
Expanding the Utility of Bioinformatic Data for the Full Stereostructural Assignments of Marinolides A and B, 24- and 26-Membered Macrolactones Produced by a Chemically Exceptional Marine-Derived Bacterium

 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
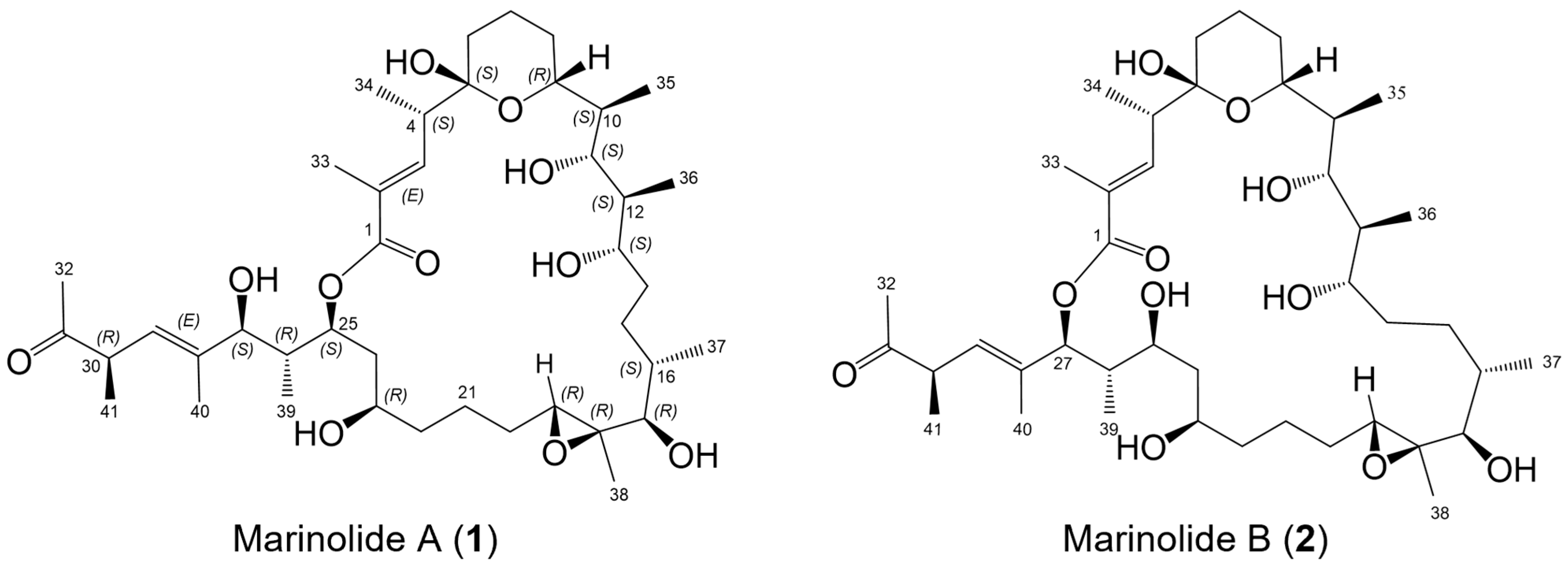
2.1. Marinolides A and B
