
New Fusarochromanone Derivatives from the Marine Fungus Fusarium equiseti UBOCC-A-117302
 , , ,
, , ,  , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Results and Discussion
2.1. FBMN Analysis and Metabolite Annotation
2.2. Structure Elucidation
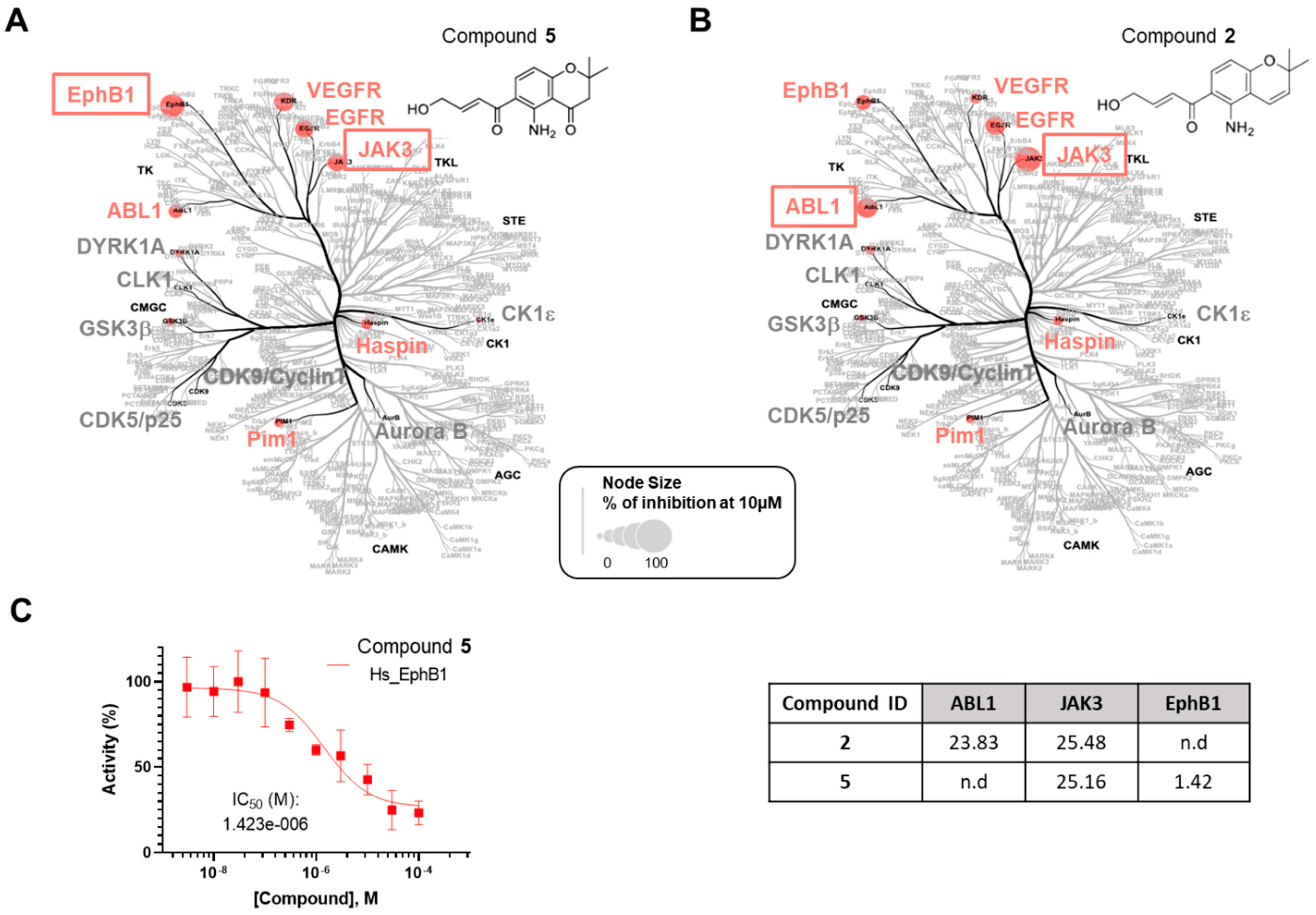
2.3. Biological Assays
3. Materials and Methods
3.1. General Experimental Procedure
3.2. Isolation and Identification of the Fungus
3.3. Fermentation
3.4. Global Natural Products Social Molecular Networking
3.5. Extraction and Purification
3.6. Kinase Inhibition Assays
3.7. Cytotoxic Assay
3.7.1. Cell Culture
3.7.2. Cell Viability
3.8. Antimicrobial Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Stien, D. Marine Microbial Diversity as a Source of Bioactive Natural Products. Mar. Drugs 2020, 18, 215. [Google Scholar] [CrossRef] [PubMed]
- Kamat, S.; Kumar, S.; Philip, S.; Kumari, M. Secondary Metabolites from Marine Fungi: Current Status and Application. In Microbial Biomolecules; Elsevier: Amsterdam, The Netherlands, 2023; pp. 181–209. [Google Scholar]
- Xie, W.; Mirocha, C.J.; Wen, Y. Isolation and Structure Identification of Two New Derivatives of the Mycotoxin Fusarochromenone Produced by Fusarium equiseti. J. Nat. Prod. 1995, 58, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.P.; Mirocha, C.J.; Pawlosky, R.J.; Wen, Y.C.; Xu, X.G. Biosynthesis of Fusarochromanone and Its Monoacetyl Derivative by Fusarium equiseti. Appl. Environ. Microbiol. 1989, 55, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.J.; Goodwin, J.T.; Fraiman, A.; Cole, R.J.; Lynn, D.G. Characterization of the Fusarium Toxin Equisetin: The Use of Phenylboronates in Structure Assignment. J. Am. Chem. Soc. 1989, 111, 8223–8231. [Google Scholar] [CrossRef]
- Huang, B.; Peng, S.; Liu, S.; Zhang, Y.; Wei, Y.; Xu, X.; Gao, C.; Liu, Y.; Luo, X. Isolation, Screening, and Active Metabolites Identification of Anti-Vibrio Fungal Strains Derived from the Beibu Gulf Coral. Front. Microbiol. 2022, 13, 930981. [Google Scholar] [CrossRef]
- Zhao, D.; Han, X.; Wang, D.; Liu, M.; Gou, J.; Peng, Y.; Liu, J.; Li, Y.; Cao, F.; Zhang, C. Bioactive 3-Decalinoyltetramic Acids Derivatives from a Marine-Derived Strain of the Fungus Fusarium equiseti D39. Front. Microbiol. 2019, 10, 1285. [Google Scholar] [CrossRef]
- Liu, S.; Gao, W.; Yang, X.; Huo, R.; Chen, F.; Cao, F.; Luo, D. Structure Determination and Cytotoxic Evaluation of Metabolites from the Entomogenous Fungus Fusarium equiseti. J. Antibiot. 2021, 74, 176–180. [Google Scholar] [CrossRef]
