2. Results and Discussion
Compound
1 was isolated as colourless crystals. Its optical rotation value was [
α]
25D + 57.8 (
c 1.0, MeOH). The IR absorption peaks were found at 3415, 2808, 1728, and 1600 cm
−1, indicating the presence of hydroxyl, aldehyde, ester carbonyl groups, and olefin, respectively. The molecular formula C
18H
26O
4 was determined by HRESIMS
m/z 307.1902 [M + H]
+ (calcd for C
18H
27O
4, 307.1904), implying six degrees of unsaturation. The
13C NMR spectrum (
Table 1) displayed the presence of eighteen carbon signals which, in combination with the HSQC spectrum, can be classified as one aldehyde carbonyl (
δC 194.0), one ester carbonyl (
δC 170.8), two protonated sp
2 (
δC 149.7 and 129.0), two non-protonated sp
2 (
δC 142.8 and 139.4), one oxyquaternary sp
3 (
δC 76.8), one quaternary sp
3 (
δC 41.1), one oxymethine sp
3 (
δC 73.8), one methine sp
3 (
δC 34.4), four methylene sp
3 (
δC 43.6, 32.2, 27.3, and 26.3), and four methyl (
δC 26.2, 22.8, 21.2, and 16.6) carbons. The
1H NMR spectrum (
Table 1), in conjunction with the HSQC spectrum, displayed a singlet of the aldehydric proton at
δH 9.35/
δC 194.0, two olefinic protons at
δH 6.21 (d,
J = 5.7, 1.9 Hz)/
δC 149.7 and
δH 5.59 (dd,
J = 5.7, 2.2 Hz)/
δC 129.0, one oxymethine at
δH 6.51 (d,
J = 5.7 Hz)/
δC 73.8, one methine at
δH 2.47, m/
δC 34.4, four pairs of methylene protons at
δH 3.07 (d,
J = 18.9 Hz) and 2.93 (d,
J =18.9 Hz)/
δC 32.2,
δH 2.14, m and 2.04, m/
δC 26.3,
δH 1.87 (d,
J = 15.8 Hz) and 1.42 (d,
J = 15.8 Hz)/
δC 43.6, and
δH 1.49, m/
δC 27.3, one methyl doublet at
δH 0.98 (d,
J = 6.7 Hz)/
δC 16.6, and three methyl singlets at
δH 2.15/
δC 21.2,
δH 1.14/
δC 26.2, and
δH 0.87/
δC 22.8.
The planar structure of
1 was elucidated by extensive analysis of
1H–
1H COSY and HMBC spectra (
Figure 2). The
1H–
1H COSY correlations revealed the presence of two spin systems from H-4 to H-5 and H-9 to H
2-10, H
2-10 to H
2-11, H
2-11 to H-12, and H-12 to H
3-15, while the HMBC spectrum showed correlations from H
3-13 to C-1, C-2, C-8, and C-12; H
3-14 to C-2, C-3, and C-4; H-16 to C-5, C-6, and C-7; and H-9 to C-1, C-7, C-10, and C-11, suggesting the presence of the 6/8 neolemnane sesquiterpene scaffold. The HMBC correlations from H-4 to C-17 and H
3-18 to C-17 indicated the existence of an acetoxyl group on C-4, while the correlations from H-16 to C-5, C-6, and C-7 revealed the presence of a formyl substituent on C-6. The presence of a formyl group on a sesquiterpene skeleton is not common.
