2. Results
Samples of
L. catalai were frozen immediately to –20 ℃ after collection and stored at that temperature before they were exhaustively extracted by acetone. The Et
2O-soluble portion of the acetone extract was subjected to repeated column chromatography (CC) (silica gel, Sephadex LH-20 and reversed-phase HPLC) to yield five new cembrane-type diterpenes lobocalines A–E (
1–
5) and four new steroids lobocaloids A–D (
9–
12), along with six known analogues (
6–
8 and
13–
15). The known compounds were rapidly characterized as sarcoboettgerol D (
6) [
20], 11,12-epoxy-1
E,3
E,7
E-cembratrien-15-ol (
7) [
21], sarcophytrol L (
8) [
22], 24
ξ-methylcholestane-3
β,5
α,6
β,25-tetrol-25-monoacetate (
13) [
23], acutumosterol B (
14) [
24], and lobophytrol B (
15) [
25], respectively, by the comparison of their NMR data and optical rotation [
α]
D values with those reported in the literature.
Compound
1, namely lobocaline A, was isolated as colorless crystals and gave the molecular formula of C
22H
34O
3 as established by HRESIMS from the protonated molecular ion peak observed at
m/
z 347.2580 [M + H]
+ (calcd. for C
22H
35O
3, 347.2581), implying six degrees of unsaturation. The characteristic peak at ν
max 1737 cm
−1 in its IR spectrum showed the presence of ester carbonyl group, which was further confirmed by the diagnostic
13C NMR chemical shift at
δC 170.6. The
1H and
13C NMR data (
Table 1 and
Table 2) of
1 along with the assistance of DEPT spectrum revealed the presence of two trisubstituted double bonds [
δH 6.06 (1H, d,
J = 10.8 Hz, H-2),
δC 118.6 (CH, C-2) and 146.9 (qC, C-1);
δH 5.97 (1H, d,
J = 10.9 Hz, H-3),
δC 135.1 (qC, C-4) and 121.7 (CH, C-3)], one exocyclic double bond [
δH 5.15 (1H, s, H-19) and
δH 5.18 (1H, s, H-19),
δC 146.8 (qC, C-8) and 113.0 (CH
2, C-19)] and one epoxy ring [
δH 2.86 (1H, dd,
J = 7.5, 4.6Hz),
δC 61.0 (CH, C-11) and
δC 61.6 (qC, C-12)]. The above-mentioned moieties accounted for five of the six degrees of unsaturation, indicating the monocyclic carbon framework for
1. A comparison of the NMR data of
1 with those of the co-occurring known compound sarcoboettgerol D (
6), which had been recently isolated from soft coral
Sarcophyton boettgeri and its absolute configuration was determined by the TDDFT-ECD calculation method [
20], which revealed that they were structural analogues, with the only difference being that the hydroxyl group at C-7 in
6 was substituted by an acetyloxy group at C-7 in
1, in agreement with the 42 mass unit difference in their molecular weights. The position of the acetyloxy group at C-7, as evidenced by the observation of the upfield shift of C-6 from
δC 32.9 in
6 to
δC 30.0 in
1. This assignment was further confirmed by strong HMBC correlations (
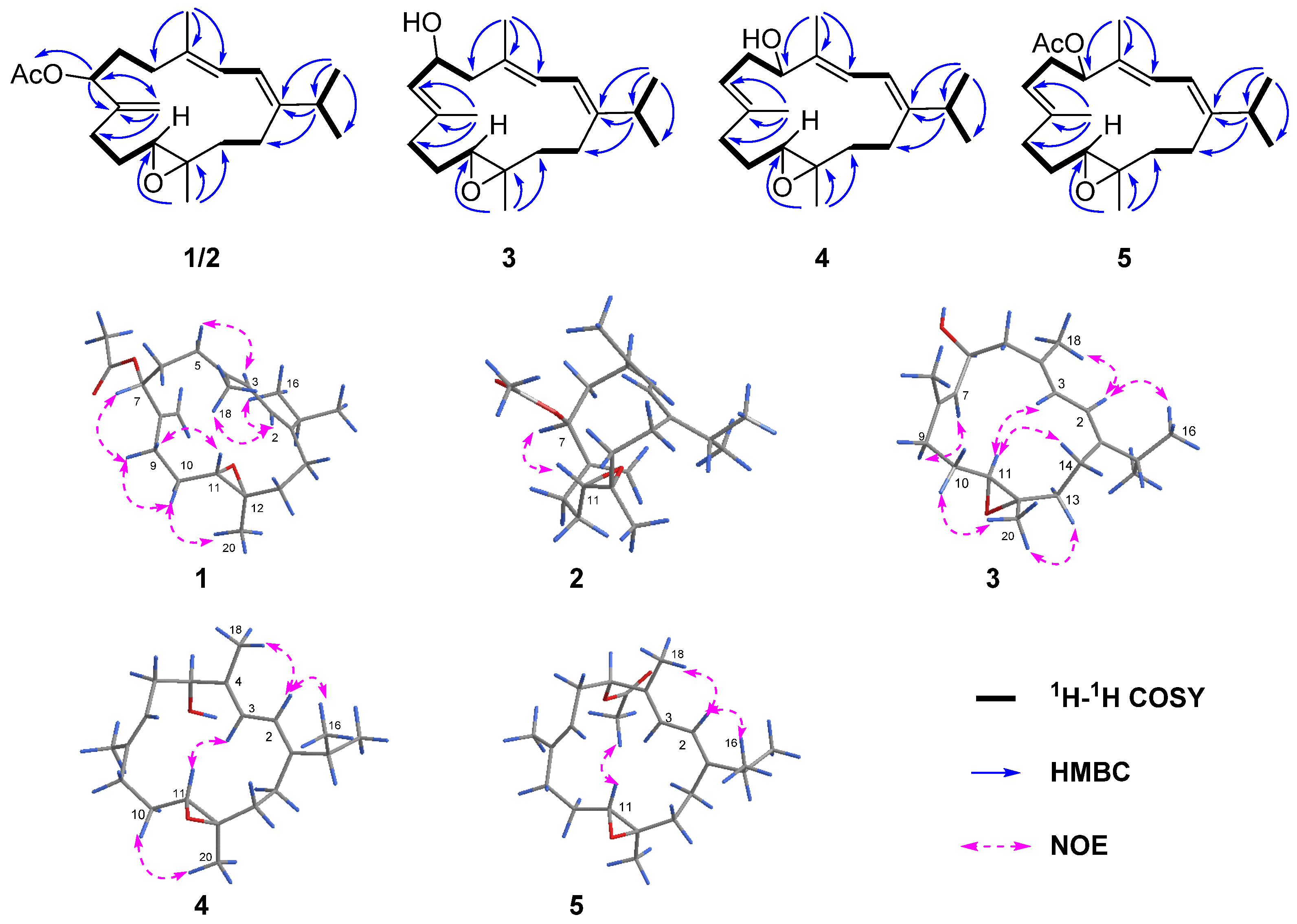
Figure 2) from H-7 (
δH 5.16) to C-19 (
δC 113.0), C-8 (
δC 146.8) and carbonyl carbon (
δC 170.6). Thus, the planar structure of
1 was determined as shown in
Figure 2.