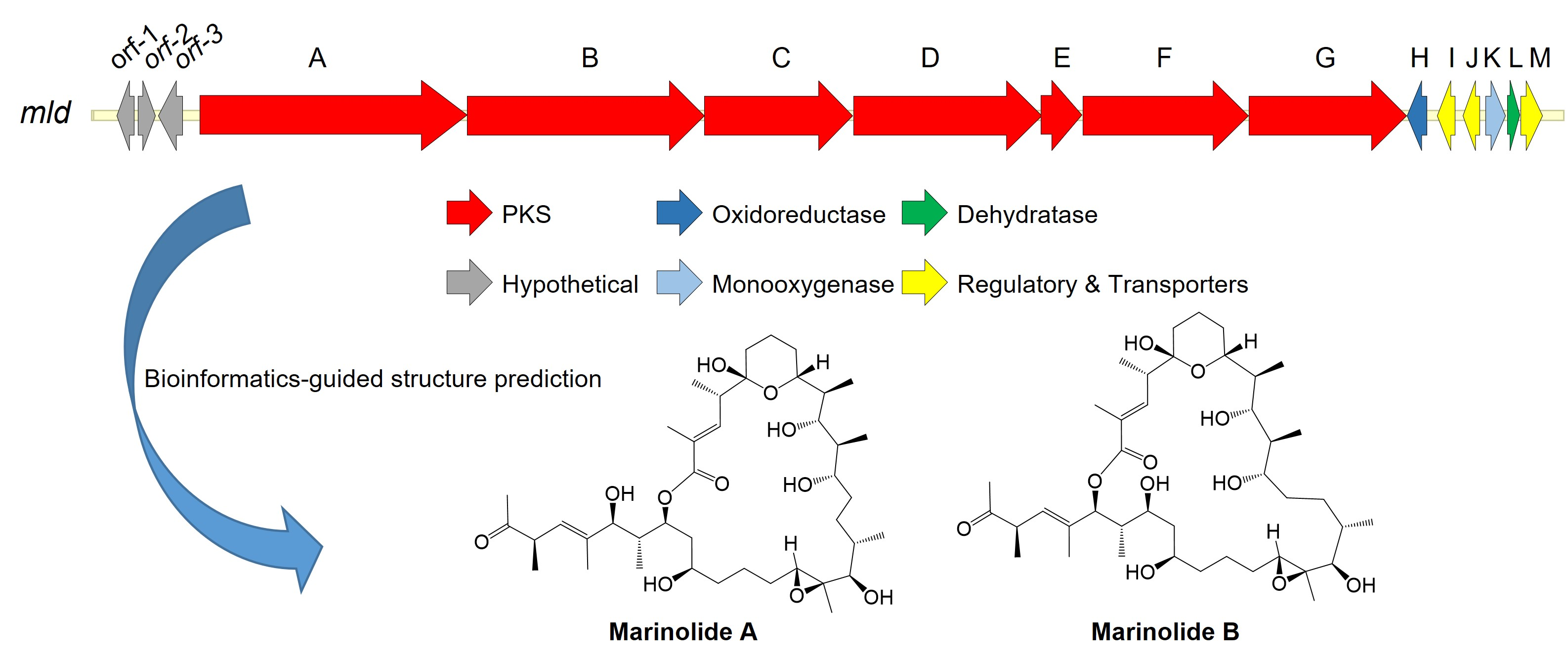
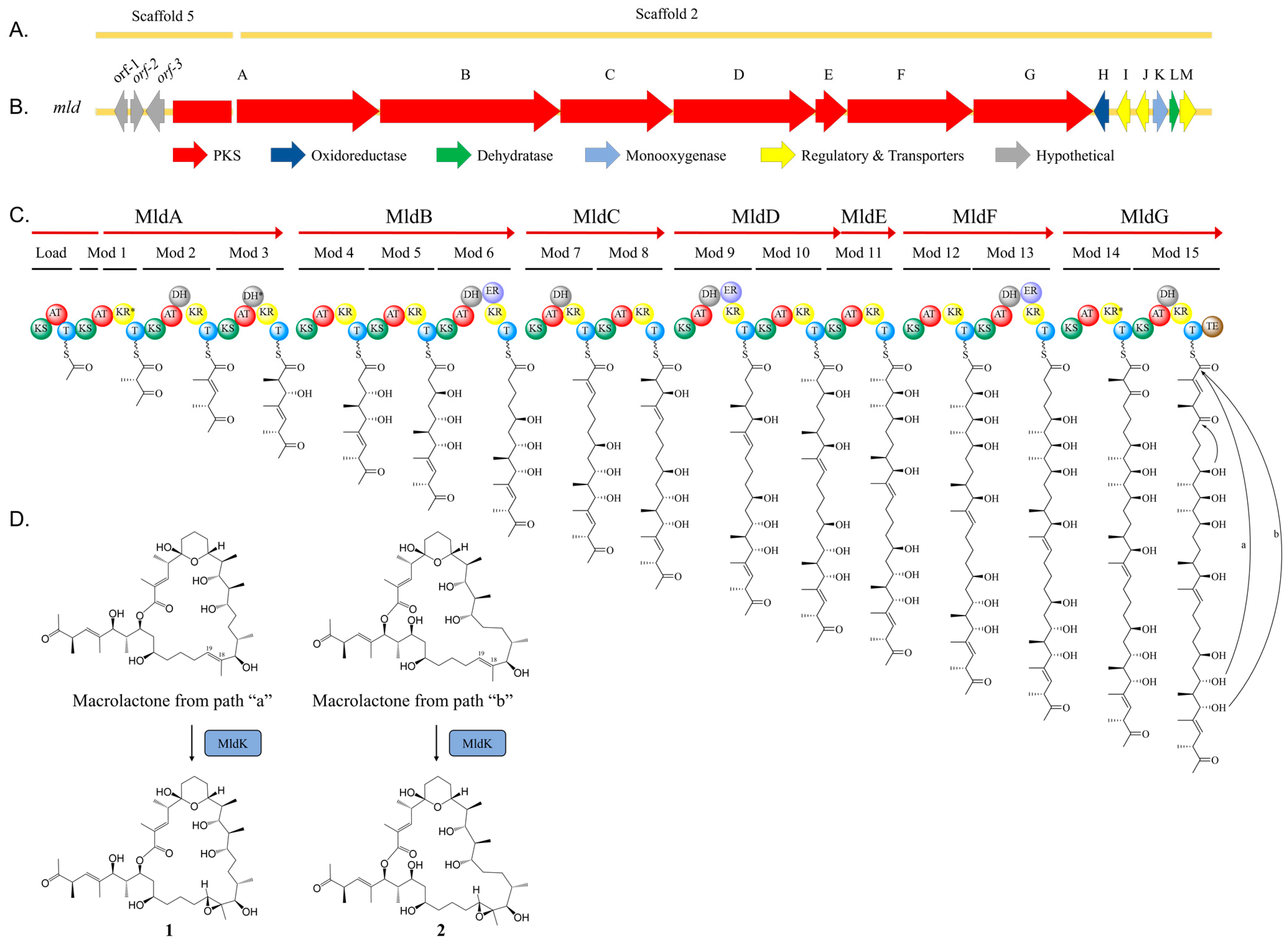
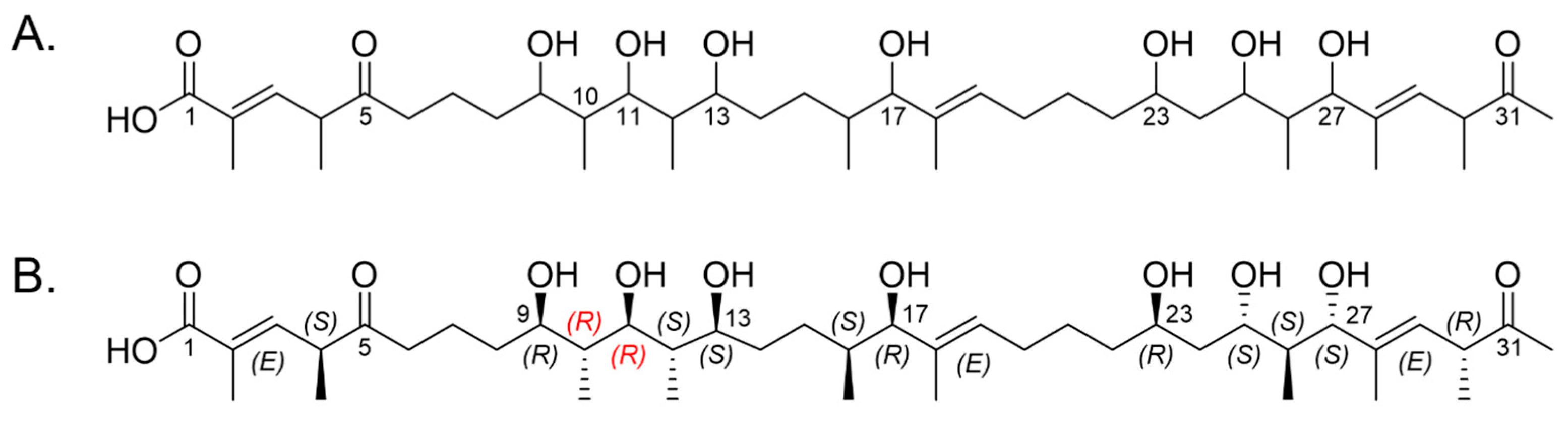
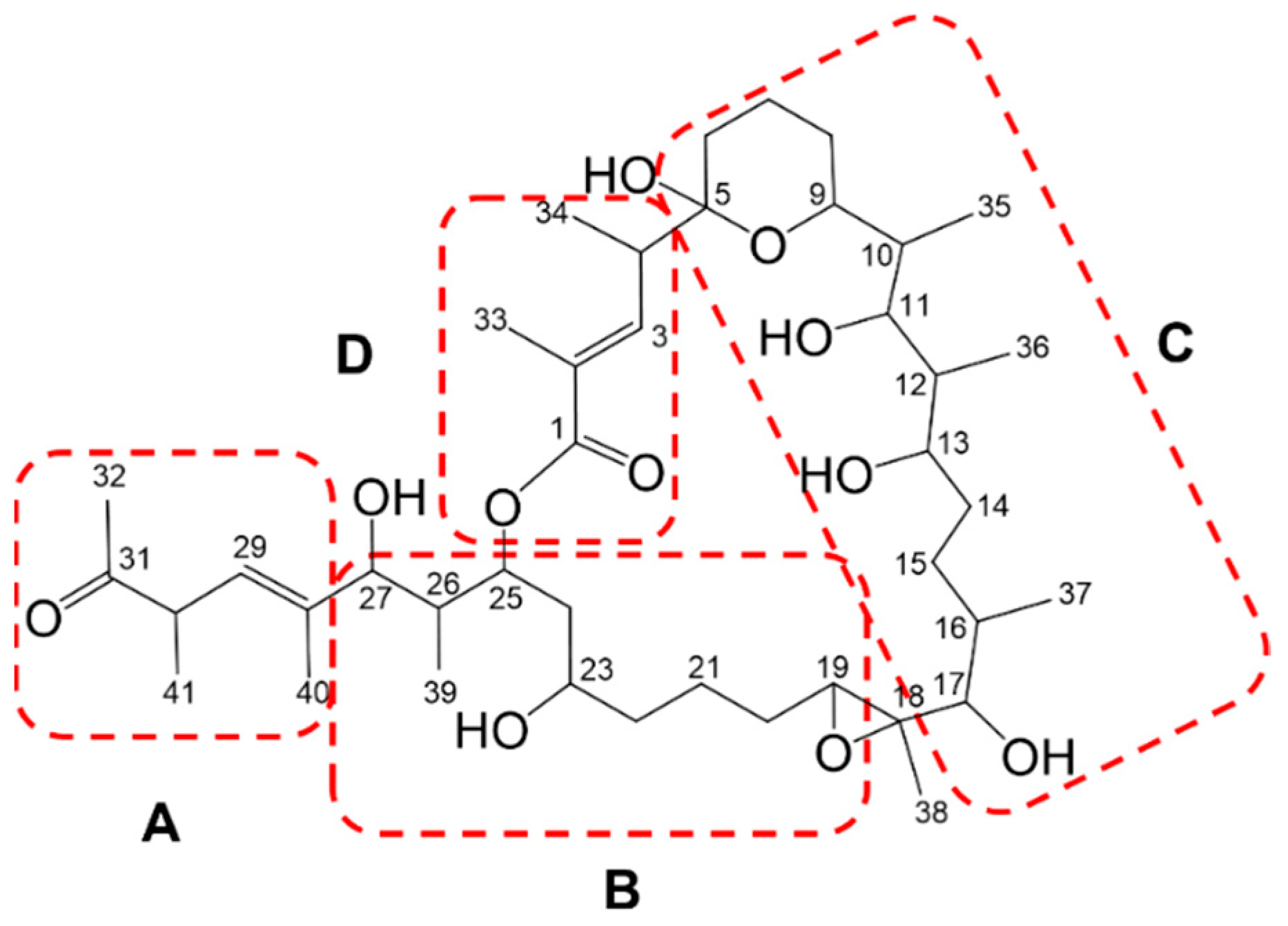
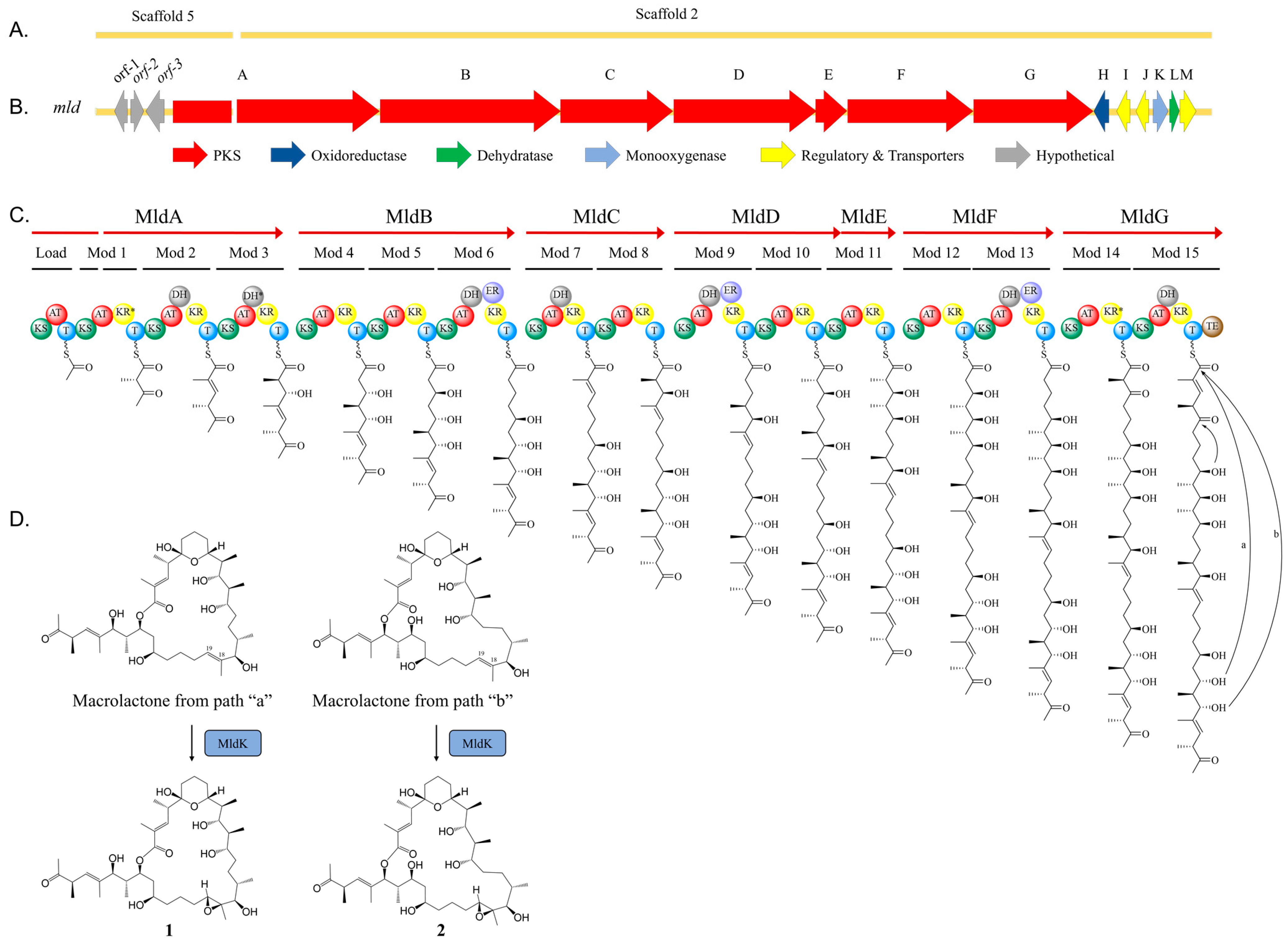
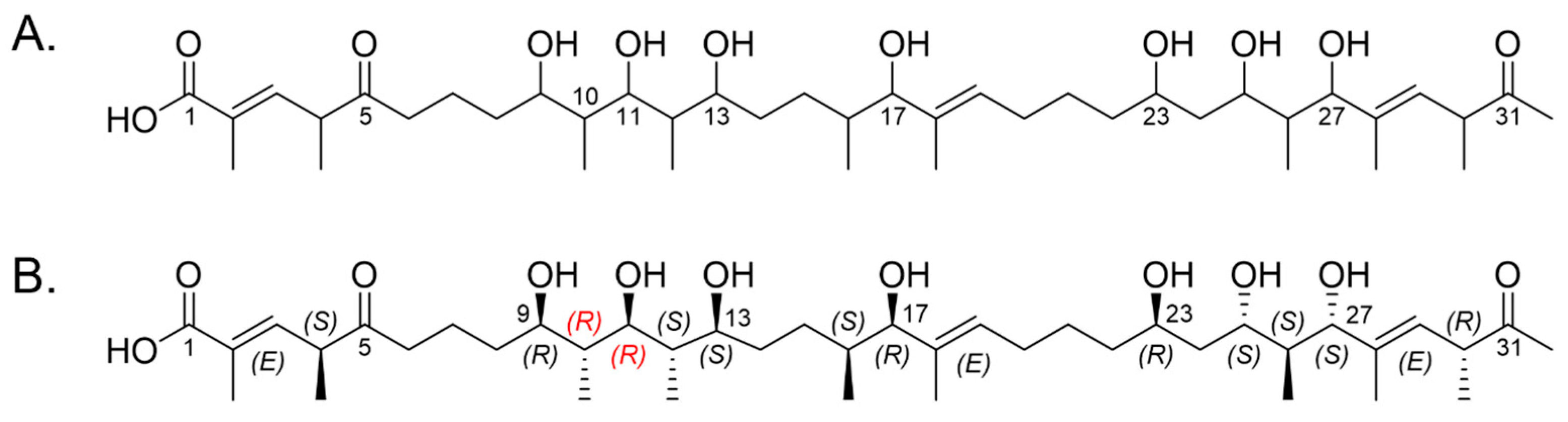
2.2. Marinolide Structure Prediction through Biosynthetic Gene Cluster Analysis