The relative configuration of
1 was established by NOESY correlations (
Figure 2). In the NOESY spectrum, H-5 displayed a correlation to H-16, confirming the
E-configuration of the Δ
5,6 double bond. NOESY cross-peaks from H
3-13 to H
3-15, H-2a, H
3-15 to H-2a, and H
3-14 to H-2a revealed that the three methyl groups were on the same face. The absence of a NOESY correlation from H-4 to H
3-14 suggested that H-4 and H
3-14 were on the opposite side. Thus, the relative configurations at C-1, C-3, C-4, and C-12 in
1 were established as 1
R*, 3
R*, 4
R*, and 12
S*. Finally, a suitable crystal of
1 was obtained for X-ray analysis using a diffractometer equipped with Cu Kα radiation. The Ortep diagram of
1 (
Figure 3) shows the absolute configurations of C-1, C-3, C-4, and C-12 1
R, 3
R, 4
R, and 12
S, which were ascertained by a flack parameter of 0.2 (7) (
Table S4).
Compound
2 was obtained as colourless crystals with an optical rotation, [
α]
25D +1.6 (
c 1.0, MeOH). The molecular formula of
2 was determined as C
18H
26O
4 by HRESIMS
m/z 307.1898 [M + H]
+ (calcd for C
18H
27O
4, 307.1904), which is the same as that of
1. The
1H and
13C NMR spectra (
Table 1) of
2 closely resembled those of
1, with some chemical shift changes around the chiral C-3, suggesting that
2 could be a stereoisomer of
1. Furthermore, the HMBC correlations and
1H–
1H COSY cross-peaks (
Figure 2) confirmed that the planar structure of
2 was the same as that of
1.
In the NOESY spectrum (
Figure 2), H-5 displayed a correlation to H-16, consistent with the
E-configuration of the Δ
5,6 double bond. The NOESY correlations from H
3-13 to H-2a, H
2-11, H
3-15 to H-2a, H
2-11, revealed that the two methyl groups were on the same face. In addition, the NOESY correlations from H-4 to H
3-14 and H-12 to H
3-14 indicated that H-4, H
3-14, and H-12 were on the same side. Thus, the relative configuration at C-3 in
2 was different from that in
1, and the relative configurations at C-1, C-3, C-4, and C-12 in
2 were established as 1
R*, 3
S*, 4
R*, and 12
S*. Ultimately, the absolute configurations at C-1, C-3, C-4 and C-12 in
2 were established as 1
R, 3
S, 4
R, and 12
S [Flack parameter of 0.02(6)] (
Table S5) by the single-crystal X-ray diffraction experiment (
Figure 3). Therefore,
1 and
2 are epimers at C-3.
Compound
3 was isolated as colourless crystals and its molecular formula C
18H
26O
5 was determined by the HRESIMS
m/z 345.1670 [M + Na]
+ (calcd for C
18H
26O
5Na, 345.1672), requiring six degrees of unsaturation. Compound
3 was levorotatory, with [
α]
25D −10.8 (
c 1.0, MeOH). The
13C NMR spectrum (
Table 2) displayed the presence of eighteen carbon signals which, in combination with the HSQC spectrum, can be classified as two ester carbonyl (
δC 173.5 and 170.3), one protonated sp
2 (
δC 132.5), one non-protonated sp
2 (
δC 128.7), one oxyquaternary sp
3 (
δC 63.1), one quaternary sp
3 (
δC 37.6), two oxymethine sp
3 (
δC 72.1 and 65.0), two methine sp
3 (
δC 43.7 and 39.5), three methylene sp
3 (
δC 32.6, 26.4, and 24.6), one methoxy (
δC 51.7) and four methyl (
δC 20.9, 18.8, 16.0, and 15.4) carbons. The
1H NMR spectrum (
Table 2), in conjunction with the HSQC spectrum, displayed a singlet of the olefinic proton at
δH 5.58/
δC 149.7, two oxymethine at
δH 5.68 (d,
J = 5.3 Hz)/
δC 72.1 and
δH 3.15, m/
δC 65.0, two methine at
δH 2.24, m/
δC 43.7 and
δH 1.06, m/
δC 39.5, three pairs of methylene protons at
δH 2.60, m/
δC 32.6,
δH 2.03, m and 1.74, m/
δC 26.4 and
δH 1.30, m and 1.08, m/
δC 24.6, one methyl doublet at
δH 0.80 (d,
J = 6.8 Hz)/
δC 15.4, and four methyl singlets at
δH 3.65/
δC 51.7,
δH 1.99/
δC 20.9,
δH 1.66/
δC 18.8, and
δH 0.92/
δC 16.0.
