Abstract
Six new nostocyclophanes and four known compounds have been isolated from Nostoc linckia (Nostocaceae) cyanobacterial strain UTEX B1932. The new compounds, nostocyclophanes E–J (1–6), were characterized by NMR and MS techniques. The known compounds were nostocyclophanes B–D, previously isolated from this strain, and dedichloronostocyclophane D. Structural modifications on the new [7.7]paracyclophane analogs 1–5, isolated from the 80% methanol fraction, range from simple changes such as the lack of methylation or halogenation to more unusual modifications such as those seen in nostocyclophane H (4), in which the exocyclic alkyl chains are of different length; this is the first time this modification has been observed in this family of natural products. In addition, nostocyclophane J (6) is a linear analog in which C-20 is chlorinated in preparation for the presumed enzymatic Friedel–Craft cyclization needed to form the final ring structure, analogous to the biosynthesis of the related cylindrocyclophanes. Nostocyclophane D, dedichloronostocyclophane D, and nostocyclophanes E-J demonstrated moderate to weak growth inhibition against MDA-MB-231 breast cancer cells.
1. Introduction
Filamentous cyanobacteria are prolific producers of biologically potent secondary metabolites [1], often with highly unusual structures [2]. One such unique class of compounds is [7.7]paracyclophanes [3] such as cylindrocyclophanes, nostocyclophanes [4], merocyclophanes [5] and carbamidocyclophanes [6] produced by cyanobacteria; compounds whose activities range from weakly cytotoxic [4] to moderately antibacterial (MRSA) [7]. Structurally unusual, even among synthetic compounds, these [7.7]paracyclophanes contain 22-membered rings formed by the head-to-tail dimerization of an alkyl resorcinol intermediate. Labelling studies by Moore and Bobzin [8] in the 1990s established the acetate origin of most of the carbons and led to a proposed biosynthesis that involved C-C bond formation between an sp2 and sp3 center that linked the monomers by an unknown mechanism using an unknown intermediate [8]. Studies 20 years later by Balskus et al. identified the biosynthetic gene cluster (BGC) for the cylindrocyclophanes based on an HMG-CoA synthase homolog [9,10]. They subsequently determined that ring formation for that series depended on cryptic halogenation of the alkyl chain by a novel halogenase (CylC) and two subsequent stereospecific Friedel–Crafts reactions. Further studies have defined the substrate scope [11] and examined the key enzymatic interactions [12,13].
Several years ago, as part of a larger NIH-funded project to provide known natural products for biological screening, we began reexamining strains within the Patterson/Moore Culture Collection of cyanobacteria at the University of Hawaii. The original investigations into many of these strains in the 80s and 90s led to the discovery of the first examples of several well-known families of cyanobacterial metabolites. Hence, the primary aim of that project was to regrow strains known to produce those specific metabolites. One of those strains was Nostoc linckia strain UTEX B1932, from which nostocyclophanes A-D had been discovered; a family distinguished from other cyanobacterial [7.7]paracyclophanes by additional chlorination on the alkyl chain, and a lack of the HMG-CoA derived methyl group. While isolating nostocyclophanes B–D [4], we also isolated dedichloronostocyclophane D originally reported from a sea hare [14] and several additional minor analogs. Here, we report the structures of these new compounds within the nostocyclophane series. The structures of nostocyclophanes E–J (1–6) were deduced using NMR and MS techniques. Of particular interest is nostocyclophane J (6), a linear analog in which one of the “cryptic” halogenations is evident, and nostocyclophane H (4), derived from two different alkyl resorcinol intermediates.
2. Results and Discussion
A cryostock of UTEX B1932 was scaled up via progressively larger cultures to a final culture volume of 240 L split over 12 × 20 L glass carboys. After 37 days, the cultures were harvested by filtration. The freeze-dried cell mass was extracted thrice, and the combined extract was separated by multiple rounds of chromatography. LCMS and NMR analyses of the resulting fractions showed the 80% MeOH fraction from a C18 flash column contained the nostocyclophanes of interest. HPLC purification of this fraction yielded the known compounds nostocyclophanes B–D [4] that had been targeted for isolation, along with dedichloronostocyclophane D [14] and six new compounds, nostocyclophanes E–J (1–6).