2.3. Isolation of Marinolides A and B (1, 2)
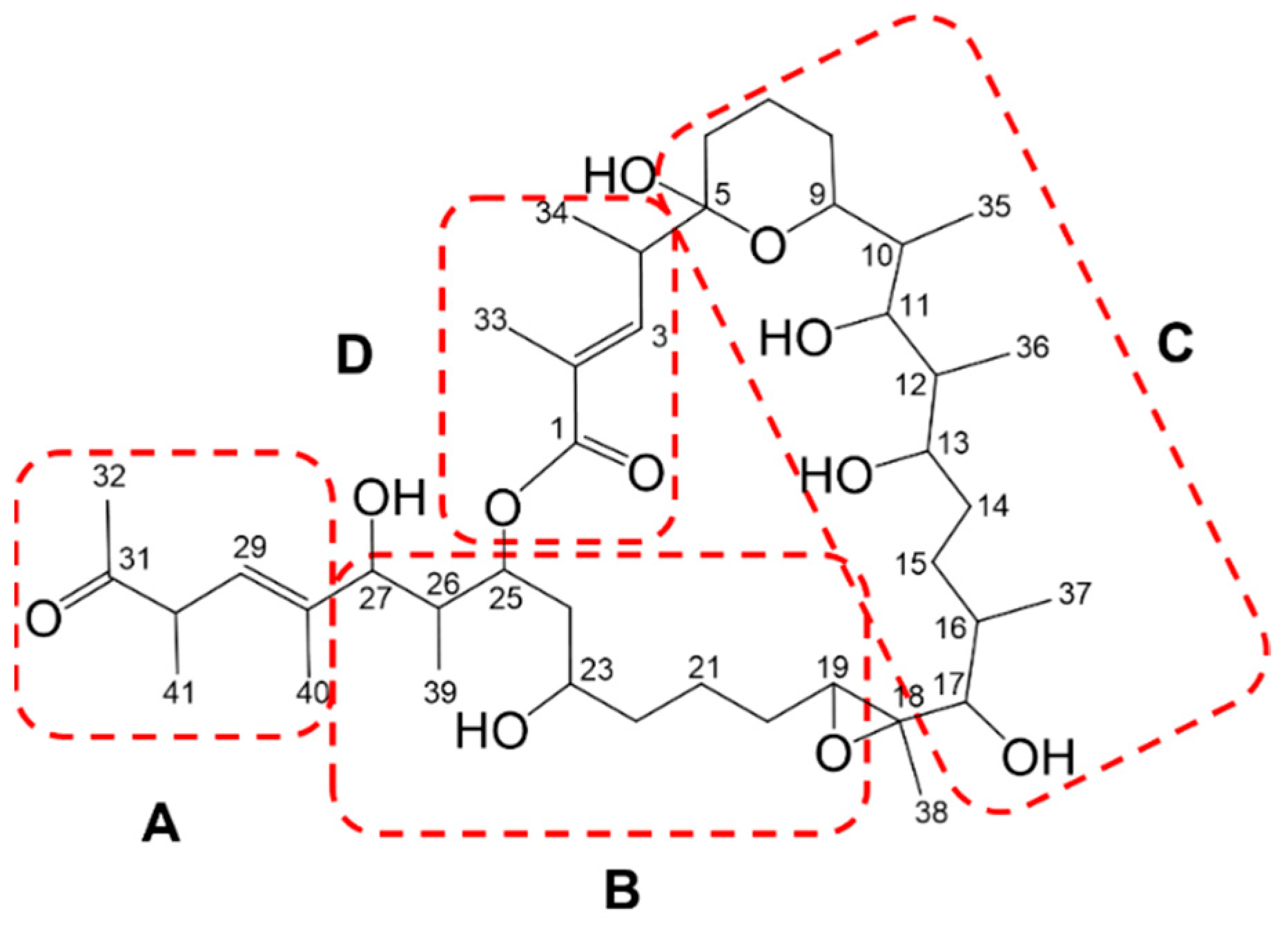
2.4. Confirmation of the Structures of Marinolides A and B by Spectroscopic Methods
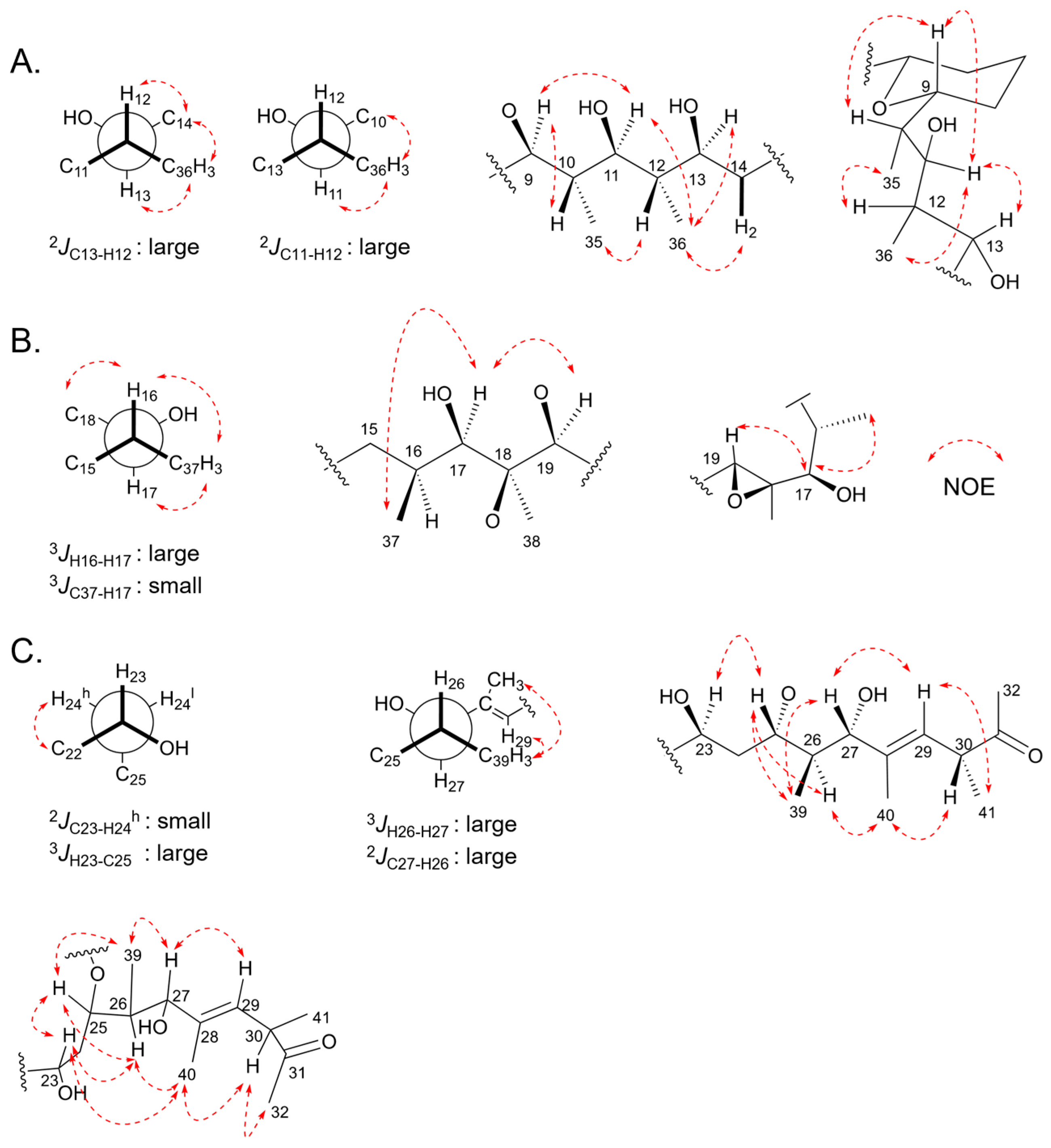
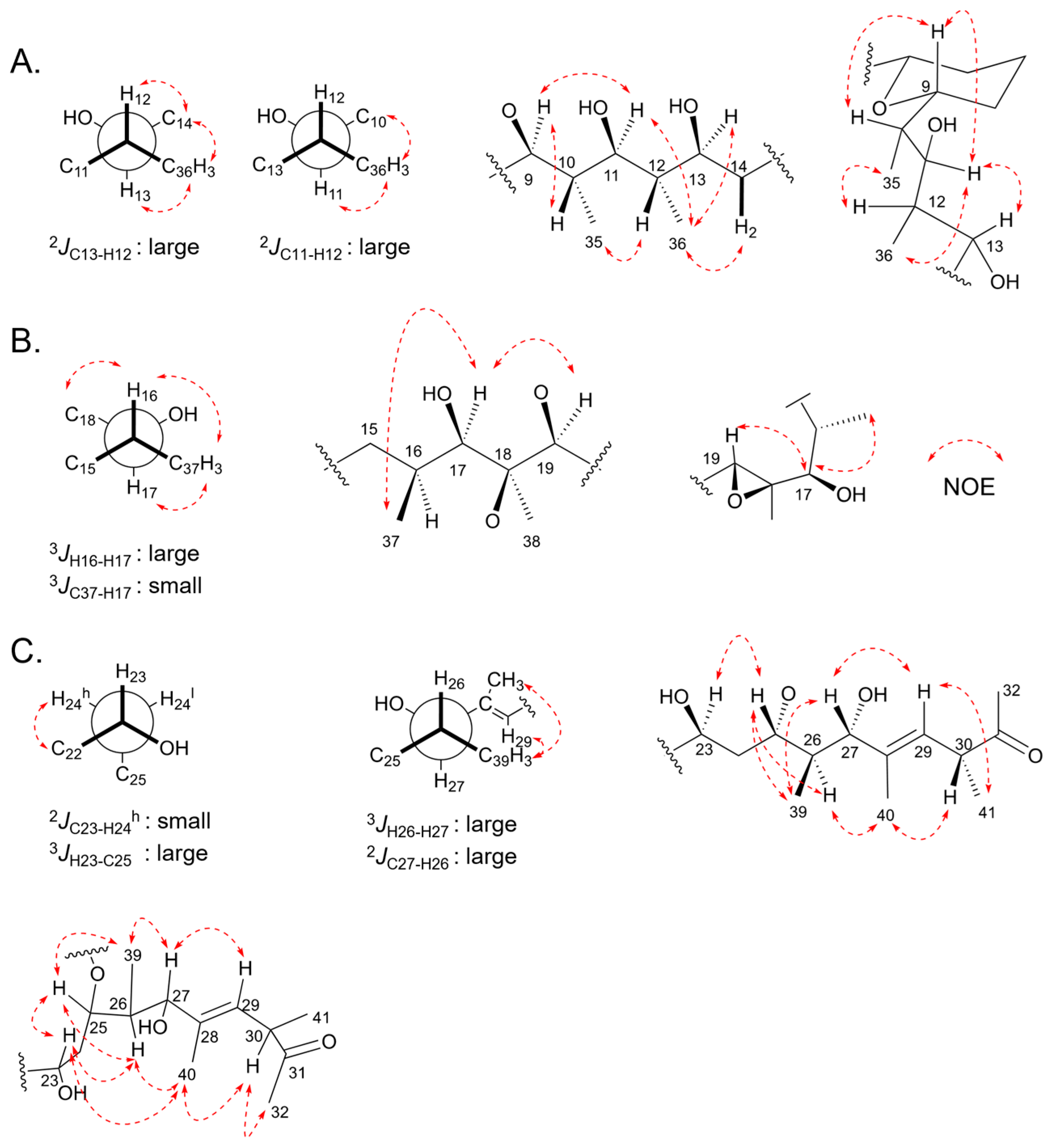
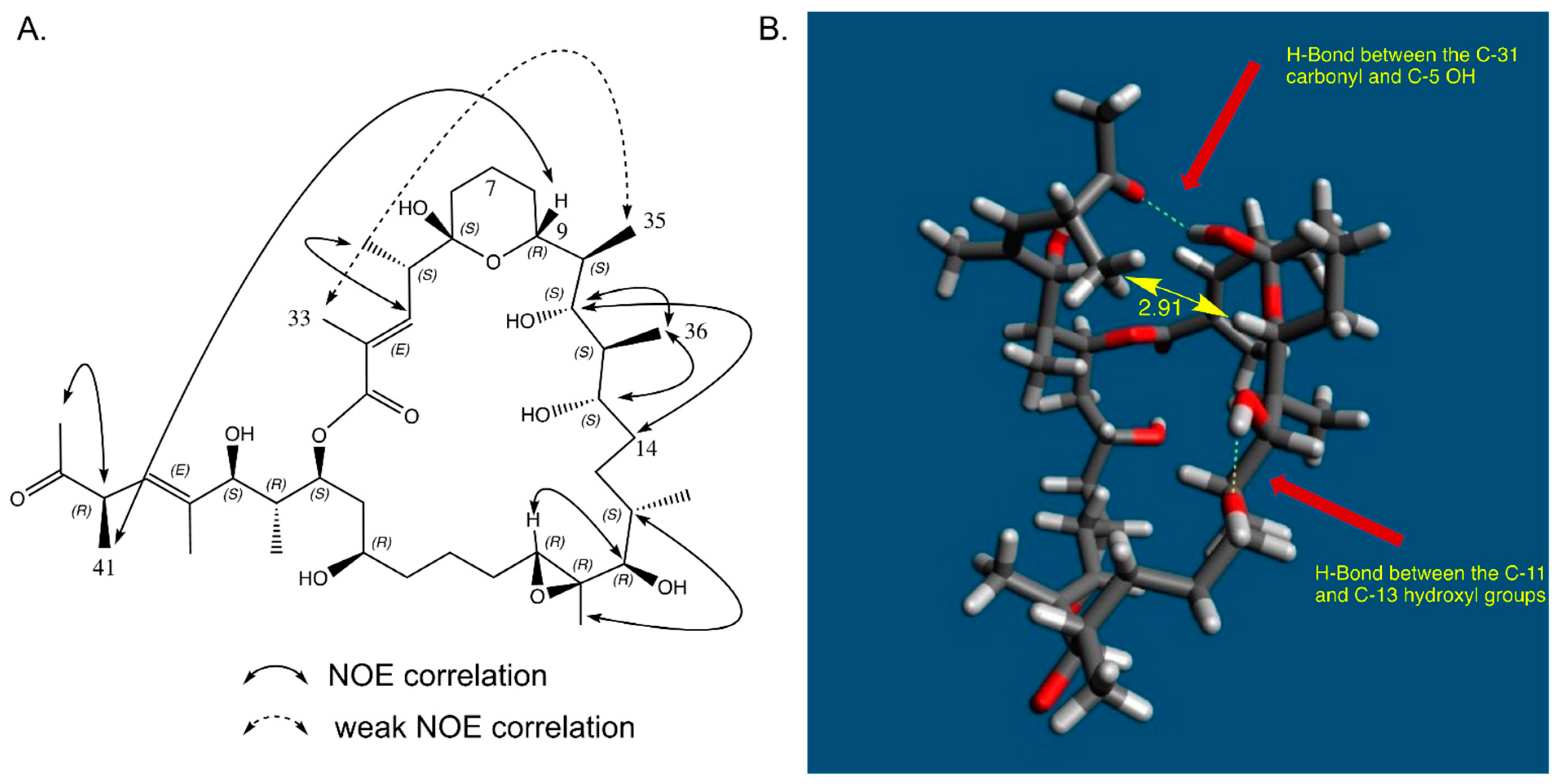