The
1H–
1H COSY correlations (
Figure 2) from H-4 to H-5, H-5 to H-6, and H-9 to H
2-10, H
2-10 to H
2-11, H
2-11 to H-12, and H-12 to H
3-15 revealed the presence of two spin systems, and together with the HMBC correlations (
Figure 2) from H
3-13 to C-1, C-2, C-8, and C-12; H
3-14 to C-2, C-3, and C-4; and H-9 to C-5 and C-8, enabling the construction of an adecalin scaffold. In addition, the HMBC correlations from H-4 to C-17 and H
3-18 to C-17 indicated the existence of an acetoxy group on C-4. The presence of a carbomethoxy on C-7 was supported by the HMBC correlations from H-6 to C-7 and H
3-16 to C-7. The established planar structure of
3 has the same decalin scaffold as other nardosinane-type sesquiterpenoids, but differs from previously reported nardosinanes by the presence of the 2-ethoxy-2-methyl-2-oxoethyl group.
The relative configurations of
3 were partially established by the NOESY spectrum (
Figure 2). The NOESY correlations from H-2 to H
3-14 indicated the
Z-configuration of the Δ
2,3 double bond. The correlation from H
3-13 to H
3-15 in the NOESY spectrum indicated that Me-13 and Me-15 are cofacial. And the NOESY correlations from H-5 to H-4, H-9, and H-9 to H-10a, and H-12 to H-10a, showed that H-4, H-5, H-9, and H-12 are on the same side. Only the relative configuration of C-8 remained undetermined by the NOESY correlations. Since
3 could be obtained as a suitable crystal, the absolute configurations at C-1, C-4, C-5, C-8, C-9, and C-12 in
3 were determined as 1
S, 4
R, 5
S, 8
S, 9
R, and 12
S by a single-crystal X-ray diffraction experiment.
Figure 3 shows the Ortep diagram of
3 with a Flack parameter of 0.04 (7) (
Table S6).
Compound
4 was obtained as colourless crystals and is dextrorotatory, [
α]
25D +181.9 (
c 1.0, MeOH). The molecular formula of
3 was established as C
15H
20O
3 by HRESIMS
m/z 249.1485 [M + H]
+ (calcd for C
15H
21O
3, 249.1485), requiring six degrees of unsaturation. The
13C NMR spectrum (
Table 3) displayed the presence of fifteen carbon signals which, in combination with the HSQC spectrum, can be classified as two ketone carbonyl (
δC 217.8 and 211.0), one protonated sp
2 (
δC 141.0), one non-protonated sp
2 (
δC 137.6), one oxyquaternary sp
3 (
δC 93.6), two quaternary sp
3 (
δC 57.6 and 51.6), one methine sp
3 (
δC 39.4), four methylene sp
3 (
δC 43.9, 42.9, 33.7, and 30.3) and three methyl (
δC 16.2, 12.1, and 11.0) carbons. The
1H NMR spectrum (
Table 3), in conjunction with the HSQC spectrum, displayed one olefinic proton at
δH 5.73 (d,
J = 1.7 Hz)/
δC 141.0, one methine at
δH 2.28, m/
δC 39.4, four pairs of methylene protons at
δH 2.73 (d,
J = 16.1 Hz) and 2.10, m/
δC 42.9,
δH 2.36, m and 2.22, m/
δC 33.7,
δH 2.22, m and 2.13, m/
δC 43.9 and
δH 2.00, m and 1.71, m/
δC 30.3, two methyl doublets at
δH 1.61 (d,
J = 1.6 Hz)/
δC 12.1 and
δH 0.95 (d,
J = 6.8 Hz)/
δC 16.2, one methyl singlet at
δH 1.04/
δC 11.0, and a singlet of the hydroxyl proton
δH 3.20. The
1H NMR signal at
δH 5.73 (d, d,
J = 1.7 Hz), along with the carbon signals at
δC 141.0, 137.6, 217.8, and 211.0 indicated the presence of one trisubstituted double bond and two ketone groups, respectively. Thus, the remaining three degrees of unsaturation indicated the existence of a tricyclic system in
4.
