst, L. Rho GTPases, dendritic structure, and mental retardation. J. Neurobiol. 2005, 64, 58–74. [Google Scholar] [CrossRef] [PubMed]
- Tada, T.; Sheng, M. Molecular mechanisms of dendritic spine morphogenesis. Curr. Opin. Neurobiol. 2006, 16, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Redmond, L.; Ghosh, A. The role of Notch and Rho GTPase signaling in the control of dendritic development. Curr. Opin. Neurobiol. 2001, 11, 111–117. [Google Scholar] [CrossRef]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; Srivastava, D.P. Convergent CaMK and RacGEF signals control dendritic structure and function. Trends Cell Biol. 2008, 18, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Ridley, A.J.; Paterson, H.F.; Johnston, C.L.; Diekmann, D.; Hall, A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell 1992, 70, 401–410. [Google Scholar] [CrossRef]
- Kozma, R.; Sarner, S.; Ahmed, S.; Lim, L. Rho family GTPases and neuronal growth cone remodelling: Relationship between increased complexity induced by Cdc42Hs, Rac1, and acetylcholine and collapse induced by RhoA and lysophosphatidic acid. Mol. Cell. Biol. 1997, 17, 1201–1211. [Google Scholar] [CrossRef]
- Li, Z.; Van Aelst, L.; Cline, H.T. Rho GTPases regulate distinct aspects of dendritic arbor growth in Xenopus central neurons in vivo. Nat. Neurosci. 2000, 3, 217–225. [Google Scholar] [CrossRef]
- Threadgill, R.; Bobb, K.; Ghosh, A. Regulation of dendritic growth and remodeling by Rho, Rac, and Cdc42. Neuron 1997, 19, 625–634. [Google Scholar] [CrossRef]
- Hayashi, M.L.; Choi, S.Y.; Rao, B.S.; Jung, H.Y.; Lee, H.K.; Zhang, D.; Chattarji, S.; Kirkwood, A.; Tonegawa, S. Altered cortical synaptic morphology and impaired memory consolidation in forebrain-specific dominant-negative PAK transgenic mice. Neuron 2004, 42, 773–787. [Google Scholar] [CrossRef]
- Zhang, H.; Webb, D.J.; Asmussen, H.; Niu, S.; Horwitz, A.F. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J. Neurosci. 2005, 25, 3379–3388. [Google Scholar] [CrossRef] [Green Version]
- Boda, B.; Nikonenko, I.; Alberi, S.; Muller, D. Central nervous system functions of PAK protein family: From spine morphogenesis to mental retardation. Mol. Neurobiol. 2006, 34, 67–80. [Google Scholar] [CrossRef]
- Hofmann, C.; Shepelev, M.; Chernoff, J. The genetics of Pak. J. Cell Sci. 2004, 117, 4343–4354. [Google Scholar] [CrossRef] [PubMed]
- Bokoch, G.M. Biology of the p21-activated kinases. Annu. Rev. Biochem. 2003, 72, 743–781. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.C.; Sanders, L.C.; Bokoch, G.M.; Gill, G.N. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat. Cell Biol. 1999, 1, 253–259. [Google Scholar] [CrossRef]
- Delorme, V.; Machacek, M.; DerMardirossian, C.; Anderson, K.L.; Wittmann, T.; Hanein, D.; Waterman-Storer, C.; Danuser, G.; Bokoch, G.M. Cofilin activity downstream of Pak1 regulates cell protrusion efficiency by organizing lamellipodium and lamella actin networks. Dev. Cell 2007, 13, 646–662. [Google Scholar] [CrossRef]
- Huettner, J.E.; Bean, B.P. Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: Selective binding to open channels. Proc. Natl. Acad. Sci. USA 1988, 85, 1307–1311. [Google Scholar] [CrossRef]
- Ogden, K.K.; Traynelis, S.F. New advances in NMDA receptor pharmacology. Trends Pharmacol. Sci. 2011, 32, 726–733. [Google Scholar] [CrossRef]