Nostocyclophane E (1) provided a negative HRESIMS ion [M − H]− at m/z 617.3599. A molecular formula of C36H55ClO6 was deduced, and this formula did not provide a good match to any known [7.7]paracyclophanes in our available database [15]. The similarity of the 1H NMR spectrum of 1 to that of nostocyclophane D indicated that 1 belonged to this family of compounds, which had also been isolated from this fraction. The 1H and 13C NMR spectra (Table 1 and Table 2) of nostocyclophane E were more complex than those of nostocyclophane D, indicating that 1 lacked symmetry, and hence many of the NMR signals were doubled. For example, in the 1H NMR spectrum of nostocyclophane E, four phenolic and four aromatic proton signals were present rather than two of each type as observed for nostocyclophane D. A detailed comparison of the 13C NMR data for nostocyclophane D and 1 revealed that C-16 was the point of asymmetry in 1 as this was no longer a chlorinated methine resonating around 60 ppm but now was a simple methylene (30.1 ppm). Analysis of the 2D NMR spectra of 1 confirmed that conclusion. Specifically, H-14 displayed HMBC correlations to one methoxy carbon, three sp2 carbons, and two aliphatic methylenes (C-15, -16). In contrast, H-1 displayed the expected HMBC correlations to a methoxy group, three sp2 carbons (C-24, -21, -27), one aliphatic methylene, and one halogenated carbon (C-3). Evidence of the expected aromatic rings were HMBC correlations from both sets of sp2 carbons, e.g., H-10 and H-12 to C-12 and C-10, respectively, and to quaternary carbons C-11, C-12, and C-13. Finally, while no HMBC correlations were visible in our data to prove the C-7/C-8 and C-20/C-21 connections, these connections are required by the number of degrees of unsaturation.

Table 1.
1H NMR Spectroscopic Data (500 MHz, δH (J in Hz)) for compounds 1–4 (DMSO-d6).
Table 2.
13C NMR Spectroscopic Data (125 MHz) for 1–4 (DMSO-d6).
Compounds 2 and 3 were simple analogs of 1 whose structures were readily deduced by comparison of their NMR spectra and MS spectrometric data with the known members of the nostocyclophane structural family (Figure 1). Briefly, nostocyclophane F (2) differed from 1 by 14 amu, which could be attributed to a missing methyl group (14-OMe) attached to the corresponding oxygen at C-14. The change in chemical shift of C-14 (73.5 ppm for F vs. 83.6 ppm for E) supports this conclusion. Finally, nostocyclophane G (3), with a molecular formula of C35H52Cl2O6 ([M − H]− = 621.3102), clearly lacked one of the methoxy groups found in nostocyclophane D. Based on analyses of 1H and 13C NMR chemical shifts, C-14 was no longer oxygenated and now corresponded to a methylene. Analyses of the 2D NMR data supported this conclusion. Specifically, the HSQC spectrum revealed an additional aliphatic methylene carbon at 31.2 ppm that correlated to a proton multiplet at 2.40 ppm. This proton signal, in turn, showed HMBC correlations to three sp2 carbons (C-10, -11, and -12), establishing the benzylic location of the modification. The types of modifications observed for 2 and 3 have been previously observed [4].
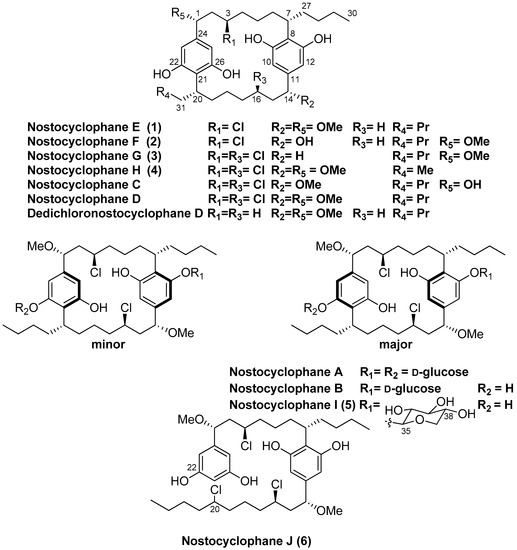
Figure 1.
Nostocyclophane structures.
Comparison of the NMR spectra and MS spectrometric data for nostocyclophane H (4) (Table 1 and Table 2) with the other nostocyclophane analogs isolated revealed an unusual modification in this compound. The molecular formula of 4 was C34H50Cl2O6 based on the ion observed at [M − H]− = 623.2897 Da. This formula indicated 4 was smaller than nostocyclophane D by C2H4, which, given the previously observed modifications, was initially attributed to the lack of two O-methylations. Further analysis of the 1H NMR spectrum indicated this assumption was incorrect, as signals for both methoxy groups were observed. Analysis of the 13C NMR spectrum also indicated two methylene carbons were missing compared to that of 1. Based on analysis of the 1H-1H COSY and HSQC spectra of 4, it was clear the exocyclic butyl group appended to C-20 in 1 had been truncated to an ethyl group as evidenced by COSY correlations between H2-31 to H-20 and H3-32. Specifically, methylene carbons at 32.2 and 22.2 ppm were absent from the HSQC spectra of 4, and one of the triplet methyl signals (H3-32) now showed an HMBC correlation to a methine at 37.0 ppm (C-20). To our knowledge, such a modification has not been observed before in the cyanobacterial [7.7]paracyclophanes. Given that these carbons originate from a fatty acid starter unit in the related cylindrocyclophanes, an octanoic starter unit rather than a decanoic acid was likely loaded for one of the monomers (Figure 2). In the case of the cylindrocyclophanes, the corresponding enzyme will accept fatty acids of variable lengths, although it has a clear preference for C-10 fatty acids. In LC-MS competition assays, the loaded enzyme contained a small amount of C-8 (<10%) when the enzyme was presented with equal amounts of C-6 through C-14 saturated fatty acids [9]. The fact that 4 was in relatively low abundance (1.5 mg) compared to the major metabolite nostocyclophane D (200 mg) is consistent with this observation.