The relative configuration of
1 was established mainly by the analysis of NOESY correlations (
Figure 2). The clear NOE correlations of H-2 (
δH 6.06)/H
3-16 (
δH 1.10), H-2/H
3-18 (
δH 1.78) and H-3 (
δH 5.97)/H-5
α (
δH 2.24) indicated the “1
E, 3
E” geometry of the two double bonds Δ
1,2 and Δ
3,4. The highly similar NOE correlations of H-7/H-9
β, H-9
β/H-10
β and H-10
β/H
3-20 and H-9α/H-11 between compounds
1 and
6 suggested that these two compounds shared the same stereochemistry. Furthermore, the suitable single crystals of
1 in MeOH were obtained. The X-ray crystallographic analysis using Cu Kα radiation (λ = 1.54178 Å) firmly disclosed the absolute configuration of
1 to be 7
R, 11
S, 12
S with the Flack parameter of 0.00 (10) (
Figure 3, CCDC 2290865). What is more is the acetylation of
6 yielding
1 further confirmed the structure and stereo-configuration of
1. The chemical structure of
1 was thus established as shown in
Figure 1.
Lobocaline B (
2), a colorless oil, gave the same molecular formula as
1 on basis of its HRESIMS ion peak at
m/
z 347.2581 [M + H]
+ (calcd. for C
22H
35O
3, 347.2581). Overall, the
1H and
13C NMR data of
2 (
Table 1 and
Table 2) were reminiscent of
1. Careful comparison of their NMR data revealed they possessed the same planar structure and only chemical shifts at C-7 (δ
C 72.5 in
1 vs. δ
C 75.9 in
2) were different, suggesting that
2 was simply the C-7 epi-isomer of
1, thus suggesting the assignment of 7
S*, 11
S*, 12
S* configuration of
2. Further analysis of 2D NMR spectra and the obvious NOESY correlations of H-7/H-11 confirmed this hypothesis (
Figure 2). Subsequently, the absolute configuration of
2 was then assigned to be 7
S, 11
S, 12
S by the comparison of similar experimental ECD spectra of compounds
1 (
Figure S1.9) and
2 (
Figure S2.9). Accordingly, the structure of
2 was determined as depicted in
Figure 1.
Lobocaline C (
3) was isolated as a colorless oil, and its molecular formula was assigned as C
20H
32O
2 by HRESIMS ion peak at
m/
z 327.2295 [M + Na]
+ (calcd. for C
20H
32O
2Na, 327.2295), indicating five degrees of unsaturation. The 1D NMR data of
3 (
Table 1 and
Table 2) showed great similarities to those of l
E,3
E,7
E,11:12-epoxy-l,3,7-cembratriene, a known cembranoid isolated from the South Andaman Coast soft coral
Lobophytum sp. [
26], with the only difference being the presence of an additional hydroxyl group at C-6 in
3, in agreement with the 16 mass unit difference between these two compounds. This assignment was further confirmed by the strong
1H–
1H COSY correlations of H-6 (δ
H 4.54)/H-7(δ
H 5.30) (
Figure 2). As mentioned above, the planar structure of
3 was determined as shown in
Figure 2. The geometries of the double bonds at Δ
1,2, Δ
3,4 and Δ
7,8 were both assigned to be
E by the NOESY correlations of H-2 (δ
H 5.94)/H
3-16 (δ
H 1.05), H-2/H
3-18 (δ
H 1.78) and H-7 (δ
H 5.30)/H-9α (δ
H 2.30) (
Figure 2), respectively. The relative configurations of C-11 and C-12 in
3 were proven to be the same as those of
1 due to the diagnostic NOESY correlations of H
3-20 (δ
H 1.25)/H-10b (δ
H 1.46), H
3-20/H-13b (δ
H 2.00), H-11 (δ
H 2.82)/H-3 (δ
H 5.92) and H-11/H-14a (δ
H 2.36). To figure out the relative configuration of
3, the QM-NMR calculation was performed to give the best match of more than 99% probability in DP4+ with the 6
R*, 11
S*, 12
S* isomer (see the details in the
Supplementary Materials). The absolute configuration of
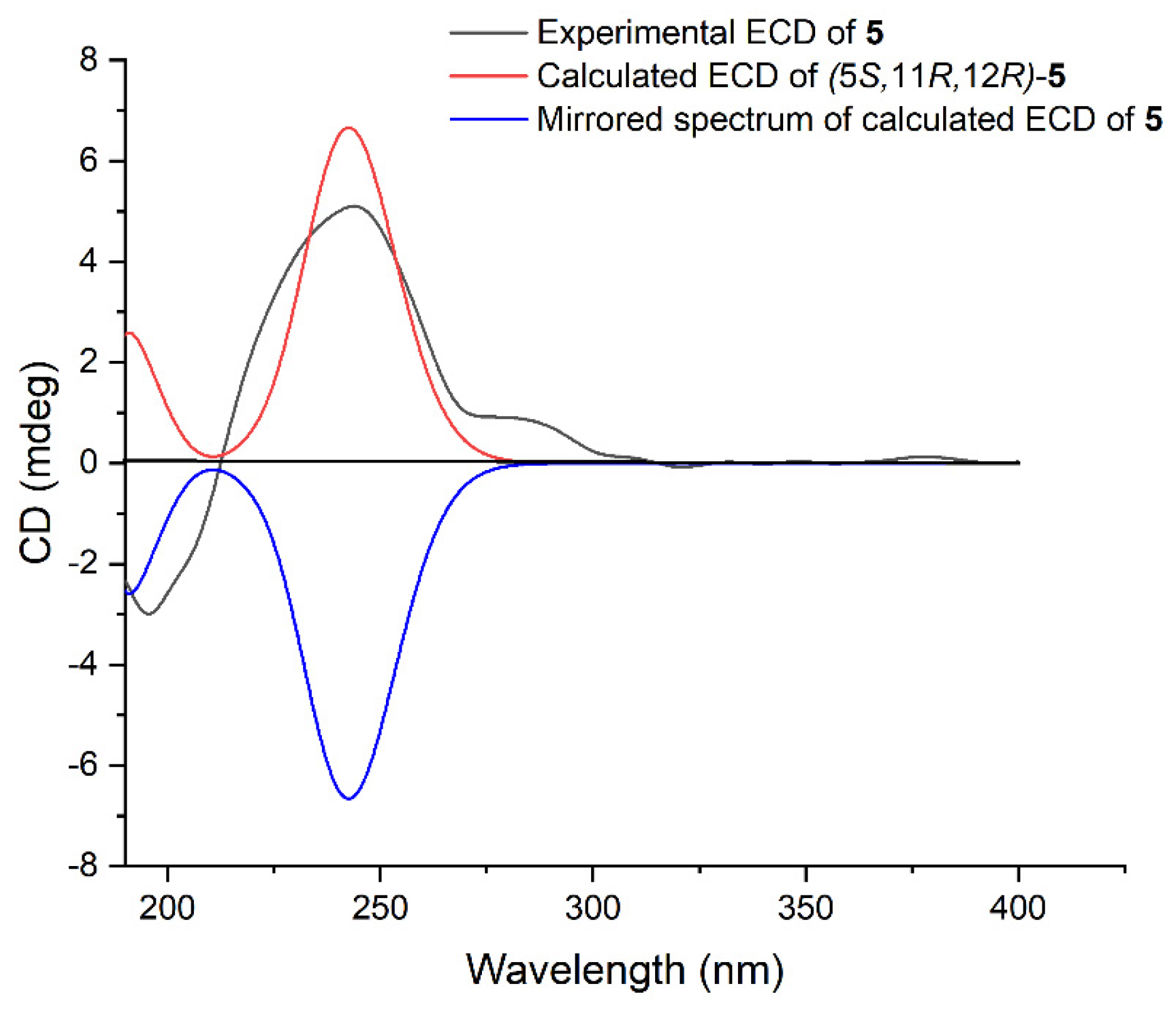
3 was determined by time-dependent density functional theory (TDDFT) ECD calculation. The theoretical ECD spectrum of
3 was calculated by the DFT method at the B3LYP/6-311G(d,p) level. As a result, the Boltzmann-averaged ECD spectrum of (6
R, 11
S, 12
S)-
3 highly matched the experimental ECD spectrum of
3, while the calculated ECD spectrum of enantiomer showed a completely opposite curve (
Figure 4). Consequently, the absolute configuration of
3 was established to be 6
R, 11
S, 12
S.