- Hansen, K.B.; Ogden, K.K.; Traynelis, S.F. Subunit-selective allosteric inhibition of glycine binding to NMDA receptors. J. Neurosci. 2012, 32, 6197–6208. [Google Scholar] [CrossRef]
- Williams, K. Ifenprodil discriminates subtypes of the N-methyl-D-aspartate receptor: Selectivity and mechanisms at recombinant heteromeric receptors. Mol. Pharmacol. 1993, 44, 851–859. [Google Scholar]
- Abrahamsson, T. Characterization of the beta 1-adrenoceptor stimulatory effects of the partial beta 1-agonists acebutolol, xamoterol, H142/08 and H201/70. Eur. J. Pharmacol. 1989, 164, 121–128. [Google Scholar] [CrossRef]
- Berman, F.W.; Murray, T.F. Brevetoxin-induced autocrine excitotoxicity is associated with manifold routes of Ca2+ influx. J. Neurochem. 2000, 74, 1443–1451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, M.; Cummings, J.; Roldan, L.A.; Jan, Y.N.; Jan, L.Y. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 1994, 368, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Ishima, T.; Hashimoto, K. Potentiation of nerve growth factor-induced neurite outgrowth in PC12 cells by ifenprodil: The role of sigma-1 and IP3 receptors. PLoS ONE 2012, 7, e37989. [Google Scholar] [CrossRef] [PubMed]
- George, J.; Baden, D.G.; Gerwick, W.H.; Murray, T.F. Bidirectional influence of sodium channel activation on NMDA receptor-dependent cerebrocortical neuron structural plasticity. Proc. Natl. Acad. Sci. USA 2012, 109, 19840–19845. [Google Scholar] [CrossRef] [PubMed]
- Sumi, M.; Kiuchi, K.; Ishikawa, T.; Ishii, A.; Hagiwara, M.; Nagatsu, T.; Hidaka, H. The newly synthesized selective Ca2+/calmodulin dependent protein kinase II inhibitor KN-93 reduces dopamine contents in PC12h cells. Biochem. Biophys. Res. Commun. 1991, 181, 968–975. [Google Scholar] [CrossRef]
- Wong, M.H.; Samal, A.B.; Lee, M.; Vlach, J.; Novikov, N.; Niedziela-Majka, A.; Feng, J.Y.; Koltun, D.O.; Brendza, K.M.; Kwon, H.J.; et al. The KN-93 Molecule Inhibits Calcium/Calmodulin-Dependent Protein Kinase II (CaMKII) Activity by Binding to Ca2+/CaM. J. Mol. Biol. 2019, 431, 1440–1459. [Google Scholar] [CrossRef]
- Rezazadeh, S.; Claydon, T.W.; Fedida, D. KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinnamyl)-N -methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J. Pharmacol. Exp. Ther. 2006, 317, 292–299. [Google Scholar] [CrossRef]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef]
- Fuentes, E.O.; Leemhuis, J.; Stark, G.B.; Lang, E.M. Rho kinase inhibitors Y27632 and H1152 augment neurite extension in the presence of cultured Schwann cells. J. Brachial Plex. Peripher. Nerve Inj. 2008, 3, 19. [Google Scholar] [CrossRef]
- Manser, E.; Leung, T.; Salihuddin, H.; Zhao, Z.S.; Lim, L. A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 1994, 367, 40–46. [Google Scholar] [CrossRef]
- Sells, M.A.; Knaus, U.G.; Bagrodia, S.; Ambrose, D.M.; Bokoch, G.M.; Chernoff, J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 1997, 7, 202–210. [Google Scholar] [CrossRef] [Green Version]
- Deacon, S.W.; Beeser, A.; Fukui, J.A.; Rennefahrt, U.E.; Myers, C.; Chernoff, J.; Peterson, J.R. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem. Biol. 2008, 15, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Hannigan, M.; Mo, Z.; Liu, B.; Lu, W.; Wu, Y.; Smrcka, A.V.; Wu, G.; Li, L.; Liu, M.; et al. Directional sensing requires G beta gamma-mediated PAK1 and PIX alpha-dependent activation of Cdc42. Cell 2003, 114, 215–227. [Google Scholar] [CrossRef]