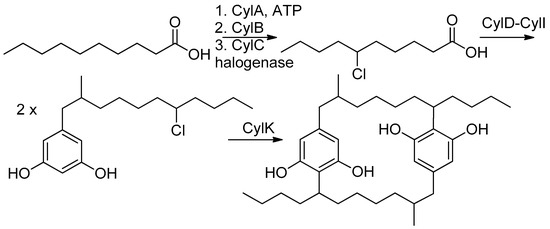
Figure 2.
Established biosynthesis for cylindrocyclophane F [16].
Nostocyclophane I (5) was obtained as a colorless powder with a molecular formula of C41H62Cl2O10 ([M − H]− = 783.3625 Da). The spectroscopic data for 5 were very similar to those of nostocyclophane B. Analysis of the 1H and 13C NMR spectra (Table 3) indicated that nostocyclophane I existed in two conformations in solution. The observation of two dominant conformers in the NMR spectra of glycosylated nostocyclophane analogs has been previously noted and provided an initial clue as to the identity of 5 [4]. Further analysis of the NMR spectra led to the conclusion that 5 possessed the typical [7.7]paracyclophane core. For example, two benzylic carbons at ~80 ppm could be easily identified by HMBC correlations from the attached methoxy groups. Those oxygenated benzylic methines showed COSY correlations to a methylene at ~4.05 ppm, which then showed COSY correlations to a methylene signal at 2.08 ppm, which further correlated to a proton signal at ~2.8 ppm. The proton chemical shift of that latter proton was consistent with a halide being attached to the corresponding carbon. The main differences in the spectra of 5 and nostocyclophane B could therefore be explained by the presence of a β-xylopyranoside in 5 rather than a D-β-glucopyranoside. Evidence in support of a xylose unit was the presence of an oxygenated methylene (δC 65.4), expected for this sugar, which showed an HMBC correlation from the anomeric proton (δH 4.93, d) and a series of consecutive COSY correlations that established the ring, i.e., from that anomeric proton to H-36, from H-36 to H-37, from H-37 to H-38, and then from H-38 to that same oxygenated methylene (H-39). As to the relative configuration of this unit, the equatorial orientations for the hydroxy groups at C-36, C-37 and C-38 were established by a network of 3JH,H couplings starting at the anomeric proton (H-35, δH 4.62 d, (7.0 Hz)) through H-38 (δH 3.29, ddd (10.9, 8.8, 5.0 Hz)). The β-configuration of the sugar was established by the observation of a 7.0 Hz coupling, consistent with the data reported for hexyl β-xylopyranoside, between the anomeric proton H-35 and H-36 as opposed to the 3.5 Hz coupling observed for the α-configuration [17]. Interestingly, this is the first report of a different sugar unit within the nostocyclophane class of [7.7]paracyclophanes, as nostocyclophanes A-B contain a D-glucose unit.
Table 3.
1H and 13C NMR Spectroscopic Data for 5 (DMSO-d6).
Another nostocyclophane analog that did not fit the typical structural pattern was isolated from the extract. An initial inspection of the NMR data revealed the compound was asymmetrical, given the number of resonances observed. In addition, the 1H NMR spectrum of 6 contained a total of five aromatic resonances rather than four as observed in the other asymmetrical analogs, e.g., nostocyclophane D. HRMS data provide another valuable clue as 6 provided an isotope pattern consistent with the incorporation of three chlorine atoms, one more than previously observed for any of the known nostocyclophanes. Analyses of 1H-1H COSY, HSQC, and HMBC spectra, which were collected in CDCl3 as it provided better dispersion in this case, established that 6 lacked the characteristic linkage between C-20 and C-21 and was, therefore, an uncyclized version of nostocyclophane D. In particular, the HSQC spectrum clearly indicated the presence of a new signal for an aromatic methine at 6.09 ppm (H-21) and a new downfield methine signal at δH 3.97 (H-20) that showed a 1JCH to a carbon at 64.5 ppm. The latter aromatic signal showed HMBC correlations to two other aromatic signals (C-22 and C-21), which in turn could be connected to a benzylic carbon (C-1) bearing one of the methoxy groups using HMBC data. In contrast, the signal at 3.94 ppm (H-20) showed COSY correlations only to signals indicative of aliphatic protons.