- Zhao, D.L.; Liu, J.; Han, X.B.; Wang, M.; Peng, Y.L.; Ma, S.Q.; Cao, F.; Li, Y.Q.; Zhang, C.S. Decalintetracids A and B, Two Pairs of Unusual 3-Decalinoyltetramic Acid Derivatives with Phytotoxicity from Fusarium equiseti D39. Phytochemistry 2022, 197, 113125. [Google Scholar] [CrossRef]
- Shiono, Y.; Shibuya, F.; Murayama, T.; Koseki, T.; Poumale, H.M.P.; Ngadjui, B.T. A Polyketide Metabolite from an Endophytic Fusarium equiseti in a Medicinal Plant. Z. Naturforsch. B 2013, 68, 289–292. [Google Scholar] [CrossRef]
- Hawas, U.W.; Al-Farawati, R.; Abou El-Kassem, L.T.; Turki, A.J. Different Culture Metabolites of the Red Sea Fungus Fusarium equiseti Optimize the Inhibition of Hepatitis C Virus NS3/4A Protease (HCV PR). Mar. Drugs 2016, 14, 190. [Google Scholar] [CrossRef]
- Dai, X.M.; Pan, H.L.; Lan, W.J.; Chen, L.P.; Feng, G.K.; Deng, R.; Zhu, X.F.; Li, H.J. Indole Alkaloids Fusarindoles A–E from Marine-Derived Fungus Fusarium equiseti LJ-1. Phytochemistry 2022, 204, 113456–113463. [Google Scholar] [CrossRef] [PubMed]
- Mahdavian, E.; Palyok, P.; Adelmund, S.; Williams-Hart, T.; Furmanski, B.D.; Kim, Y.J.; Gu, Y.; Barzegar, M.; Wu, Y.; Bhinge, K.N.; et al. Biological Activities of Fusarochromanone: A Potent Anti-Cancer Agent. BMC Res. Notes 2014, 7, 601. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Barzegar, M.; Chen, X.; Wu, Y.; Shang, C.; Mahdavian, E.; Salvatore, B.A.; Jiang, S.; Huang, S. Fusarochromanone-Induced Reactive Oxygen Species Results in Activation of JNK Cascade and Cell Death by Inhibiting Protein Phosphatases 2A and 5. Oncotarget 2015, 6, 42322–42333. [Google Scholar] [CrossRef] [PubMed]
- Dreau, D.; Foster, M.; Hogg, M.; Culberson, C.; Nunes, P.; Wuthier, R.E. Inhibitory Effects of Fusarochromanone on Melanoma Growth. Anticancer Drugs 2007, 18, 897–904. [Google Scholar] [CrossRef]
- Mahdavian, E.; Marshall, M.; Martin, P.M.; Cagle, P.; Salvatore, B.A.; Quick, Q.A. Caspase-Dependent Signaling Underlies Glioblastoma Cell Death in Response to the Fungal Metabolite, Fusarochromanone. Int. J. Mol. Med. 2014, 34, 880–885. [Google Scholar] [CrossRef]
- Minervini, F.; Lucivero, G.; Visconti, A.; Bottalico, C. Immunomodulatory Effects of Fusarochromanones TDP-1 and TDP-2. Nat. Toxins 1992, 1, 15–18. [Google Scholar] [CrossRef]
- Mahdavian, E.; Salvatore, B.; Clifford, J. Novel Derivatives of Fusarochromanone: Potential Therapeutic Compounds. Cancer Res. 2007, 67 (Suppl. S9), 3975. [Google Scholar]
- Nothias, L.F.; Petras, D.; Schmid, R.; Duhrkop, K.; Rainer, J.; Sarvepalli, A.; Protsyuk, I.; Ernst, M.; Tsugawa, H.; Fleischauer, M.; et al. Feature-Based Molecular Networking in the GNPS Analysis Environment. Nat. Methods 2020, 17, 905–908. [Google Scholar] [CrossRef]