Lobocaline D (
4), a colorless oil, has the same molecular formula (C
20H
32O
2) as that of
3, implying they are isomers. Comparison of
1H and
13C chemical shifts of
4 and
3 (
Table 1 and
Table 2) followed by a detailed analysis of 2D NMR data revealed that the structure of
4 was similar to that of
3 with a different substituted position of the hydroxyl group. This conclusion was further supported by HMBC correlations from H
3-18 (δ
H 1.74) to C-5 (δ
C 78.1) and
1H–
1H COSY correlations of H-5 (δ
H 4.12)/H-6α (δ
H 2.39)/H-7 (δ
H 5.19). Thus, the planar structure of
4 was determined. The relative configurations of C-11 and C-12 in
4 were assigned to be the same as those of
1 due to the similar
13C NMR data. Similarly, the relative configuration of
4 was also elucidated via QM-NMR calculations using the DP4+ protocol. The best match was observed for the 5
R*, 11
S*, 12
S* relative configuration with a DP4+ probability over 99%. Moreover, the absolute configuration of
4 was further determined by the TDDFT-ECD approach. As shown in
Figure 5, the Boltzmann-averaged ECD curve calculated for the (5
S, 11
R, 12
R)-
4 enantiomer matched very well with the experimental ECD spectrum of
4. Accordingly, the absolute configuration of
4 was determined to be 5
S, 11
R, 12
R.
Lobocaline E (
5) was obtained as a colorless oil. Its molecular formula, C
22H
34O
3, was deduced from the ion peak observed at
m/
z 369.2397 [M + Na]
+ (calcd. for C
22H
34O
3Na, 369.2400) in its HRESIMS spectrum. The
1H and
13C NMR data (
Table 1 and
Table 2) of
5 were extremely similar to those of the co-occurring compound
4, indicating that they are structural analogues. In fact, the only difference between these two compounds is the replacement of the hydroxyl group at C-5 in
4 by the acetyl group at C-5 in
5, in agreement with the 42 mass unit difference between compounds
4 and
5. To determine the relative configuration of
5, the theoretical and experimental NMR data were correlated and their corresponding DP4+ probabilities were estimated. Consequently, the candidate structure
5a (
Figure S11.3) showed the dominant probability of 99.52%, indicating the 5
R*, 11
S*, 12
S* relative configuration of
5. In this case, the TDDFT-ECD calculation was also employed to determine the absolute stereochemistry of
5. As shown in
Figure 6, the calculated ECD spectrum of (5
S, 11
R, 12
R)-
5 fairly matched with the experimental one, allowing us to assign the absolute configuration of
5 to be 5
S, 11
R, 12
R. Moreover, the hydrolysis reaction of
5 was also performed, but unfortunately, no product was detected due to the low number of isolated compounds. The chemical structure of
5 was thus established as depicted in
Figure 1.
Lobocaloid A (
9) was isolated as a white powder. Its molecular formula was determined as C
29H
50O
6 by HRESIMS
m/
z 517.3504 [M + Na]
+ (calcd. for C
29H
50O
6Na, 517.35), appropriate for five degrees of unsaturation. The
1H NMR data of
9 (
Table 3) showed the general characters of polyhydroxylated sterols, including an olefinic proton resonating at δ
H 5.70 (1H, dd,
J = 3.5, 1.5 Hz, H-16), two oxymethines at δ
H 4.27(1H, s, H-3), 3.82 (1H, dd,
J = 12.2, 4.9 Hz, H-6), a hydroperoxyl group signal at δ
H 9.20 (br s), and seven methyls at δ
H 0.94 (3H, s, Me-18), 0.95 (3H, s, Me-19), 1.30 (3H, s, Me-21), 1.20 (3H, s, Me-26), 1.19 (3H, s, Me-27), 0.86 (3H, d,
J = 7.3 Hz, Me-28), 0.93 (3H, d,
J = 7.1 Hz, Me-29). Moreover, the
13C NMR data (
Table 3) and DEPT spectrum of
9 indicated the presence of 29 signals, which comprised seven methyls, eight methylenes, eight methines (including one olefinic at δ
C 126.9, and seven sp
3 hybridized at δ
C 67.9, 72.0, 32.5, 43.5, 58.0, 30.2, 50.3) and six quaternary carbons (including an olefinic at δ
C 158.3 and five sp
3 hybridized at δ
C 78.2, 41.3, 47.5, 85.7, 75.6). These above data suggest the presence of a trisubstituted double bond, which accounted for one degree of unsaturation. The remaining four degrees of unsaturation were attributed to a tetracyclic system in the molecule. In the
1H–
1H COSY spectrum, it was possible to identify three different structural units extending from C-1 to C-4; from C-6 to both C-12 and C-16 through C-8; and from C-22 to both C-28 and C-29 through C-23 (
Figure 7). From the HMBC spectrum, the correlations from H
3-19 to C-1, C-5, C-9 and C-10; from H
3-18 to C-12, C-13, C-14 and C-17; from H-6 to C-4 and C-5; from H-16 to C-20; from H
3-21 to C-17, C-20 and C-22; from both H
3-26 and H
3-27 to C-24; and from H
3-28 to C-25 permitted the establishment of the carbon skeleton of a 23,24-dimethycholestane (
Figure 7). The hydroperoxyl group substituted at C-20 was confirmed by the HMBC correlation of the hydroperoxyl proton δ
H 9.20 (br s) to the oxygenated carbon at δ
C 86.1 (C-20). An extensive survey of the literature revealed that the structure of
9 closely resembled that of michosterols A (
16), a polyoxygenated steroid isolated from the soft coral
Lobophytum michaelae [
27]. A comparison of the NMR data of
9 with
16, revealed that they were structural analogues, and the only difference occurred at the position C-25, where the acetyloxy group in
16 was replaced by a hydroxyl group in
9, in agreement with the 42 mass unit difference between compounds
9 and
16. As mentioned above, the planar structure of
9 was thus established.