The relative and absolute configurations of 1–6 are assumed to be analogous to those of the other members of this series, also isolated from this strain. The configuration of nostocyclophane D produced by this strain (UTEX B1932) was established by X-ray crystallographic analysis [3], as NOE analysis is expected to provide no useful transannular information due to the distance between each set of isolated stereocenters. It was subsequently shown by ECD analysis that nostocyclophanes A-D exhibited similar ECD curves, indicating that the other analogs had identical configurations to nostocyclophane D [4]. Not surprisingly, the ECD curves for nostocyclophane D and 1–5 have identical Cotton effects as well (See Figure S33), with a strong negative Cotton effect at around 214, a shoulder at 230 nm, and two broad weakly negative effects between 260 and 280 nm. The ECD spectrum of the linear analog 6 (see Figure S34) shows similar Cotton effects to 1–5, consistent with the report of the similarity between the ECD spectra of linear and cyclic forms of the cylindrofridins [7]. Based on their shared biosynthesis, 1–6 are expected to share the same configurations as nostocyclophane D at the conserved chiral centers. Regarding the final chiral center in 6, at this time, the configuration of C-20 has not been established. In the cylindrocyclophane biosynthesis, an R-configuration is required at this position of the fatty acid unit, and this sp3 center undergoes an inversion of configuration during the cyclization process [16]. A similar mechanism likely operates in the nostocyclophane biosynthesis, but it is also possible that the linear analog 6 possesses the “incorrect” configuration at C-20 and is, therefore, unable to cyclize. The corresponding cylindrocyclophane enzyme cannot cyclize fatty acids with an S-configuration [16]. Thus, C-20 in 6 remains unassigned, as it is for the other linear halogenated analog cylindrofridin B [7]. Due to the isolated nature of this center, the chemical shift differences between the C-20 diastereomers are likely to be minor [18], so determining that center’s configuration would require synthesis or X-ray crystallography rather than computational approaches relying on NMR spectroscopy.
Known cyanobacterial [7.7]paracyclophanes are divided into three basic structural families. The cylindrocyclophanes generally have hydroxylation and methylation at C-1 and C-2, respectively. In contrast, the core of the merocyclophanes is only decorated with methyl groups at C-1/C-14. Finally, the nostocyclophanes have hydroxy and chloro groups at C-1/C-14 and C-3/C-16, respectively. Linear analogs within the cylindrocyclophane family have been previously reported by Preisitsch et al. [7]. In that case, cylindrofridin A is the monomeric halogenated alkyl resorcinol, i.e., the monomer, while B and C result from a single coupling between the aromatic ring and the alkyl chain similar to nostocyclophane J, i.e., dimeric but uncyclized. The linear cylindrofridins were the first examples of this likely biosynthetic intermediate within the [7.7.]paracyclophanes structural family.
Dedichloronostocyclophane D, which notably lacked any chlorine atoms, has been previously reported from the sea slug, Planaxis sulcatus [14] and had not been reported from any cyanobacterial strain before. That report represents the first time that naturally occurring [7.7]paracyclophanes have been reported from a sea slug. Its presence is likely due to the slug feasting on cyanobacteria, as observed for other compounds [19,20].
Nostocyclophane C and 1–6 displayed moderate to weak growth inhibition against breast epithelial adenocarcinoma MDA-MB-231 cells (Table 4). The observed activity and level of potency are generally consistent with previous observations for naturally occurring [7.7]paracyclophanes. For example, nostocyclophanes A–C were cytotoxic at 1–2 μg/mL, while nostocyclophane D was slightly more potent (GI50 = ~0.76 μM) against KB (human nasopharyngeal carcinoma) and LoVo (human colon adenocarcinoma) cells [4], similar to the cylindrocyclophanes A–F (0.5–5 μg/mL). Related compounds, the merocyclophanes, were inhibitory to HT-29, NCI-H460, SF268, and MCF7 cancer cells with GI50 values of ~1–10 μM.
Table 4.
Assay Data against MDA-MB-231.