- Rust, M.B.; Gurniak, C.B.; Renner, M.; Vara, H.; Morando, L.; Gorlich, A.; Sassoe-Pognetto, M.; Banchaabouchi, M.A.; Giustetto, M.; Triller, A.; et al. Learning, AMPA receptor mobility and synaptic plasticity depend on n-cofilin-mediated actin dynamics. EMBO J. 2010, 29, 1889–1902. [Google Scholar] [CrossRef]
- Gu, J.; Lee, C.W.; Fan, Y.; Komlos, D.; Tang, X.; Sun, C.; Yu, K.; Hartzell, H.C.; Chen, G.; Bamburg, J.R.; et al. ADF/cofilin-mediated actin dynamics regulate AMPA receptor trafficking during synaptic plasticity. Nat. Neurosci. 2010, 13, 1208–1215. [Google Scholar] [CrossRef]
- Chen, L.Y.; Rex, C.S.; Casale, M.S.; Gall, C.M.; Lynch, G. Changes in synaptic morphology accompany actin signaling during LTP. J. Neurosci. 2007, 27, 5363–5372. [Google Scholar] [CrossRef]
- Bosch, M.; Castro, J.; Saneyoshi, T.; Matsuno, H.; Sur, M.; Hayashi, Y. Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 2014, 82, 444–459. [Google Scholar] [CrossRef]
- Yu, X.M.; Salter, M.W. Gain control of NMDA-receptor currents by intracellular sodium. Nature 1998, 396, 469–474. [Google Scholar] [CrossRef]
- Rose, C.R.; Konnerth, A. NMDA receptor-mediated Na+ signals in spines and dendrites. J. Neurosci. 2001, 21, 4207–4214. [Google Scholar] [CrossRef]
- Groc, L.; Heine, M.; Cousins, S.L.; Stephenson, F.A.; Lounis, B.; Cognet, L.; Choquet, D. NMDA receptor surface mobility depends on NR2A-2B subunits. Proc. Natl. Acad. Sci. USA 2006, 103, 18769–18774. [Google Scholar] [CrossRef]
- Gambrill, A.C.; Storey, G.P.; Barria, A. Dynamic regulation of NMDA receptor transmission. J. Neurophysiol. 2011, 105, 162–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gambrill, A.C.; Barria, A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc. Natl. Acad. Sci. USA 2011, 108, 5855–5860. [Google Scholar] [CrossRef] [PubMed]
- Holtmaat, A.; Wilbrecht, L.; Knott, G.W.; Welker, E.; Svoboda, K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature 2006, 441, 979–983. [Google Scholar] [CrossRef] [PubMed]
- Henle, F.; Dehmel, M.; Leemhuis, J.; Fischer, C.; Meyer, D.K. Role of GluN2A and GluN2B subunits in the formation of filopodia and secondary dendrites in cultured hippocampal neurons. Naunyn-Schmiedebergs Arch. Pharmacol. 2012, 385, 171–180. [Google Scholar] [CrossRef]
- Konur, S.; Ghosh, A. Calcium signaling and the control of dendritic development. Neuron 2005, 46, 401–405. [Google Scholar] [CrossRef]
- Redmond, L.; Kashani, A.H.; Ghosh, A. Calcium regulation of dendritic growth via CaM kinase IV and CREB-mediated transcription. Neuron 2002, 34, 999–1010. [Google Scholar] [CrossRef]
- Bayer, K.U.; De Koninck, P.; Leonard, A.S.; Hell, J.W.; Schulman, H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature 2001, 411, 801–805. [Google Scholar] [CrossRef]
- Bayer, K.U.; LeBel, E.; McDonald, G.L.; O’Leary, H.; Schulman, H.; De Koninck, P. Transition from reversible to persistent binding of CaMKII to postsynapti sites and NR2B. J. Neurosci. 2006, 26, 1164–1174. [Google Scholar] [CrossRef]
- Goshima, Y.; Ohsako, S.; Yamauchi, T. Overexpression of Ca2+/calmodulin-dependent protein kinase II in Neuro2a and NG108-15 neuroblastoma cell lines promotes neurite outgrowth and growth cone motility. J. Neurosci. 1993, 13, 559–567. [Google Scholar] [CrossRef]
- Sogawa, Y.; Yoshimura, Y.; Otaka, A.; Yamauchi, T. Ca2+-independent activity of Ca2+/calmodulin-dependent protein kinase II involved in stimulation of neurite outgrowth in neuroblastoma cells. Brain Res. 2000, 881, 165–175. [Google Scholar] [CrossRef]