- Marshall, J.W.; de Mattos-Shipley, K.M.J.; Ghannam, I.A.Y.; Munawar, A.; Killen, J.C.; Lazarus, C.M.; Cox, R.J.; Willis, C.L.; Simpson, T.J. Fusarochromene, a Novel Tryptophan-Derived Metabolite from Fusarium sacchari. Org. Biomol. Chem. 2021, 19, 182–187. [Google Scholar] [CrossRef]
- Niederer, D.; Tamm, C.; Zürcher, W. Nitrogen-Containing Metabolites of Fusarium sambucinum. Tetrahedron Lett. 1992, 33, 3997–4000. [Google Scholar] [CrossRef]
- Knestrick, M.A. From Florida to Antarctica: Dereplication Strategies and Chemical Investigations of Marine Organisms. Ph.D. Thesis, University of South Florida, Tampa, FL, USA, 2018. [Google Scholar]
- Dasgupta, Y.; Koptyra, M.; Hoser, G.; Kantekure, K.; Roy, D.; Gornicka, B.; Nieborowska-Skorska, M.; Bolton-Gillespie, E.; Cerny-Reiterer, S.; Muschen, M.; et al. Normal ABL1 Is a Tumor Suppressor and Therapeutic Target in Human and Mouse Leukemias Expressing Oncogenic ABL1 Kinases. Blood 2016, 127, 2131–2143. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.J.; Malaviya, R.; Yang, C.; Argentieri, R.; Wang, B.; Chen, X.; Murray, W.V.; Cavender, D. Synthetic Staurosporines via a Ring-Closing Metathesis Strategy as Potent JAK3 Inhibitors and Modulators of Allergic Responses. Bioorg. Med. Chem. Lett. 2009, 19, 3333–3338. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Properties of FDA-Approved Small Molecule Protein Kinase Inhibitors: A 2020 Update. Pharmacol. Res. 2020, 152, 104609. [Google Scholar] [CrossRef]
- Han, D.C.; Shen, T.L.; Miao, H.; Wang, B.; Guan, J.L. EphB1 Associates with Grb7 and Regulates Cell Migration. J. Biol. Chem. 2002, 277, 45655–45661. [Google Scholar] [CrossRef] [PubMed]
- Metz, K.S.; Deoudes, E.M.; Berginski, M.E.; Jimenez-Ruiz, I.; Aksoy, B.A.; Hammerbacher, J.; Gomez, S.M.; Phanstiel, D.H. Coral: Clear and Customizable Visualization of Human Kinome Data. Cell Syst. 2018, 7, 347–350.e1. [Google Scholar] [CrossRef]
- Furmanski, B.D.; Dreau, D.; Wuthier, R.E.; Fuseler, J.W. Differential Uptake and Selective Permeability of Fusarochromanone (FC101), a Novel Membrane-Permeable Anticancer Naturally Fluorescent Compound in Tumor and Normal Cells. Microsc. Microanal. 2009, 15, 545–557. [Google Scholar] [CrossRef]
- Gu, Y.; Chen, X.; Shang, C.; Singh, K.; Barzegar, M.; Mahdavian, E.; Salvatore, B.A.; Jiang, S.; Huang, S. Fusarochromanone Induces G1 Cell Cycle Arrest and Apoptosis in COS7 and HEK293 Cells. PLoS ONE 2014, 9, e112641. [Google Scholar] [CrossRef]
- Pathre, S.V.; Gleason, W.B.; Lee, Y.-W.; Mirocha, C.J. The Structure of Fusarochromanone: A New Mycotoxin from Fusarium roseum “Graminearum”. Can. J. Chem. 1986, 64, 1258–1261. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, S.; Liu, X.; Lin, W.; Zhu, K. Equisetin Restores Colistin Sensitivity Against Multi-Drug Resistant Gram-Negative Bacteria. Antibiotics 2021, 10, 1263. [Google Scholar] [CrossRef]