In conclusion, the structures of six new nostocyclophanes were determined by NMR and MS techniques. Of particular interest is nostocyclophane J, a linear analog in which one of the “cryptic” halogenations is evident, and nostocyclophane H, derived from two different alkyl resorcinol intermediates. These compounds provide interesting insights into the biosynthesis and substrate tolerance for Nostoc sp. B1932 and the way in which it compares to other reported [7.7]paracyclophanes. Compounds 1–6 mildly inhibited the growth of MDA-MB-231 cells, a level of activity consistent with what has been reported for other naturally occurring [7.7]paracyclophanes.
3. Materials and Methods
3.1. General Experimental Procedures
Optical rotations were measured on a Jasco-DIP-700 polarimeter at the sodium line (589 nm). UV spectra were obtained on a Hewlett-Packard 8453 spectrophotometer and IR spectra were measured as a thin film on a CaF2 disc using a Perkin Elmer 1600 series FTIR. ECD measurements were obtained on a Chirascan Circular Dichroism Spectrometer with the samples dissolved in MeOH and placed in a 1 cm quartz cuvette with a solvent subtraction for baseline correction. NMR spectra were acquired on a Varian Inova Unity 500 MHz spectrometer operating at 500 (1H) or 125 (13C) MHz using the residual solvent signals as an internal reference. Samples were in 3 mm Shigemi tubes during NMR analyses. High-resolution mass spectrometry data were obtained on an Agilent LC-TOF or LC-QTOF with ES ionization. Gradient separations used a Shimadzu system consisting of LC-20AT Solvent Delivery Modules, an SPD-M20A VP Diode Photodiode Array Detector, and a SCL-20A VP System Controller. TLC analyses were performed on Si60F254 plates and visualized under UV or by heating after spraying with a 1% anisaldehyde solution in acetic acid:H2SO4 (50:1). Samples were weighted on a Mettler Toledo analytical balance.
3.2. Chemicals and Reagents
ACS grade solvents (Fisher Scientific, Waltham, MA, USA) were distilled and filtered before use. HP-20 resin, flash column stationary phases, and HPLC columns were purchased from Supelco (St. Louis, MO, USA), Sorbent Technologies (Atlanta, GA, USA), and Phenomenex (Torrance, CA, USA), respectively.
3.3. Biological Material
Cyanobacterial sample UTEX B1932, Nostoc linckia, was originally purchased from the University of Texas Culture Collection. UTEX identified the strain before our purchase. After recovery from cryopreservation, the bacterium was cultured at a temperature of 24 ± 1 °C in 20 L glass carboys containing A3M7 media [21]. Cultures were continuously illuminated at an incident intensity of 100–200 µmol·m−2·s−1 from banks of fluorescent tubes and aerated at 0.20–0.25 times the culture volume per minute (4–5 L/min) with 0.5% CO2 in air. After 37 days, the cyanobacterial cultures were harvested through 210 µm plastic mesh by recirculating the culture via a peristaltic pump until the filtrate was clear. The retained cyanobacterial cell mass was then freeze-dried, yielding between 0.11 and 0.36 g/L of biomass.
3.4. Extraction and Isolation of Metabolites
Freeze-dried alga (35 g) was extracted three times with 2 L of 7:3 EtOH:H2O for 24 h at room temperature. The combined dark green extract (10 g) was concentrated in vacuo to 350 mL and the concentrate applied to a 25 × 4.6 cm column of ODS silica gel. The column was eluted with 1:1 MeOH:H2O (1.5 L), 4:1 MeOH:H2O (2 L), and MeOH (2 L). The 4:1 MeOH:H2O fraction was evaporated. The residue (1.3 g) was further purified by RP-HPLC (Luna C18, 250 × 10 mm, a linear gradient from 50 to 100% MeCN in water over 40 min and then 100% MeCN for 20 min) to afford 40 mg of nostocyclophane B (0.4% yield, 95.6% purity, tR 8.1 min), 5.0 mg of nostocyclophane I (0.05% yield, 98.6% purity, tR 10.1 min), 2.0 mg of nostocyclophane F (0.02% yield, 93.7% purity, tR 12.5 min), 6 mg of nostocyclophane C (0.06% yield, 97.6% purity, tR 14.2 min), 1.0 mg of nostocyclophane H (0.01% yield, 97.6% purity, tR 16.0 min), 3 mg of dedichloronostocyclophane D (0.03% yield, 98.9% purity, tR 17.2 min), 20 mg of nostocyclophane E (0.2% yield, 99.2% purity, tR 19.5 min), 200 mg of nostocyclophane D (2% yield, 95.2% purity, tR 22.5 min), 1.0 mg of nostocyclophane G (0.01% yield, 95.2% purity, tR 26.5 min), and 1.5 mg of nostocyclophane J (0.015% yield, 95.7% purity, tR 27.5 min).