The relative configuration of
9 was established mainly by analysis of the NOESY correlations. As depicted in
Figure 7, it was found that the NOE interactions displayed by both H
3-18/H-8, H
3-19/H-8, H
3-19/H-6 and H-6/H-8, assuming the β-orientation of H
3-19, H-6, H-8 and H
3-18. Moreover, the NOESY correlation of H-9/H-14 assigned the α-orientation of H-9 and H-14. Further, the chemical shifts of C-20, C-21, C-22, C-23, C-28 and C-29 on the side chain are similar to that of
16, suggested the same relative configuration of the chair centers on the side chain. In addition, the stereochemistry of C-5 and C-6 was assigned to be 5
S*, 6
S* by the comparison of chemical shifts of C-5 (δ
C 78.4) and C-6 (δ
C 73.2) with the model known compound 5β-cholestane-3β,5,6α-triol, which was previously isolated from the soft coral
Sinularia sp. [
28,
29]. Consequently, the relative configuration of
9 was determined. Furthermore, based on the biogenetic consideration of the biosynthetic pathway of steroids, Me-18 and Me-19 should be positioned on the
β-orientation; thus, the absolute configuration of
9 was elucidated as shown in
Figure 1.
Lobocaloid B (
10) was isolated as a white powder with the molecular formula of C
31H
52O
6 on the basis of the HRESIMS ion peak at
m/
z 543.3655 [M + Na]
+ (calcd. for C
31H
52O
6Na, 543.3656), corresponding to six degrees of unsaturation. The
13C NMR and DEPT spectra of
10 displayed 31 carbon signals, including eight methyls, eight methylenes, eight methines (including one olefinic at δ
C 123.6) and seven quaternary carbons (including an olefinic at δ
C 161.2 and one carbonyl at δ
C 170.6), accounting for two out of six degrees of unsaturation, thus requiring four extra rings in the molecule structure of
10. The
13C NMR data (
Table 3) of
10 closely resembled that of michosterols A (
16), suggesting that they were also structural analogues. In fact, the only difference between
10 and
16 appeared at C-20, where the hydroperoxyl group in
16 was substituted by a hydroxyl group in
10, in agreement with a 16 mass unit difference between their molecular weights. This assignment was supported by not only the carbon chemical shift of C-20 significantly upfield shifted from δ
C 85.6 ppm to δ
C 75.7 ppm, but also the chemical shift of the distinctive olefinic proton H-16 shifted from δ
H 5.70 (d,
J = 2.0 Hz) to δ
H 5.49 (dd,
J = 3.4, 1.5 Hz). The relative stereochemistry of
10 was determined by the analysis of NOE correlations (
Figure 7) and by comparison of NMR spectroscopic data with those of model molecule sarcophytosterol, a known steroid isolated from the Dongsha atoll soft coral
Lobophytum sarcophytoides, whose structure has been unambiguously determined by X-ray diffraction analysis [
30]. On the basis of the above findings, the structure of compound
10 was determined as shown in
Figure 1.
The molecular formula of lobocaloid C (
11) was the same as michosterols A (
16), which was determined by HRESIMS ion peak at
m/
z 559.3609 [M + Na]
+ (calcd. for C
31H
52O
7Na, 559.3605). The proton and carbon resonances of
11 showed a high degree of similarity to those of
16. By comparison of the
1H and
13C NMR data (
Table 4) of
11 and
16, the differences were found in the chemical shifts of carbons in rings A and B [C-1 (δ
C 32.3), C-2 (δ
C 31.0), C-4 (δ
C 40.8), C-5 (δ
C 76.3) and C-6 (δ
C 76.2) in
11 and C-1 (δ
C 25.2), C-2 (δ
C 27.7), C-4 (δ
C 30.0), C-5 (δ
C 78.1) and C-6 (δ
C 71.8) in
16)]. The following detailed analysis of
1H–
1H COSY and HMBC correlations assigned the planar structure of
11, the same as
16, which suggested that
11 was the isomer of
16. Literature checking revealed that the NMR data of rings A and B in
11 were strongly reminiscent of the known compound 23,24-dimethylcholest-16-ene-3β,5α,6β,11α,20(
R)-pentol-3-monoacetate [
31], indicating that the rings’ structures were identical. Thus, the absolute stereochemistry of C-3, C-5, and C-6 in
11 was speculatively assigned as 3
S, 5
R, 6
R.
Lobocaloid D (
12), which was obtained as a white powder, gave the molecular formula C
31H
52O
6 on the basis of its HRESIMS ion peak at
m/
z 543.3653 [M + Na]
+ (calcd. for C
31H
52O
6Na, 543.3656), 16 mass units less than that of
11. A comparison of the NMR data of
12 with
11 (
Table 4), revealed that they were structural analogues, with the only difference being the presence of a hydroxyl group substituted at C-20 on the side chain of
12 instead of a hydroperoxyl group in
11, in agreement with the 16 mass unit difference between compounds
11 and
12. The hydroxyl group was substituted at C-20, as evidenced by the observation of the upfield shifting of C-20 from δ
C 86.1 in
11 to δ
C 75.8 in
12. Similarly, the relative configuration of
12 was determined by analyzing NOE correlations and comparing its NMR data with those of compounds
10 and
11. As mentioned above, the structure of
12 was assigned as shown in
Figure 1.
Compound
15 was readily identified as lobophytrol B, a capnosane-type diterpenoid previously reported to be isolated from
Lobophytum sp. by our group [
23]. By comparing the nuclear magnetic and optical rotation data, they proved to be exactly the same. In this study, the suitable crystals of
15 were eventually obtained, and they were then sent for X-ray crystallographic analysis using Cu Ka radiation (λ = 1.54178 Å). Analysis of the X-ray data unambiguously determined the planar structure and absolute configuration of
15 with the Flack parameter of 0.12 (7) (CCDC 2290866). Surprisingly, the X-ray result disclosed that the correct structure of lobophytrol B is
15a, instead of
15 (
Figure 8). Compared to the mass spectrum, the molecular weight is 18 more than before, probably due to the presence of different molecular fragmentation peaks. We carefully and analyzed in depth the data to determine why the structure of
15 was incorrectly assigned, realizing that the error was triggered by the chemical shift of carbon substituted by the hydroxyl group and the same oxygen ring established at positions C-8 and C-12, as well as the existence of various molecular fragment peaks in the mass spectrum. Careful re-examination of the HRESIMS spectrum revealed wrong identification of the molecular ion peak at
m/
z HREIMS 363.2517 [M + Na]
+ (calcd for C
20H
36O
4Na, 363.2506), while the cluster of quasi-molecular ion peaks centered at
m/
z 323.2581 [M + H]
+ (calcd for C
20H
35O
3, 323.2581) was overlooked (
Figure S10.1).