- Chen, S.; Liu, D.; Zhang, Q.; Guo, P.; Ding, S.; Shen, J.; Zhu, K.; Lin, W. A Marine Antibiotic Kills Multidrug-Resistant Bacteria Without Detectable High-Level Resistance. ACS Infect. Dis. 2021, 7, 884–893. [Google Scholar] [CrossRef]
- Larson, E.C.; Lim, A.L.; Pond, C.D.; Craft, M.; Cavuzic, M.; Waldrop, G.L.; Schmidt, E.W.; Barrows, L.R. Pyrrolocin C and Equisetin Inhibit Bacterial Acetyl-CoA Carboxylase. PLoS ONE 2020, 15, e0233485. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Chen, S.; Liu, F.; Zhu, Q.; Shen, J.; Lin, W.; Zhu, K. Equisetin Targets Intracellular Staphylococcus aureus through a Host-Acting Strategy. Mar. Drugs 2022, 20, 656. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Zhang, C.; Chen, G.; Wu, M.; Liu, W.; Ding, C.; Dong, Q.; Fan, E.; Liu, Q. Reactive Oxygen Species Inhibit Biofilm Formation of Listeria monocytogenes. Microb. Pathog. 2019, 127, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.W.; Wu, M.; Liu, W.K.; Xie, M.M.; Zhang, W.S.; Fan, E.G.; Liu, Q. Reactive Oxygen Species Inhibit Listeria monocytogenes Invasion into HepG2 Epithelial Cells. Food Sci. Nutr. 2018, 6, 1501–1507. [Google Scholar] [CrossRef]
- O’Donnell, K.; Kistler, H.C.; Cigelnik, E.; Ploetz, R.C. Multiple Evolutionary Origins of the Fungus Causing Panama Disease of Banana: Concordant Evidence from Nuclear and Mitochondrial Gene Genealogies. Proc. Natl. Acad. Sci. USA 1998, 95, 2044–2049. [Google Scholar] [CrossRef]
- Quemener, M.; Dayras, M.; Frotte, N.; Debaets, S.; Le Meur, C.; Barbier, G.; Edgcomb, V.; Mehiri, M.; Burgaud, G. Highlighting the Biotechnological Potential of Deep Oceanic Crust Fungi Through the Prism of Their Antimicrobial Activity. Mar. Drugs 2021, 19, 411. [Google Scholar] [CrossRef]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular Framework for Processing, Visualizing, and Analyzing Mass Spectrometry-Based Molecular Profile Data. BMC Bioinformatics 2010, 11, 395. [Google Scholar] [CrossRef]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T.; et al. Sharing and Community Curation of Mass Spectrometry Data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A Software Environment for Integrated Models of Biomolecular Interaction Networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Zegzouti, H.; Zdanovskaia, M.; Hsiao, K.; Goueli, S.A. ADP-Glo: A Bioluminescent and Homogeneous ADP Monitoring Assay for Kinases. Assay Drug Dev. Technol. 2009, 7, 560–572. [Google Scholar] [CrossRef]