- Nostocyclophane E (1): amorphous white power; [α]D22 +5.7 (c 2.0, MeOH); UV (MeOH) λmax (log ε) 209 (2.5), 214 (2.5), 227 (1.3), 277 (0.2), 283 (0.2) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 206 (0.80), 218 (−0.71), 240 (−0.20), 261 (−0.80), 284 (−0.090) nm; IR (film) νmax 3325, 2926, 2854, 1646, 1595, 1431, 1083, 1021 cm−1; see Table 1 and Table 2 for tabulated NMR spectroscopic data; HRESIMS m/z 617.3599 [M − H]− (calcd for C36H5435ClO6, 617.3609; Δ = −1.6 ppm).
- Nostocyclophane F (2): amorphous white power; [α]D22 −6.5 (c 0.2, MeOH); UV (MeOH) λmax (log ε) 210 (2.5), 214 (2.5), 227 (1.4), 277 (0.3), 283 (0.2) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 205 (1.10), 217 (−1.80), 235 (−0.67), 272 (−0.26), 280 (−0.25) nm; IR (film) νmax 3345, 2958, 2864, 1650, 1431, 1020 cm−1; see Table 1 and Table 2 for tabulated NMR spectroscopic data; HRESIMS m/z 603.3454 [M − H]− (calcd for C35H5235ClO6−, 603.3453; Δ = 0.3 ppm).
- Nostocyclophane G (3): amorphous white power; [α]D22 −25.6 (c 2.0, MeOH); UV (MeOH) λmax (log ε) 208 (0.9), 229 (0.3), 274 (0.1) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 202 (0.9), 217 (−0.20), 237 (−0.89), 285 (−0.02) nm; IR (film) νmax 3390, 2932, 2859, 1588, 1429, 1085 cm−1; see Table 1 and Table 2 for tabulated NMR spectroscopic data; HRESIMS m/z 621.3102 [M − H]− (calcd for C35H5135Cl2O5−, 621.3114; Δ = −1.9 ppm).
- Nostocyclophane H (4): amorphous white power; [α]D22 −0.2 (c 2.0, MeOH); UV (MeOH) λmax (log ε) 209 (2.4), 227 (0.9), 274 (0.2), 283 (0.2) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 207 (0.22), 218 (−0.25), 233 (−0.09), 273 (−0.35), 282 (−0.37) nm; IR (film) νmax 3399, 2932, 2873, 1589, 1428, 1089 cm−1; see Table 1 and Table 2 for tabulated NMR spectroscopic data; HRESIMS m/z 623.2901 [M − H]− (calcd for C34H4935Cl2O6−, 623.2906; Δ = −0.8 ppm).
- Nostocyclophane I (5): amorphous white power; [α]D22 +2.0 (c 1.0, MeOH); UV (MeOH) λmax (log ε) 209 (2.0), 227 (0.6), 275 (0.1), 283 (0.1) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 201 (4.9), 215 (−5.0), 233 (−1.45), 271 (−0.35), 283 (−0.34) nm; IR (film) νmax 3341, 2927, 2863, 1614, 1595, 1429, 1093 cm−1; see Table 3 for tabulated NMR spectroscopic data; HRESIMS m/z 783.3642 [M − H]− (calcd for C41H6135Cl2O10−, 783.3642; Δ = −2.1 ppm).
- Nostocyclophane J (6): amorphous white power; [α]D22 +5.0 (c 1.0, MeOH); UV (MeOH) λmax (log ε) 214 (2.6), 228 (1.7), 278 (0.4), 282 (0.4) nm; ECD (c 0.2 MeOH) λmax (Δ ε) 207 (2.6), 217 (−2.2), 228 (−0.8), 273 (−0.44), 293 (−0.37) nm. IR (film) νmax 3374, 2950, 2864, 1604, 1081 cm−1; see Table 5 for tabulated NMR spectroscopic data; HRESIMS m/z 687.2973 [M − H]− (calcd for C36H5435Cl3O6−, 687.2986; Δ = −1.9 ppm).
Table 5. 1H and 13C NMR Spectroscopic Data for 6.