In the in vitro bioassay, the isolated compounds were tested for their antibacterial, cytotoxic and anti-inflammatory effects. In the antibacterial bioassays (
Table 5), compound
2 showed moderate antibacterial activities against the fish pathogenic bacteria
Streptococcus parauberis KSP28 (MIC 8.7 μg/mL) and
Phoyobacterium damselae FP2244 (MIC 17.3 μg/mL). Compounds
7–
9 showed weak antibacterial activities against
S. parauberis KSP28 (MIC 30.4, 32.2, 49.4 μg/mL) and
P. damselae FP2244 (MIC 30.4, 16.1, 49.4 μg/mL). All the steroids exhibited antibacterial activities against
S. parauberis KSP28 with MIC values ranging from 12.3 to 53.6 µg/mL. Furthermore, compounds
10 and
13 exhibited significant antibacterial activities against a variety of strains; compound
13 especially displayed potent inhibitory activity against
P. damselae FP2244,
S. parauberis SPOF3K and
S. agalactiae WR10 with MIC value of 6.2 µg/mL, 12.3 µg/mL and 12.3 µg/mL, respectively. Furthermore, compound
13 showed the growth inhibitory activities against the vancomycin-resistant
Enterococcus faecium G1 and G8 with the MIC values of 12.3 μg/mL and 12.3 μg/mL, respectively, comparable with that of positive control levofloxacin hydrochloride (MIC > 39.78 µg/mL), ampicillin sodium (MIC > 37.14 µg/mL) and vancomycin hydrochloride (MIC > 297 µg/mL and 74.25 µg/mL).
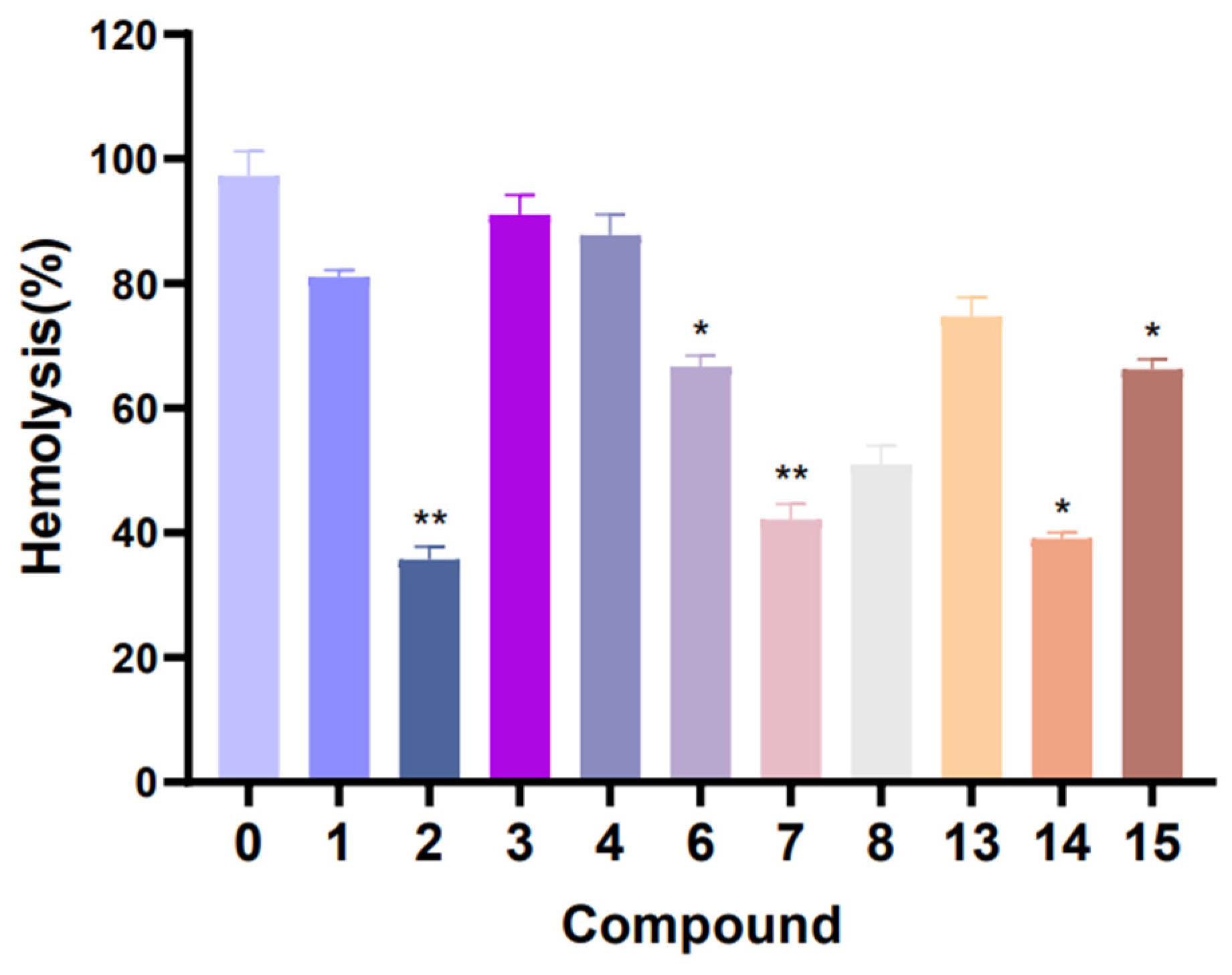
In hemolytic activity, the results showed that the hemolytic activity of
S. aureus treated with compounds
1–
4,
6–
8 and
13–
15 were obviously reduced when compared with the control (without compound treatment). In particular, the hemolysis percentage of compound
2,
7 and
14 test groups reached about 35.7%, 42.2% and 39.1% (
Figure 9). These results suggest that compounds
2,
7 and
14 have a strong inhibitory effect on the hemolysin production of
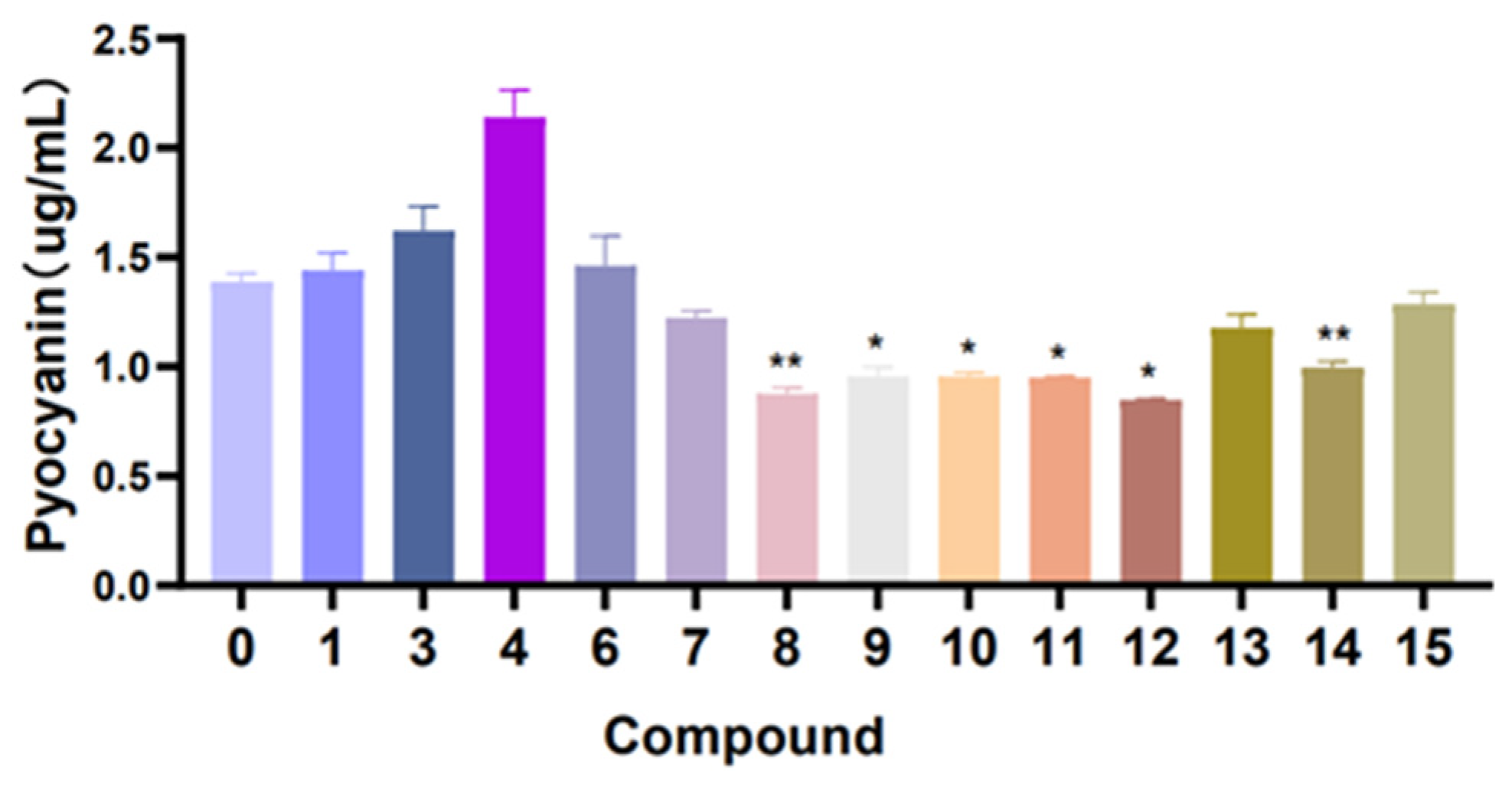
S. aureus. Furthermore, in the pyocyanin quantitation assay experiment (
Figure 10), compounds
8–
12 have medium inhibition effects on the pyocyanin production in
P. aeruginosa, compared with the control (without compound treatment).
3. Materials and Methods
3.1. General Experimental Procedures
Melting points were measured on an X-4 digital micro melting point apparatus. Optical rotations were measured on a Perkin-Elmer 241MC polarimeter (PerkinElner, Fremont, CA, USA). IR spectra were measured on a Nicolet 6700 spectrometer (Thermo Scientific, Waltham, MA, USA), and peaks are reported in cm