Firstly, the
1H–
1H COSY correlations (
Figure 2) from H
2-11 to H-12 and from H-12 to H
3-15 and the HMBC correlations (
Figure 2) from H
3-13 to C-1, C-2, C-8 and C-12; H
3-14 to C-2, C-3, and C-4; and H-9 to C-1, C-8, C-10, and C-11 constructed a 6/5 ring system. Furthermore, the
1H−
1H COSY correlations from H-6 to H-7, in combination with the HMBC correlations from H-6 to C-5 and C-8 and 4-OH to C-4, C-5, and C-8 indicated the presence of a cyclopentanone ring fused with the cyclohexene ring. Consequently, the planar structure of
4 was elucidated as a 6/5/5 ring system.
The NOESY spectrum (
Figure 2) showed correlations from H
3-13 to H
3-15, suggesting that H
3-13 and H
3-15 are on the same face of the molecule. Moreover, the NOESY correlation between H
3-13 and H
2-7 revealed that H
3-13 and H
2-7 are on the same face. The absolute configurations of C-1, C-4, C-8, and C-12 in
4 were established as 1
S, 4
R, 8
S, and 12
S which was confirmed by a single-crystal X-ray diffraction experiment (
Figure 3) with the Flack parameter of −0.1(2) (
Table S7).
Based on the structural features of
1–
4, we postulated that these compounds were originated from the same precursor
5 (
Scheme 1). First, the methylation of
5 led to the formation of intermediate
1a. Then,
1a was converted to the intermediate
1c by the reduction of the C-5 carbonyl of
1a to the 5-hydroxyl group in
1b, followed by dehydration of
1b to give a double bond in
1c. The hydroxylation of Me-16 of
1c led to the formation of a primary alcohol in
1d which, after oxidation, gave an aldehyde functionality at C-6 in
1e. Finally, hydroxylation of the C2/C3 double bond in
1e led to the formation of
1 and
2. This was due to the fact that C-3 could form a more stable carbocation (tertiary) than C-2, and nucleophilic addition would occur via the SN1 mechanism, resulting in the formation of two isomers with opposite configurations. Compound
3 originated from the intermediate
3a, a hydroxylation product of
5 at C-7. Compound
3a was oxidized at C-7 to the form
3b, which was converted to
3c by the epoxidation of C-8/C-9. The dehydrogenation of
3c at C-4 was cyclized at C-7/C-4 to form
3d. Then,
3d was converted to the lactone product
3e via Baeyer–Villiger oxidation. Compound
3e was converted to
3f by hydrolysis, followed by the dehydration and reduction of
3f to form the intermediate
3g. Finally, the methylation of
3g led to the formation of
3, while in the proposed biosynthetic pathway of compound
4, the precursor
4a was the hydroxylation product of
5 at C-10. Then,
4a was converted to the intermediate
4b by the oxidation of C-10. The dehydrogenation of
4b at C-4 induced subsequent cyclization of C-4/C-8 which led to the formation of the intermediate
4c, which was converted to the form
4 by the deacetylation of C-4.