3.5. Growth Inhibition Assays
MDA-MB-231 cell lines were maintained in DMEM media supplemented with 10% premium fetal bovine serum and 50 U/mL penicillin and 50 μg/mL streptomycin. One day before treatment, cancer cells were seeded at 2000 cells per well into a 96-well tissue culture plate. Twenty four hours post-seeding, the serially diluted compounds and vehicle (DMSO) controls were added to the cells for the cytotoxicity assay, and incubated at 37 °C with 5% CO2 for 72 h. Then, equal volume of MTT dye (Alfa Aesar, Teksbury, MA, USA) was added to each well and incubated with 5% CO2 at 37 °C for 60 min. Next, media were aspirated and 100 μL of DMSO were added to each well to dissolve formazan crystals. Cell viability data were collected with a Victor Nivo Multimode Microplate Reader (Perkin Elmer, Waltham, MA, USA) at 570 nm and GI50 curves were generated using GraphPad Prism 5. For GI50 determination, samples were tested in triplicate on the same plate at each concentration, and the results averaged. Three consecutive biological replicates were collected and average GI50 values were calculated. Tamoxifen (1 μM final) and Gemcitabine (1 μM final) were used as positive controls for each replicate. The MDA-MB 231 cells were obtained from the University of Hawaii Cancer Center which they obtained as part of the NCI-60 set of cell lines from National Cancer Insti-tute.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/md21020101/s1, Figures S1–S34: Copies of the NMR, MS, and ECD spectra for all new compounds.
Author Contributions
The authors contributed to the research in the following ways: Conceptualization, P.G.W.; software, C.S.P.; analysis, W.Y.Y., J.D., C.W. and P.G.W.; investigation, C.W., W.Y.Y., C.S.P., J.D. and P.G.W.; data curation, W.Y.Y. and P.G.W.; writing—original draft preparation, J.D. and P.G.W.; writing—review and editing, J.D., W.Y.Y., P.G.W., C.W. and C.S.P.; supervision, P.G.W.; project administration, P.G.W.; funding acquisition, P.G.W. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by NIH grant 5P41GM103484 to P.W. Funds for the upgrades of the NMR instrumentation were provided by the CRIF program of the National Science Foundation (CHE9974921), the Elsa Pardee Foundation, and the University of Hawaii at Manoa. The purchase of the Agilent TOF LC-MS was funded by grant W911NF-04-1-0344 from the Department of Defense, and the purchase of the Agilent QTOF LC-MS was funded by MRI grant 1532310 from the National Science Foundation.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material; further inquiries can be directed to the corresponding author.
Acknowledgments
We thank E. Haglund, J. Simien and G. Orellana for the assistance with ECD measurements.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Salvador-Reyes, L.A.; Luesch, H. Biological targets and mechanisms of action of natural products from marine cyanobacteria. Nat. Prod. Rep. 2015, 32, 478–503. [Google Scholar] [CrossRef] [PubMed]
- Prinsep, M.R.; Caplan, F.R.; Moore, R.E.; Patterson, G.M.L.; Smith, C.D. Tolyporphin, A Novel Multidrug Resistance Reversing Agent from the Blue-green Alga Tolypothrix nodosa. J. Am. Chem. Soc. 1992, 114, 385–387. [Google Scholar] [CrossRef]
- Moore, B.S.; Chen, J.L.; Patterson, G.M.L.; Moore, R.E.; Brinen, L.S.; Kato, Y.; Clardy, J. [7.7]Paracyclophanes from blue-green algae. J. Am. Chem. Soc. 1990, 112, 4061–4063. [Google Scholar] [CrossRef]
- Chen, J.L.; Moore, R.E.; Patterson, G.M.L. Structures of Nostocyclophanes A–D. J. Org. Chem. 1991, 56, 4360–4364. [Google Scholar] [CrossRef]
- May, D.S.; Chen, W.-L.; Lantvit, D.D.; Zhang, X.; Krunic, A.; Burdette, J.E.; Eustaquio, A.; Orjala, J. Merocyclophanes C and D from the Cultured Freshwater Cyanobacterium Nostoc sp. (UIC 10110). J. Nat. Prod. 2017, 80, 1073–1080. [Google Scholar] [CrossRef]