- Dickson, B.J. Rho GTPases in growth cone guidance. Curr. Opin. Neurobiol. 2001, 11, 103–110. [Google Scholar] [CrossRef]
- Govek, E.E.; Newey, S.E.; Van Aelst, L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005, 19, 1–49. [Google Scholar] [CrossRef] [PubMed]
- Nobes, C.D.; Hall, A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell 1995, 81, 53–62. [Google Scholar] [CrossRef]
- Tolias, K.F.; Bikoff, J.B.; Burette, A.; Paradis, S.; Harrar, D.; Tavazoie, S.; Weinberg, R.J.; Greenberg, M.E. The Rac1-GEF Tiam1 couples the NMDA receptor to the activity-dependent development of dendritic arbors and spines. Neuron 2005, 45, 525–538. [Google Scholar] [CrossRef]
- Hayashi-Takagi, A.; Takaki, M.; Graziane, N.; Seshadri, S.; Murdoch, H.; Dunlop, A.J.; Makino, Y.; Seshadri, A.J.; Ishizuka, K.; Srivastava, D.P.; et al. Disrupted-in-Schizophrenia 1 (DISC1) regulates spines of the glutamate synapse via Rac1. Nat. Neurosci. 2010, 13, 327–332. [Google Scholar] [CrossRef]
- Sala, C.; Cambianica, I.; Rossi, F. Molecular mechanisms of dendritic spine development and maintenance. Acta Neurobiol. Exp. (Wars) 2008, 68, 289–304. [Google Scholar]
- Hou, H.; Chavez, A.E.; Wang, C.C.; Yang, H.; Gu, H.; Siddoway, B.A.; Hall, B.J.; Castillo, P.E.; Xia, H. The Rac1 inhibitor NSC23766 suppresses CREB signaling by targeting NMDA receptor function. J. Neurosci. 2014, 34, 14006–14012. [Google Scholar] [CrossRef]
- Jakobi, R.; Moertl, E.; Koeppel, M.A. p21-activated protein kinase gamma-PAK suppresses programmed cell death of BALB3T3 fibroblasts. J. Biol. Chem. 2001, 276, 16624–16634. [Google Scholar] [CrossRef]
- Kiosses, W.B.; Hood, J.; Yang, S.; Gerritsen, M.E.; Cheresh, D.A.; Alderson, N.; Schwartz, M.A. A dominant-negative p65 PAK peptide inhibits angiogenesis. Circ. Res. 2002, 90, 697–702. [Google Scholar] [CrossRef]
- Manabe, R.; Kovalenko, M.; Webb, D.J.; Horwitz, A.R. GIT1 functions in a motile, multi-molecular signaling complex that regulates protrusive activity and cell migration. J. Cell Sci. 2002, 115, 1497–1510. [Google Scholar] [CrossRef]
- Sells, M.A.; Boyd, J.T.; Chernoff, J. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J. Cell Biol. 1999, 145, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Chen, Z.; Ambrose, D.; Liu, J.; Gibbs, J.B.; Chernoff, J.; Field, J. Kinase-deficient Pak1 mutants inhibit Ras transformation of Rat-1 fibroblasts. Mol. Cell. Biol. 1997, 17, 4454–4464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.S.; Lim, J.P.; Ng, Y.W.; Lim, L.; Manser, E. The GIT-associated kinase PAK targets to the centrosome and regulates Aurora-A. Mol. Cell 2005, 20, 237–249. [Google Scholar] [CrossRef]
- Cau, J.; Faure, S.; Comps, M.; Delsert, C.; Morin, N. A novel p21-activated kinase binds the actin and microtubule networks and induces microtubule stabilization. J. Cell Biol. 2001, 155, 1029–1042. [Google Scholar] [CrossRef] [PubMed]
- Duffney, L.J.; Wei, J.; Cheng, J.; Liu, W.; Smith, K.R.; Kittler, J.T.; Yan, Z. Shank3 deficiency induces NMDA receptor hypofunction via an actin-dependent mechanism. J. Neurosci. 2013, 33, 15767–15778. [Google Scholar] [CrossRef]
- Sundberg-Smith, L.J.; Doherty, J.T.; Mack, C.P.; Taylor, J.M. Adhesion stimulates direct PAK1/ERK2 association and leads to ERK-dependent PAK1 Thr212 phosphorylation. J. Biol. Chem. 2005, 280, 2055–2064. [Google Scholar] [CrossRef]