−1. NMR spectra were measured in CDCl
3 with a Bruker DRX 400, 500, 600 and 800 MHz spectrometer (Bruker Biospin AG, Fällanden, Germany). Chemical shifts are reported in parts per million (δ) in CDCl
3 (δ
H reported referred to CHCl
3 at 7.26 ppm; δ
C reported referred to CDCl
3 at 77.2 ppm), and coupling constants (
J) are expressed in Hz. HRESIMS spectra were recorded on an Agilent 1290-6545 UHPLC-QTOF mass spectrometer. Commercial silica gel (Qingdao Haiyang Chemical Group Co., Ltd., Qingdao, China, 200–300 and 300–400 mesh) and Sephadex LH-20 gel (Amersham Biosciences, Amersham, UK) were used for column chromatography. Precoated silica gel plates (Yan Tai Zi Fu Chemical Group Co., Yantai, China, G60 F-254) were used for analytical TLC. Reversed-phase (RP) HPLC was performed on an Agilent 1260 series liquid chromatography equipped with a DAD G1315D detector at 210 and 254 nm. A semi-preparative ODS-HG-5 column [5 µm, 250 × 9.4 mm] was employed for the purifications. All solvents used for CC and HPLC were of analytical grade (Shanghai Chemical Reagents Co., Ltd., Shanghai, China) and chromatographic grade (Dikma Technologies Inc., Foothill Ranch, CA, USA), respectively. X-ray crystallographic analysis was carried out on a Bruker D8 Venture diffractometer with Cu Kα radiation (λ = 1.54178 Å) at 100 K. The collected data integration and reduction were processed with SAINT V8.37A software, and multi-scan absorption corrections were performed using the SADABS program. The structure was solved with the SHELXT structure solution program using intrinsic phasing and refined with the SHELXL refinement package using least-squares minimization. Copies of these data can be obtained free of charge via
www.ccdc.cam.ac.uk. or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB21EZ, UK. [Fax: (+44) 1223-336-033. E-mail:
[email protected]].
3.2. Animal Materials
Specimens of L. catalai, identified by Prof. Xiu-Bao Li from Hainan University, were collected by scuba along the coast off Yalong Bay (18°15′ N, 109°30′ E), Hainan Province, China, in 2006, at a depth of −15 m. A voucher specimen (YAL-65) was deposited and is available for inspection at the Bohai Rim Advanced Research Institute for Drug Discovery.