2.5. Relative Configurations of Marinolides by Spectroscopic Methods
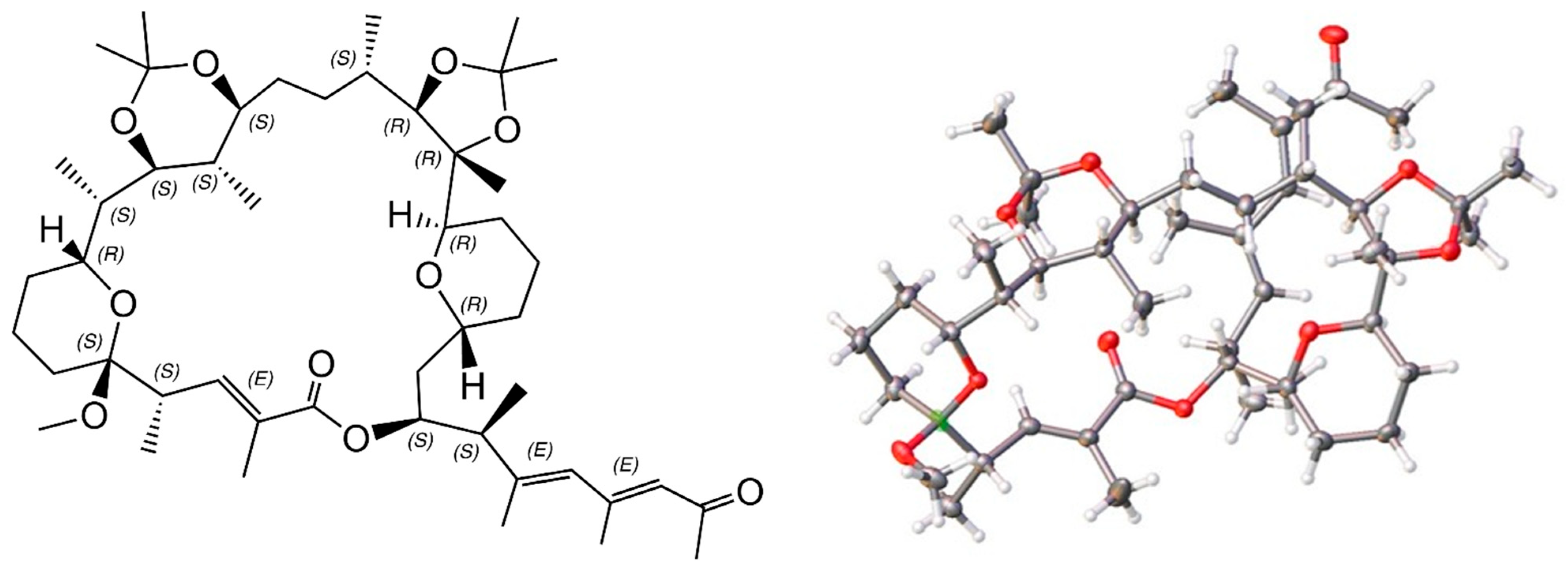
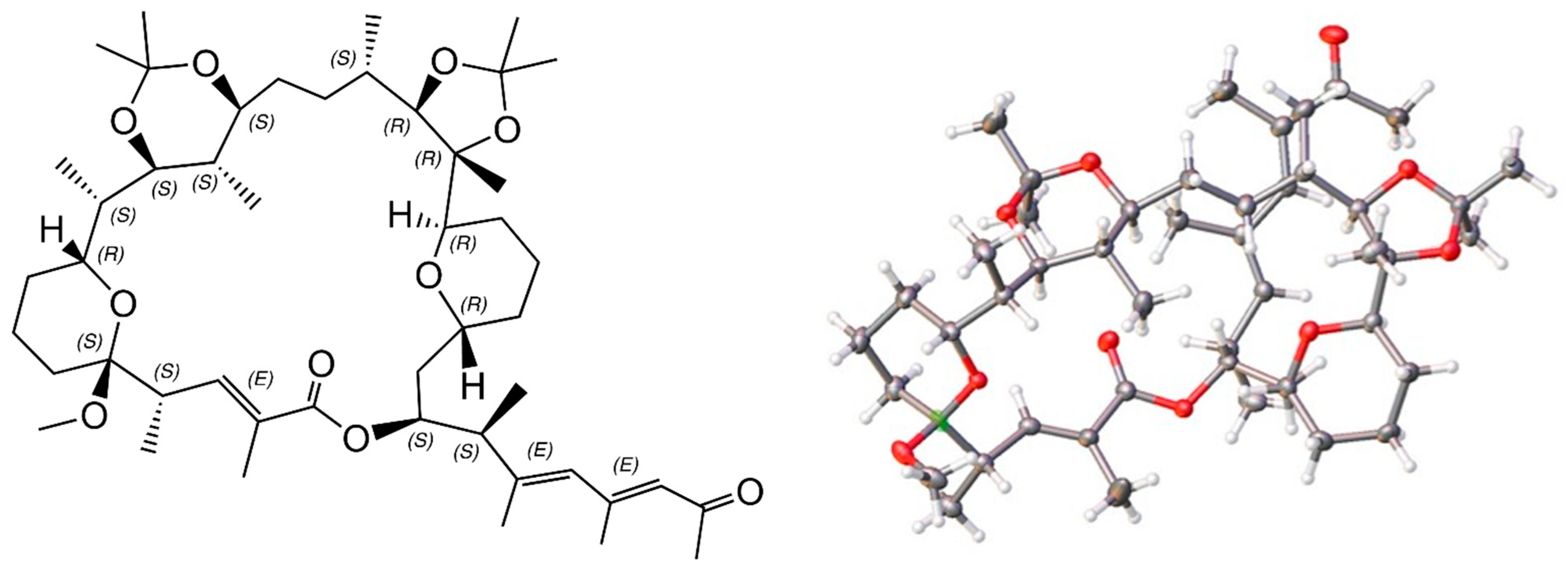
2.6. X-ray Structure of a Bis-Acetonide Derivative (1a) of Marinolide A
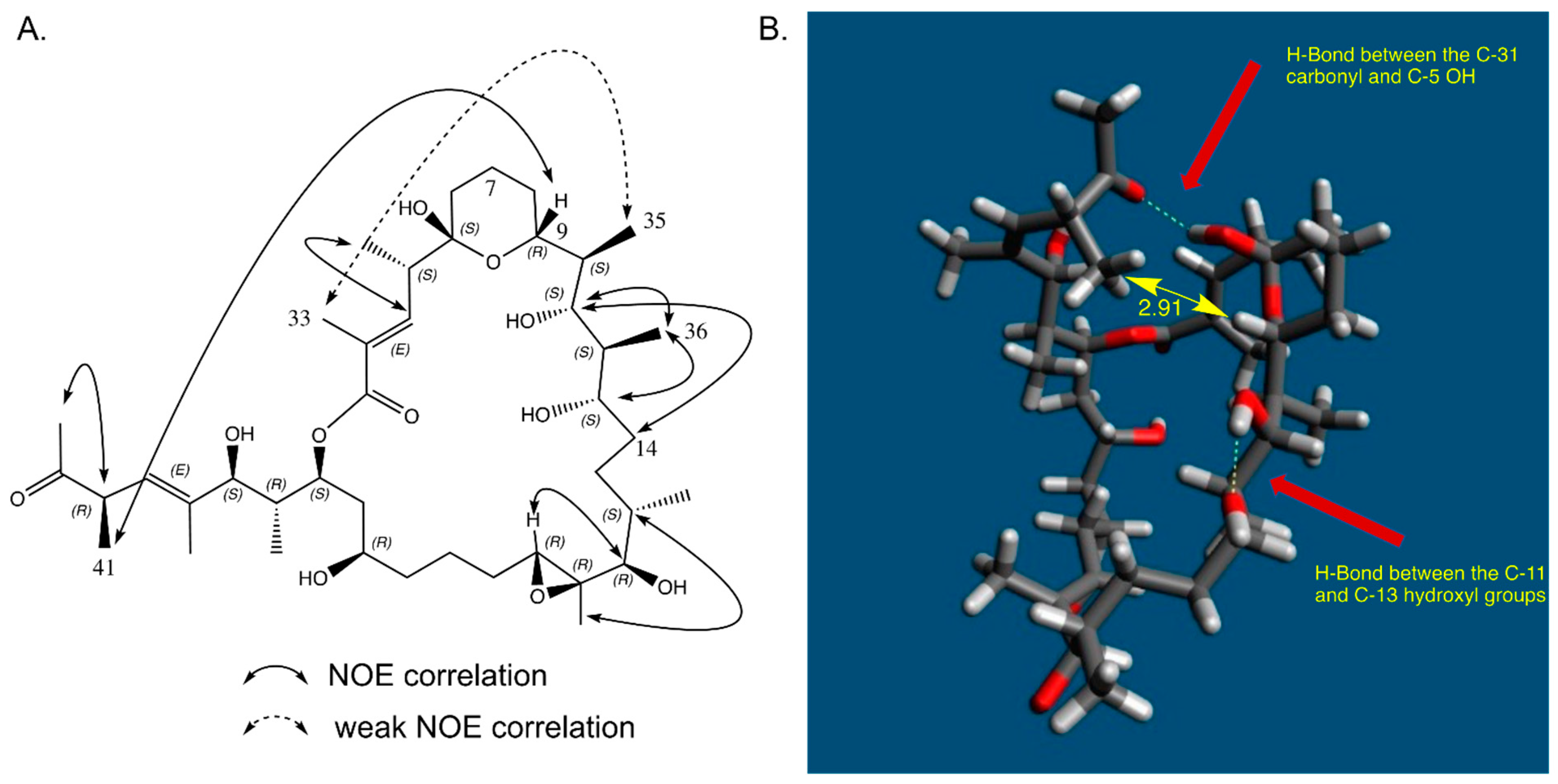
2.7. In Solution Conformation of Marinolide A (1)
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Isolation and Identification of Strain AJS-327
3.3. Cultivation, and Extraction of Strain AJS-327
3.4. Isolation of Marinolides A (1) and B (2)
3.5. Preparation of Bis-Acetonide 1a
3.6. Cytotoxicity Bioassay Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dinos, G.P. The macrolide antibiotic renaissance. Br. J. Pharm. 2017, 174, 2967–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farzam, K.; Nessel, T.A.; Quick, J. Erythromycin. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2023. Available online: https://www.ncbi.nlm.nih.gov/books/NBK532249 (accessed on 19 May 2023).