- Preisitsch, M.; Heiden, S.E.; Beerbaum, M.; Niedermeyer, T.H.J.; Schneefeld, M.; Herrmann, J.; Kumpfmüller, J.; Thürmer, A.; Neidhardt, I.; Wiesner, C.; et al. Effects of Halide Ions on the Carbamidocyclophane Biosynthesis in Nostoc sp. CAVN2. Mar. Drugs 2016, 14, 21. [Google Scholar] [CrossRef]
- Preisitsch, M.; Niedermeyer, T.H.J.; Heiden, S.E.; Neidhardt, I.; Kumpfmüller, J.; Wurster, M.; Harmrolfs, K.; Wiesner, C.; Enke, H.; Müller, R.; et al. Cylindrofridins A–C, Linear Cylindrocyclophane-Related Alkylresorcinols from the Cyanobacterium Cylindrospermum stagnale. J. Nat. Prod. 2016, 79, 106–115. [Google Scholar] [CrossRef]
- Bobzin, S.C.; Moore, R.E. Biosynthetic Origin of [7.7]paracyclophanes from Cyanobacteria. Tetrahedron 1993, 49, 7615–7626. [Google Scholar] [CrossRef]
- Nakamura, H.; Hamer, H.A.; Sirasani, G.; Balskus, E.P. Cylindrocyclophane Biosynthesis Involves Functionalization of an Unactivated Carbon Center. J. Am. Chem. Soc. 2012, 134, 18518–18521. [Google Scholar] [CrossRef]
- Moore, B.S.; Chen, J.L.; Patterson, G.M.L.; Moore, R.E. Structures of Cylindrocyclophanes A-F. Tetrahedron 1992, 48, 3001–3006. [Google Scholar] [CrossRef]
- Schultz, E.E.; Braffman, N.R.; Luescher, M.U.; Hager, H.H.; Balskus, E.P. Biocatalytic Friedel–Crafts Alkylation Using a Promiscuous Biosynthetic Enzyme. Angew. Chem. Int. Ed. 2019, 58, 3151–3155. [Google Scholar] [CrossRef] [PubMed]
- Braffman, N.R.; Ruskoski, T.B.; Davis, K.M.; Glasser, N.R.; Johnson, C.; Okafor, C.D.; Boal, A.K.; Balskus, E.P. Structural Basis for an Unprecedented Enzymatic Alkylation in Cylindrocyclophane Biosynthesis. eLife 2022, 11, e75761. [Google Scholar] [CrossRef]
- Wang, H.-Q.; Mou, S.-B.; Xiao, W.; Zhou, H.; Hou, X.-D.; Wang, S.-J.; Wang, Q.; Gao, J.; Wei, Z.; Liu, L.; et al. Structural Basis for the Friedel–Crafts Alkylation in Cylindrocyclophane Biosynthesis. ACS Catal. 2022, 12, 2108–2117. [Google Scholar] [CrossRef]
- Thanh, N.V.; Thao, N.P.; Phong, N.V.; Cuong, N.X.; Nam, N.H.; Minh, C.V. A New [7.7]paracyclophane from Vietnamese Marine Snail Planaxis sulcatus (Born, 1780). Nat. Prod. Res. 2020, 34, 261–268. [Google Scholar] [CrossRef] [PubMed]
- Van Santen, J.A.; Poynton, E.F.; Iskakova, D.; McMann, E.; Alsup, T.A.; Clark, T.N.; Fergusson, C.H.; Fewer, D.P.; Hughes, A.H.; McCadden, C.A.; et al. The Natural Products Atlas 2.0: A Database of Microbially-derived Natural Products. Nucleic Acids Res. 2021, 50, D1317–D1323. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Schultz, E.E.; Balskus, E.P. A New Strategy for Aromatic Ring Alkylation in Cylindrocyclophane Biosynthesis. Nat. Chem. Biol. 2017, 13, 916–921. [Google Scholar] [CrossRef]
- Sekine, M.; Kimura, T.; Katayama, Y.; Takahashi, D.; Toshima, K. The direct and one-pot transformation of xylan into the biodegradable surfactants, alkyl xylosides, is aided by an ionic liquid. RSC Adv. 2013, 3, 19756–19759. [Google Scholar] [CrossRef]
- Lee, J.; Kobayashi, Y.; Tezuka, K.; Kishi, Y. Toward Creation of a Universal NMR Database for the Stereochemical Assignment of Acyclic Compounds: Proof of Concept. Org. Lett. 1999, 1, 2181–2184. [Google Scholar] [CrossRef] [PubMed]
- Luesch, H.; Moore, R.E.; Paul, V.J.; Mooberry, S.L.; Corbett, T.H. Isolation of Dolastatin 10 From The Marine Cyanobacterium Symploca Species VP642 And Total Stereochemistry And Biological Evaluation Of Its Analogue Symplostatin 1. J. Nat. Prod. 2001, 64, 907–910. [Google Scholar] [CrossRef] [PubMed]
- Pettit, G.R.; Kamano, Y.; Herald, C.L.; Tuinman, A.A.; Boettner, F.E.; Kizu, H.; Schmidt, J.M.; Baczynskyj, L.; Tomer, K.B.; Bontems, R.J. The Isolation and Structure Of A Remarkable Marine Animal Antineoplastic Constituent: Dolastatin 10. J. Am. Chem. Soc. 1987, 109, 6883–6885. [Google Scholar] [CrossRef]
- Moore, R.E.; Cheuk, C.; Yang, X.Q.G.; Patterson, G.M.L.; Bonjouklian, R.; Smitka, T.A.; Mynderse, J.S.; Foster, R.S.; Jones, N.D. Hapalindoles, Antibacterial and Antimycotic Alkaloids from the Cyanophyte Hapalosiphon fontinalis. J. Org. Chem. 1987, 52, 1036–1043. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).