3.3. Extraction and Isolation
The frozen animals (260.5 g, dry weight after extraction) were cut into pieces and extracted exhaustively with acetone at room temperature (2.0 L × 4). The organic extract was evaporated to give a brown residue, which was then partitioned between Et2O and H2O. The Et2O solution was evaporated to give a dark brown residue (4.0 g). The obtained residue was subjected to gradient silica gel (200–300 mesh) column chromatography (CC) [Et2O/petroleum ether (PE) 0→100%] and yielded nine fractions (Fr. 1–9). Fr. 2 was divided into three subfractions (Fr. 2A–2C) by Sephadex LH-20 CC (PE/CH2Cl2/MeOH, 2:1:1). Following two-stage purification including Sephadex LH-20 CC (CH2Cl2, 100%) and silica gel CC (300–400 mesh, PE/Et2O 100:1→10:1), the subfraction Fr. 2C13 was purified by semi-preparative HPLC (MeCN, 100%, 3.0 mL/min) and analytical HPLC (MeOH/H2O, 80:20, 1.0 mL/min) to yield compounds 1 (2.0 mg, tR = 16.4 min), 2 (0.3 mg, tR = 18.4 min) and 4 (1.0 mg, tR = 15.3 min), respectively. Fr. 6 was split by Sephadex LH-20 CC (PE/CH2Cl2/MeOH, 2:1:1) to give two subfractions (Fr. 6A and Fr. 6B). Next, Fr. 6B was purified by Sephadex LH-20 CC (CH2Cl2, 100%), yielding subfraction Fr. 6BA. Final purification of Fra. 6BA was achieved by semi-preparative HPLC (MeOH/H2O, 80:20, 2.8 mL/min) to afford compounds 7 (0.5 mg, tR = 24.5 min) and 6 (1.0 mg, tR = 22.0 min) and the mixture of compounds 3 and 5. The mixture was further purified by analytical HPLC (MeCN/H2O, 60:40, 1.0 mL/min) to yield pure 3 (1.1 mg, tR = 13.0 min) and 5 (1.0 mg, tR = 14.6 min), respectively. Fr. 9 was subjected to a column of Sephadex LH-20 eluted with CH2Cl2/MeOH, 1:1, to yield two subfractions (Fr. 9A and 9B). Fr. 9A was first split by Sephadex LH-20 column chromatography (PE/CH2Cl2/MeOH, 2:1:1) to give five subfractions (Fr. 9AA–Fr. 9AE). Fr. 9AE was purified by RP-HPLC (MeCN/H2O, 60:40, 3.0 mL/min), yielding a subfraction (Fr. 9AEC, tR = 8.0 min). Since there are two points observed on the thin-layer chromatography (TLC), Fr. 9AEC was purified by silica gel CC (300–400 mesh, CH2Cl2/MeOH, 96:4) to give compounds 12 (1.0 mg) and 14 (0.5 mg). Fr. 9AD was purified with silica gel CC (300–400 mesh, Et2O/PE, 1:1), followed by semi-preparative HPLC (MeCN/H2O, 60:40, 3.0 mL/min) to afford 13 (5.4mg, tR = 2.1 min), 11 (3.2mg, tR = 8.8 min) and fraction 9ADFG (tR = 13.7min). In a similar manner, Fr. 9ADFG was purified by silica gel CC (300–400 mesh, CH2Cl2/MeOH, 98:2) to give compound 10 (2.6 mg). Moreover, compound 9 (2.7 mg, tR = 7.3 min) was obtained from Fr. 9B through Sephadex LH-20 CC (PE/CH2Cl2/MeOH, 2:1:1) followed by RP-HPLC (MeCN/H2O, 60:40, 3.0 mL/min), while compound 15 (2.7 mg, tR = 7.3 min) was obtained from the Fr. 9B through Sephadex LH-20 CC (PE/CH2Cl2/MeOH, 2:1:1), silica gel CC (200–300 mesh, Et2O/PE 50%→100%), followed by RP-HPLC (MeCN/H2O, 60:40, 3.0 mL/min).
3.3.1. Lobocaline A (1)
Colorless crystal, m.p. 68.9–71.6 °C;
+97.3 (c 0.20, CHCl
3); IR (KBr): ν
max 3446, 2956, 2924, 2854, 1737, 1458, 1369, 1238, 1026 cm
−1; For
1H and
13C NMR spectroscopic data, see
Table 1 and
Table 2; HRESIMS
m/
z 347.2580 [M + H]
+ (calcd. for C
22H
35O
3, 347.2581).
3.3.2. Lobocaline B (2)
Colorless oil,
+3.9 (c 0.03, MeOH); IR (KBr): ν
max 3444, 2956, 2917, 2849, 1731, 1030 cm
−1; For
1H and
13C NMR spectroscopic data, see
Table 1 and
Table 2; HRESIMS
m/
z 347.2581 [M + H]
+ (calcd. for C
22H
35O
3, 347.2581).
3.3.3. Lobocaline C (3)
Colorless oil,
+139.5 (c 0.10, MeOH); IR (KBr): ν
max 3438, 2959, 2926, 1457, 1382, 1087, 1022 cm
−1; For
1H and
13C NMR spectroscopic data, see
Table 1 and
Table 2; HRESIMS
m/
z 327.2295 [M + Na]
+ (calcd. for C
20H
32O
2Na, 327.2295).
3.3.4. Lobocaline D (4)
Colorless oil,
+162.7 (c 0.10, CHCl
3); IR (KBr): ν
max 3438, 2959, 2926, 1457, 1382, 1087, 1022 cm
−1; For
1H and
13C NMR spectroscopic data, see
Table 1 and
Table 2; HRESIMS
m/
z 327.2293 [M + Na]
+ (calcd. for C
20H
32O
2Na, 327.2295).
3.3.5. Lobocaline E (5)
Colorless oil,
+17.2 (c 0.10, CHCl
3); IR (KBr): ν
max 3359, 2922, 2851, 1736, 1238, 1141, 1031 cm
−1; For
1H and
13C NMR spectroscopic data, see
Table 1 and
Table 2; HRESIMS
m/
z 369.2397 [ M+ Na]
+ (calcd. for C
22H
34O
3Na, 369.2400).
3.3.6. Lobocaloid A (9)
White amorphous powder,
+9.8 (c 0.27, MeOH); IR (KBr): ν
max 3436, 2929, 2871, 1375, 1089, 1050 cm
−1; For
1H and
13C NMR spectroscopic data, see
Table 3; HRESIMS
m/
z 517.3504 [M + Na]
+ (calcd. for C
29H
50O
6Na, 517.35).
3.3.7. Lobocaloid B (10)
White amorphous powder,
−8.3 (c 0.26, MeOH); IR (KBr): ν
max 3444, 2930, 1730, 1369, 1260, 1050, 800 cm
−1; For
1H and
13C NMR spectroscopic data, see
Table 3; HRESIMS
m/
z 543.3655 [M + Na]
+ (calcd. for C
31H
52O
6Na, 543.3656).
3.3.8. Lobocaloid C (11)
White amorphous powder,
−16.7 (c 0.32, MeOH); IR (KBr): ν
max 3444, 2931, 2864, 1374, 1030 cm
−1; For
1H and
13C NMR spectroscopic data, see
Table 4; HRESIMS
m/
z 559.3609 [M + Na]
+ (calcd. for C
31H
52O
7Na, 559.3605).