- Zhong, J.L.; Banerjee, M.D.; Nikolic, M. Pak1 and its T212 phosphorylated form accumulate in neurones and epithelial cells of the developing rodent. Dev. Dyn. 2003, 228, 121–127. [Google Scholar] [CrossRef]
- Sananbenesi, F.; Fischer, A.; Wang, X.; Schrick, C.; Neve, R.; Radulovic, J.; Tsai, L.H. A hippocampal Cdk5 pathway regulates extinction of contextual fear. Nat. Neurosci. 2007, 10, 1012–1019. [Google Scholar] [CrossRef]
- Pacico, N.; Mingorance-Le Meur, A. New in vitro phenotypic assay for epilepsy: Fluorescent measurement of synchronized neuronal calcium oscillations. PLoS ONE 2014, 9, e84755. [Google Scholar] [CrossRef]
- Turrigiano, G.G.; Leslie, K.R.; Desai, N.S.; Rutherford, L.C.; Nelson, S.B. Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 1998, 391, 892–896. [Google Scholar] [CrossRef]
- dos Remedios, C.G.; Chhabra, D.; Kekic, M.; Dedova, I.V.; Tsubakihara, M.; Berry, D.A.; Nosworthy, N.J. Actin binding proteins: Regulation of cytoskeletal microfilaments. Physiol. Rev. 2003, 83, 433–473. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.Y.; DerMardirossian, C.; Bokoch, G.M. Cofilin phosphatases and regulation of actin dynamics. Curr. Opin. Cell Biol. 2006, 18, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Mandal, M.; Yan, Z. Phosphatidylinositol (4,5)-bisphosphate regulation of N-methyl-D-aspartate receptor channels in cortical neurons. Mol. Pharmacol. 2009, 76, 1349–1359. [Google Scholar] [CrossRef] [PubMed]
- Aizawa, H.; Wakatsuki, S.; Ishii, A.; Moriyama, K.; Sasaki, Y.; Ohashi, K.; Sekine-Aizawa, Y.; Sehara-Fujisawa, A.; Mizuno, K.; Goshima, Y.; et al. Phosphorylation of cofilin by LIM-kinase is necessary for semaphorin 3A-induced growth cone collapse. Nat. Neurosci. 2001, 4, 367–373. [Google Scholar] [CrossRef]
- Jabba, S.V.; Prakash, A.; Dravid, S.M.; Gerwick, W.H.; Murray, T.F. Antillatoxin, a novel lipopeptide, enhances neurite outgrowth in immature cerebrocortical neurons through activation of voltage-gated sodium channels. J. Pharmacol. Exp. Ther. 2010, 332, 698–709. [Google Scholar] [CrossRef]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehrotra, S.; Pierce, M.L.; Dravid, S.M.; Murray, T.F. Stimulation of Neurite Outgrowth in Cerebrocortical Neurons by Sodium Channel Activator Brevetoxin-2 Requires Both N-Methyl-D-aspartate Receptor 2B (GluN2B) and p21 Protein (Cdc42/Rac)-Activated Kinase 1 (PAK1). Mar. Drugs 2022, 20, 559. https://doi.org/10.3390/md20090559
Mehrotra S, Pierce ML, Dravid SM, Murray TF. Stimulation of Neurite Outgrowth in Cerebrocortical Neurons by Sodium Channel Activator Brevetoxin-2 Requires Both N-Methyl-D-aspartate Receptor 2B (GluN2B) and p21 Protein (Cdc42/Rac)-Activated Kinase 1 (PAK1). Marine Drugs. 2022; 20(9):559. https://doi.org/10.3390/md20090559
Chicago/Turabian StyleMehrotra, Suneet, Marsha L. Pierce, Shashank M. Dravid, and Thomas F. Murray. 2022. "Stimulation of Neurite Outgrowth in Cerebrocortical Neurons by Sodium Channel Activator Brevetoxin-2 Requires Both N-Methyl-D-aspartate Receptor 2B (GluN2B) and p21 Protein (Cdc42/Rac)-Activated Kinase 1 (PAK1)" Marine Drugs 20, no. 9: 559. https://doi.org/10.3390/md20090559
APA StyleMehrotra, S., Pierce, M. L., Dravid, S. M., & Murray, T. F. (2022). Stimulation of Neurite Outgrowth in Cerebrocortical Neurons by Sodium Channel Activator Brevetoxin-2 Requires Both N-Methyl-D-aspartate Receptor 2B (GluN2B) and p21 Protein (Cdc42/Rac)-Activated Kinase 1 (PAK1). Marine Drugs, 20(9), 559. https://doi.org/10.3390/md20090559