- Flynn, E.H.; Sigal, M.V., Jr.; Wiley, P.F.; Gerzon, K. Erythromycin. I. Properties and degradation studies. J. Am. Chem. Soc. 1954, 76, 3121–3131. [Google Scholar] [CrossRef]
- Zhang, H.; Zou, J.; Yan, X.; Chen, J.; Cao, X.; Wu, J.; Liu, Y.; Wang, T. Marine-derived macrolides 1990–2020: An overview of chemical and biological diversity. Mar. Drugs 2021, 19, 180. [Google Scholar] [CrossRef] [PubMed]
- Hochmuth, T.; Piel, J. Polyketide synthases of bacterial symbionts in sponges–evolution-based applications in natural products research. Phytochemistry 2009, 70, 1841–1849. [Google Scholar] [CrossRef]
- Shah, S.A.A.; Akhter, N.; Auckloo, B.N.; Khan, I.; Lu, Y.; Wang, K.; Wu, B.; Guo, Y.W. Structural diversity, biological properties and applications of natural products from cyanobacteria, a review. Mar. Drugs 2017, 15, 354. [Google Scholar] [CrossRef] [Green Version]
- Laureti, L.; Song, L.; Huang, S.; Corre, C.; Leblond, P.; Challis, G.L.; Aigle, B. Identification of a bioactive 51-membered macrolide complex by activation of a silent polyketide synthase in Streptomyces ambofaciens. Proc. Natl. Acad. Sci. USA 2011, 108, 6258–6263. [Google Scholar] [CrossRef] [Green Version]
- Rho, J.-R.; Subramaniam, G.; Choi, H.; Kim, E.-H.; Ng, S.P.; Yoganathan, K.; Ng, S.; Buss, A.D.; Butler, M.S.; Gerwick, W.H. Gargantulide A, a complex 52-membered macrolactone showing antibacterial activity from Streptomyces sp. Org. Lett. 2015, 17, 1377–1380. [Google Scholar] [CrossRef] [Green Version]
- Carretero-Molina, D.; Ortiz-López, F.J.; Gren, T.; Oves-Costales, D.; Martín, J.; Román-Hurtado, F.; Sparholt Jørgensen, T.; de la Cruz, M.; Diaz, C.; Vicente, F.; et al. Discovery of gargantulides B and C, new 52-membered macrolactones from Amycolatopsis sp. Complete absolute stereochemistry of the gargantulide family. Org. Chem. Front. 2022, 9, 462–470. [Google Scholar] [CrossRef]
- Hayakawa, Y.; Shinya, K.; Furihata, K.H. Structure of a novel 60-membered macrolide, quinolidomicin A1. J. Am. Chem. Soc. 1993, 115, 3014–3015. [Google Scholar] [CrossRef]
- Kim, M.C.; Winter, J.M.; Cullum, R.; Li, Z.; Fenical, W. Complementary genomic, bioinformatic, and chemical approaches facilitate the absolute structure assignment of ionostatin, a linear polyketide from a rare marine-derived actinomycete. ACS Chem. Biol. 2020, 15, 2507–2515. [Google Scholar] [CrossRef]
- Kim, M.C.; Cullum, R.; Smith, A.; Yang, I.; Fenical, W. Photopiperazines A-D, cytotoxic interconverting photo-sensitive diketopiperazines produced by a rare, marine-derived actinomycete bacterium of the family Streptomycetaceae. J. Nat. Prod. 2019, 82, 2262–2267. [Google Scholar] [CrossRef]
- Hashimoto, T.; Hashimoto, J.; Kozone, I.; Amagai, K.; Kawahara, T.; Takahashi, S.; Ikeda, H.; Shin-ya, K. Biosynthesis of quinolidomicin, the largest known macrolide of terrestrial origin: Identification and heterologous expression of a biosynthetic gene cluster over 200 kb. Org. Lett. 2018, 20, 7996–7999. [Google Scholar] [CrossRef]
- Kim, M.C.; Machado, H.; Jang, K.H.; Trzoss, L.; Jensen, P.R.; Fenical, W. Integration of genomic data with NMR analysis enables assignment of the full stereostructure of neaumycin B, a potent inhibitor of glioblastoma from a marine-derived Micromonospora. J. Am. Chem. Soc. 2018, 140, 10775–10784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, P.; Li, Y.; Zhu, J.; Shen, Y.; Wang, H. Targeted discovery of the polyene macrolide hexacosalactone a from Streptomyces by reporter-guided selection of fermentation media. J. Nat. Prod. 2021, 84, 1924–1929. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, M.; Wu, C.; Tan, Y.; Li, J.; Hao, X.; Duan, Y.; Guan, Y.; Shang, X.; Wang, Y.; et al. Identification and proposed relative and absolute configurations of niphimycins C-E from the marine-derived Streptomyces sp. IMB7-145 by genomic analysis. J. Nat. Prod. 2018, 81, 178–187. [Google Scholar] [CrossRef] [PubMed]
- An, J.S.; Lee, J.Y.; Kim, E.; Ahn, H.; Jang, Y.-J.; Shin, B.; Hwang, S.; Shin, J.; Yoon, Y.J.; Lee, S.K.; et al. Formicolides A and B, antioxidative and antiangiogenic 20-membered macrolides from a wood ant gut bacterium. J. Nat. Prod. 2020, 83, 2776–2784. [Google Scholar] [CrossRef]
- Kim, M.C.; Winter, J.M.; Asolkar, R.N.; Boonlarppradab, C.; Cullum, R.; Fenical, W. Marinoterpins A–C: Rare linear merosesterterpenoids from marine-derived actinomycete bacteria of the family Streptomycetaceae. J. Org. Chem. 2021, 86, 11140–11148. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. antiSmash 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. Blast+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Nivina, A.; Yuet, K.P.; Hsu, J.; Khosla, C. Evolution and diversity of assembly-line polyketide synthases. Chem. Rev. 2019, 119, 12524–12547. [Google Scholar] [CrossRef] [Green Version]
- Del Vecchio, F.; Petkovic, H.; Kendrew, S.G.; Low, L.; Wilkinson, B.; Lill, R.; Cortes, J.; Rudd, B.A.M.; Staunton, J.; Leadlay, P.F. Active-site residue, domain and module swaps in modular polyketide synthases. J. Ind. Microbiol. Biotechnol. 2003, 30, 489–494. [Google Scholar] [CrossRef] [PubMed]
- Yadav, G.; Gokhale, R.S.; Mohanty, D. Computational approach for prediction of domain organization and substrate specificity of modular polyketide synthases. J. Mol. Biol. 2003, 328, 335–363. [Google Scholar] [CrossRef] [PubMed]
- Haydock, S.F.; Aparicio, J.F.; Molnar, I.; Schwecke, T.; Khaw, L.E.; Konig, A.; Marsden, A.F.; Galloway, I.S.; Staunton, J.; Leadlay, P.F. Divergent sequence motifs correlated with the substrate specificity of (methyl) malonyl-CoA: Acyl carrier protein transacylase domains in modular polyketide synthases. FEBS Lett. 1995, 374, 246–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunn, B.J.; Cane, D.E.; Khosla, C. Mechanism and specificity of an acyltransferase domain from a modular polyketide synthase. Biochemistry 2013, 52, 1839–1841. [Google Scholar] [CrossRef] [Green Version]