3.3.9. Lobocaloid D (12)
White amorphous powder;
−43.3 (c 0.10, MeOH); IR (KBr): ν
max 3443, 2928, 2851, 1712, 1368, 1036 cm
−1; For
1H and
13C NMR spectroscopic data, see
Table 4; HRESIMS
m/
z 543.3653 [M + Na]
+ (calcd. for C
31H
52O
6Na, 543.3656).
3.4. Calculation Section
Conformational search was performed by using the torsional sampling (MCMM) approach and OPLS_2005 force field within an energy window of 21 kJ/mol. Conformers above 1% Boltzmann populations were re-optimized at the B3LYP/6-311G(d,p) level with the IEFPCM solvent model for chloroform. Frequency analysis was also carried out to confirm that the re-optimized geometries were at the energy minima. Subsequently, NMR calculations were performed at the PCM/mPW1PW91/6-31G(d) level, as recommended for DP4+. NMR shielding constants were calculated using the GIAO method. Finally, shielding constants were averaged over the Boltzmann distribution obtained for each stereoisomer and correlated with the experimental data. ECD spectra were obtained by TDDFT calculations with the B3LYP/6-311G(d,p) level with the IEFPCM solvent model for CH3CN. At last, the Boltzmann-averaged ECD spectra of the compounds were obtained with SpecDis (Version 1.71).
3.5. Acetylation of Compound 6
Compound
6 (0.5 mg) was dissolved in dry pyridine (2.0 mL) and mixed with 50 mL of Ac
2O. The mixtures were stirred at room temperature overnight, and the reaction was detected on the TLC by heating after spraying with vanillin H
2SO
4 reagent. The crude acetylated product, after evaporating the solvent in vacuo, was purified by silica gel CC (petroleum ether/ether, 9:1) to afford a colorless crystal compound
1a (0.5 mg, 87% yield), which was identical to the natural sample of
1 in all respects (
Figure S1.10).
3.6. X-ray Crystallographic Analysis for Compounds 1 and 15a
Lobocaline A (1): colorless crystals, m.p. 68.9–71.6 °C; monoclinic, 2(C22H34O3), Mr = 692.98, crystal size: 0.11 × 0.05 × 0.04 mm3, space group P21, a = 10.6135(3) Å, b = 16.2226(5) Å, c = 11.8888(4) Å, V = 2044.73(11) Å3, Z = 2, ρcalc = 1.126 g/cm3, F (000) = 760.0. Independent reflections: 7619 with Rint = 0.0385, Rsigma = 0.0475. Final R1 = 0.0479, wR2 = 0.1174 reflections with I ≥ 2σ (I), R1 = 0.0523 (wR2 = 0.1215) for all unique data, Flack parameter: 0.00(10). The crystals of 1 were recrystallized from MeOH at room temperature. Crystallographic data for 1 were deposited at the Cambridge Crystallographic Data Centre (Deposition nos. CCDC 2290865).
Lobophytrol B (15a): colorless crystals, m.p. 119.1–177.0 °C; monoclinic, C20H36O4, Mr = 340.49, crystal size 0.15 × 0.08 × 0.05 mm3, space group I4, a = 14.6887(3) Å, b = 14.6887(3) Å, c = 37.2232(11) Å, V = 8031.2(4) Å3, Z = 16, ρcalc = 1.126 g/cm3, F(000) = 3008.0, 57940 collected reflections, 8240 independent reflections (Rint = 0.0620, Rsigma = 0.0327), final R1 = 0.0399 (wR2 = 0.1022) reflections with I ≥ 2σ (I), R1 = 0.0431, wR2 = 0.1051 for all unique data, Flack parameter: 0.12(7). The crystals of 15a were crystallized from acetone at room temperature. Crystallographic data for 15a were deposited at the Cambridge Crystallographic Data Centre (Deposition nos. CCDC 2290866).
3.7. Antibacterial Activity Bioassays
The strains Streptococcus parauberis KSP28, Streptococcus parauberis SPOF3K, Phoyoba cteriumdamselae FP2244, Aeromonas salmonicida AS42, Photobacterium halotolerans LMG 22194T, Enterococcus faecium 5270 MDR8 and Lactococcus garvieae FP MP5245 were provided by National Fisheries Research & Development Institute, Korea. The vancomycin-resistant Enterococcus faecium bacteria G1, G4, G7, G8 and G13 were provided by Ruijin Hospital, Shanghai Jiao Tong University School of Medicine. The strain Streptococcus agalactiae WR10 and Edwardsiella piscicida TH1 were provided by Chinese Academy of Tropical Agricultural Sciences. The MIC values for all antimicrobial agents were measured by the 96-well micro-dilution method. Mueller–Hinton II broth (cation-adjusted, BD 212322) was used for MIC value determination. Generally, compounds were dissolved with DMSO to 20 mM as stock solutions. All samples were diluted with culture broth to 500 µM as the initial concentration. Further 1:2 serial dilutions were performed by addition of culture broth to reach concentrations ranging from 500 µM to 0.24 µM. A total of 100 µL of each dilution was distributed in 96-well plates, as well as sterile controls, growth controls (containing culture broth plus DMSO, without compounds) and positive controls (containing culture broth plus control antibiotics, such as tetracycline). Each test and growth control well was inoculated with 5 µL of an exponential-phase bacterial suspension (about 105 CFU/well). The 96-well plates were incubated at 37 ℃ for 24 h. MIC values of these compounds were defined as the lowest concentration to inhibit the bacterial growth completely. All MIC values were interpreted according to recommendations of the Clinical and Laboratory Standards Institute (CLSI).
3.8. Antihemolytic Activity Bioassays
Blood was collected from the eye sockets of SD rats and stood for 30 to 60 min. After centrifugation (800 rpm, 5 min), the red blood cells were cleaned twice with 0.9% normal saline and then added to create a 4% red blood cells suspension. S. aureus suspension was incubated with compounds (100 µM) in a centrifuge tube for 12 h at 37 ℃. After centrifugation (6000 rpm, 15 min), 500 µL of the supernatant was taken from each tube, filtered by a 0.2 μm filter membrane and incubated with 500 µL of freshly red blood cells suspension at 37 ℃ for 2 h. The incubation of S. aureus and red blood cells suspension was used as the positive control, and the incubation of LB liquid medium and red blood cells suspension served as the negative control. After centrifugation (800 rpm, 5 min), the absorbance of supernatants at 540 nm was examined. The percentage of hemolysis value was calculated by comparing it with the positive control (100% hemolysis).
3.9. Pyocyanin Quantitation Assay
The P. aeruginosa strain was mixed with the compounds (100 µM) at 37 ℃ and 140 rpm/min for 24 h, and the supernatant was mixed with 1 mL chloroform. Then, the lower chloroform phase was mixed with 200 µL of 0.2 N HCl; after shaking and centrifugal (4500 rpm, 10 min), the color layer was removed and measured at 520 nm. Concentrations, expressed as micrograms of pyocyanin produced per milliliter of culture supernatant, were determined by multiplying the optical density at 520 nm (OD520) by 17.072.


 ,
, 











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}