- Brikci-Nigassa, N.M.; Bentabed-Ababsa, G.; Erb, W.; Chevallier, F.; Picot, L.; Vitek, L.; Fleury, A.; Thiéry, V.; Souab, M.; Robert, T. 2-Aminophenones, a Common Precursor to N-Aryl Isatins and Acridines Endowed with Bioactivities. Tetrahedron Lett. 2018, 74, 1785–1801. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Position | 1 | 2 | ||
|---|---|---|---|---|
| 1H [δ, mult. (J in Hz)] | 13C (δ) | 1H [δ, mult. (J in Hz)] | 13C (δ) | |
| 2 | - | 77.6 | - | 77.5 |
| 3 | 5.65 d (10.0) | 128.9 | 5.64 d (10.0) | 128.8 |
| 4 | 6.58 d (10.0) | 117.1 | 6.59 d (10.0) | 117.3 |
| 5 | - | 150.2 | - | 150.6 |
| 6 | - | 112.8 | - | 113.9 |
| 7 | 7.61 d (9.0) | 133.9 | 7.70 d (9.0) | 134.5 |
| 8 | 6.12 d (9.0) | 106.9 | 6.11 d (9.0) | 106.6 |
| 9 | - | 159.9 | - | 159.7 |
| 10 | - | 107.6 | - | 107.6 |
| 11 | 1.40 s | 28.0 | 1.40 s | 28.0 |
| 12 | 1.40 s | 28.0 | 1.41 s | 28.0 |
| 1′ | - | 198.0 | - | 192.2 |
| 2′ | 3.36 m 3.21 dd (18.0, 8.3) | 38.5 | 7.23 dt (15.2, 2.0) | 126.1 |
| 3′ | 3.77 m | 51.2 | 6.90 dt (15.2, 4.1) | 145.6 |
| 4′ | 3.82 m 3.67 dd (10.7, 5.5) | 62.7 | 4.33 dd (4.1, 2.2) | 62.6 |
| Compound | EC50 (µM) | ||
|---|---|---|---|
| RPE-1 | HCT-116 | U2OS | |
| 1 | 0.176 | 0.087 | 0.896 |
| 2 | 10.030 | 13.730 | 13.180 |
| 3 | 23.140 | 62.950 | 35.090 |
| 4 | 16.700 | 84.380 | 39.790 |
| 5 | 5.222 | 8.036 | 7.351 |
| 6 | 0.058 | 0.170 | 0.232 |
| 7 | >25 | >25 | >25 |
| 8 | >25 | >25 | >25 |
| Staurosporine | 1.900 | 25.700 | 9.900 |
| MBC/MIC (µM) | ||||
|---|---|---|---|---|
| Compound | 2 | 4 | 8 | Erythromycin |
| L. monocytogenes SOR 100 | 125* | 125 * | 31.25 ** | 1.36 |
| E. faecalis CIP A 186 | (−) | (−) | 31.25 * | 5.44 |
| B. cereus ATCC 6464 | (−) | (−) | 7.8 ** | 5.45 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pham, G.N.; Josselin, B.; Cousseau, A.; Baratte, B.; Dayras, M.; Le Meur, C.; Debaets, S.; Weill, A.; Robert, T.; Burgaud, G.; et al. New Fusarochromanone Derivatives from the Marine Fungus Fusarium equiseti UBOCC-A-117302. Mar. Drugs 2024, 22, 444. https://doi.org/10.3390/md22100444
Pham GN, Josselin B, Cousseau A, Baratte B, Dayras M, Le Meur C, Debaets S, Weill A, Robert T, Burgaud G, et al. New Fusarochromanone Derivatives from the Marine Fungus Fusarium equiseti UBOCC-A-117302. Marine Drugs. 2024; 22(10):444. https://doi.org/10.3390/md22100444
Chicago/Turabian StylePham, Giang Nam, Béatrice Josselin, Arnaud Cousseau, Blandine Baratte, Marie Dayras, Christophe Le Meur, Stella Debaets, Amélie Weill, Thomas Robert, Gaëtan Burgaud, and et al. 2024. "New Fusarochromanone Derivatives from the Marine Fungus Fusarium equiseti UBOCC-A-117302" Marine Drugs 22, no. 10: 444. https://doi.org/10.3390/md22100444