- Dunn, B.J.; Khosla, C. Engineering the acyltransferase substrate specificity of assembly line polyketide synthases. J. R. Soc. Interface 2013, 10, 20130297. [Google Scholar] [CrossRef] [Green Version]
- Bailey, C.B.; Pasman, M.E.; Keatinge-Clay, A.T. Substrate structure-activity relationships guide rational engineering of modular polyketide synthase ketoreductases. Chem. Commun. 2016, 52, 792–795. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Garg, A.; Keatinge-Clay, A.T.; Khosla, C.; Cane, D.E. Epimerase and reductase activities of polyketide synthase ketoreductase domains utilize the same conserved tyrosine and serine residues. Biochemistry 2016, 55, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- Caffrey, P. Conserved amino acid residues correlating with ketoreductase stereospecificity in modular polyketide synthases. Chembiochem 2003, 4, 654–657. [Google Scholar] [CrossRef]
- Valenzano, C.R.; Lawson, R.J.; Chen, A.Y.; Khosla, C.; Cane, D.E. The biochemical basis for stereochemical control in polyketide biosynthesis. J. Am. Chem. Soc. 2009, 131, 18501–18511. [Google Scholar] [CrossRef] [Green Version]
- Keatinge-Clay, A.T. A tylosin ketoreductase reveals how chirality is determined in polyketides. Chem. Biol. 2007, 14, 898–908. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.; Taylor, C.A.; Piasecki, S.K.; Keatinge-Clay, A.T. Structural and functional analysis of A-type ketoreductases from the amphotericin modular polyketide synthase. Structure 2010, 18, 913–922. [Google Scholar] [CrossRef] [Green Version]
- Xie, X.; Garg, A.; Khosla, C.; Cane, D.E. Mechanism and stereochemistry of polyketide chain elongation and methyl group epimerization in polyether biosynthesis. J. Am. Chem. Soc. 2017, 139, 3283–3292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keatinge-Clay, A. Crystal structure of the erythromycin polyketide synthase dehydratase. J. Mol. Biol. 2008, 384, 941–953. [Google Scholar] [CrossRef] [Green Version]
- Kwan, D.H.; Sun, Y.; Schulz, F.; Hong, H.; Popovic, B.; Sim-Stark, J.C.C.; Haydock, S.F.; Leadlay, P. Prediction and manipulation of the stereochemistry of enoylreduction in modular polyketide synthases. Chem. Biol. 2008, 15, 1231–1240. [Google Scholar] [CrossRef] [PubMed]
- Kwan, D.H.; Leadlay, P.F. Mutagenesis of modular polyketide synthase enoylreductase domain reveals insights into catalysis and stereospecificity. ACS Chem. Biol. 2010, 5, 829–838. [Google Scholar] [CrossRef]
- Huijbers, M.M.E.; Montersino, S.; Westphal, A.H.; Tischler, D.; van Berkel, W.J.H. Flavin dependent monooxygenases. Arch. Biochem. Biophys. 2014, 544, 2–17. [Google Scholar] [CrossRef] [PubMed]
- Luhavaya, H.; Dias, M.V.B.; Williams, S.R.; Hong, H.; de Oliveira, L.G.; Leadlay, P.F. Enzymology of pyran ring a formation in salinomycin biosynthesis. Angew. Chem. Int. Ed. 2015, 54, 13622–13625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, M.-C.; Zou, Y.; Watanabe, K.; Walsh, C.T.; Tang, Y. Oxidative cyclization in natural product biosynthesis. Chem. Rev. 2017, 117, 5226–5333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meng, S.; Tang, G.-L.; Pan, X.-X. Enzymatic formation of oxygen-containing heterocycles in natural product biosynthesis. Chembiochem 2018, 19, 2002–2022. [Google Scholar] [CrossRef]
- Bowen, J.I.; Wang, L.; Crump, M.P.; Willis, C.L. Synthetic and biosynthetic methods for selective cyclisations of 4,5-epoxy alcohols to tetrahydropyrans. Org. Biomol. Chem. 2022, 20, 1150–1175. [Google Scholar] [CrossRef]
- Berkhan, G.; Hahn, F. A dehydratase domain in ambruticin biosynthesis displays additional activity as a pyran-forming cyclase. Angew. Chem. Int. Ed. Engl. 2014, 53, 14240–14244. [Google Scholar] [CrossRef] [PubMed]
- Matsumori, N.; Kaneno, D.; Murata, M.; Nakamura, H.; Tachibana, K. Stereochemical determination of acyclic structures based on carbon-proton spin-coupling constants. A method of configuration analysis for natural products. J. Org. Chem. 1999, 64, 866–876. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, M.C.; Winter, J.M.; Cullum, R.; Smith, A.J.; Fenical, W. Expanding the Utility of Bioinformatic Data for the Full Stereostructural Assignments of Marinolides A and B, 24- and 26-Membered Macrolactones Produced by a Chemically Exceptional Marine-Derived Bacterium. Mar. Drugs 2023, 21, 367. https://doi.org/10.3390/md21060367
Kim MC, Winter JM, Cullum R, Smith AJ, Fenical W. Expanding the Utility of Bioinformatic Data for the Full Stereostructural Assignments of Marinolides A and B, 24- and 26-Membered Macrolactones Produced by a Chemically Exceptional Marine-Derived Bacterium. Marine Drugs. 2023; 21(6):367. https://doi.org/10.3390/md21060367
Chicago/Turabian StyleKim, Min Cheol, Jaclyn M. Winter, Reiko Cullum, Alexander J. Smith, and William Fenical. 2023. "Expanding the Utility of Bioinformatic Data for the Full Stereostructural Assignments of Marinolides A and B, 24- and 26-Membered Macrolactones Produced by a Chemically Exceptional Marine-Derived Bacterium" Marine Drugs 21, no. 6: 367. https://doi.org/10.3390/md21060367
APA StyleKim, M. C., Winter, J. M., Cullum, R., Smith, A. J., & Fenical, W. (2023). Expanding the Utility of Bioinformatic Data for the Full Stereostructural Assignments of Marinolides A and B, 24- and 26-Membered Macrolactones Produced by a Chemically Exceptional Marine-Derived Bacterium. Marine Drugs, 21(6), 367. https://doi.org/10.3390/md21060367