Compounds
1–
4 were first screened for cytotoxic and anti-inflammatory properties. However, none of the compounds showed significant anti-inflammatory activity in CuSO
4-induced transgenic fluorescent zebrafish, or cytotoxic activity. Furthermore,
1–
4 were evaluated for anti-AD activity using the transgenic
Caenorhabditis elegans model. Memantine was used as a positive control due to its ability to reduce amyloid
β (A
β) peptide levels in human neuroblastoma cells, as well as to inhibit A
β oligomer-induced synaptic loss [
21], which effectively alleviates AD-like symptoms in worms [
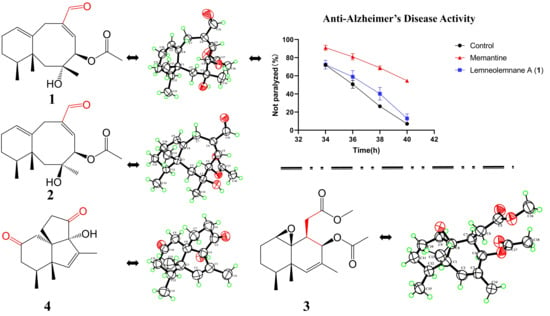
22]. Surprisingly, at a concentration of 100 μM/L,
1 significantly alleviated AD-like symptoms in the worm model (
p < 0.05) (
Figure 4). This result suggests that
1 has the potential to be a candidate for the development of an anti-AD agent based on its observed biological activity.
3. Materials and Methods
3.1. General Experimental Procedures
Melting points were measured on a microscopic melting point apparatus (Shanghai Zhuoguang Technology Company Limited, Shanghai, China). Optical rotations were measured on a Jasco P-1020 digital polarimeter (Jasco, Tokyo, Japan). The UV spectra were recorded on a Beckman DU640 spectrophotometer (Beckman Ltd., Shanghai, China). CD spectra were obtained on a Jasco J-810 spectropolarimeter (Jasco, Tokyo, Japan). IR (KBr) spectra were taken on a Nicolet NEXUS 470 spectrophotometer in KBr discs (Thermo Scientific, Beijing, China). NMR spectra were recorded using the Agilent 500 MHz (Agilent, Beijing, China), (1H, 500 MHz; 13C, 125 MHz) or the Bruker AVANCE NEO 400 (1H, 400 MHz; 13C, 100 MHz), (Bruker, Faellanden, Switzerland). The 7.26 ppm and 77.16 ppm resonances of CDCl3 were used as internal references for the 1H and 13C NMR spectra, respectively. HRESIMS spectra were measured on Micromass Q-Tof Ultima GLOBAL GAA076LC mass spectrometers (Autospec-Ultima-TOF, Waters, Shanghai, China). The crystallographic data were obtained on a Bruker APEX-II CCD diffractometer (Bruker, Beijing, China) equipped with graphite-monochromatized Cu Kα radiation. Semi-preparative HPLC was performed using a Waters 1525 pump (Waters, Singapore) equipped with a 2998 photodiode array detector and a SilGreen C18 column (SilGreen, 10 × 250 mm, 5 μm). Silica gel (200–300 mesh and 300–400 mesh), (Qingdao Marine Chemical Factory, Qingdao, China) was used for column chromatography. Chiral HPLC analysis and resolution were conducted on a chiral analytical column (Daicel, Shanghai, China).
3.2. Animal Material
The soft coral Lemnalia sp. was collected from Xisha Island (Yagong Island) of the South China Sea in 2014, and was frozen immediately after collection. The specimen was identified by Prof Ping-Jyun Sung, Institute of Marine Biotechnology, National Museum of Marine Biology & Aquarium, Pingtung 944, Taiwan. The voucher specimen (No. xs-yg-12) was deposited at the State Key Laboratory of Marine Drugs, Ocean University of China, People’s Republic of China.
3.3. Extraction and Isolation
A frozen specimen of Lemnalia sp. (7.20 kg, wet weight) was thawed, homogenized, and then exhaustively extracted with CH3OH six times (3 days each time) at room temperature. The combined solutions were concentrated in vacuo and were subsequently desalted by redissolving in CH3OH to yield a residue (175.18 g). The crude extract was subjected to silica gel vacuum column chromatography and eluted with a gradient of petroleum ether/acetone (100:1 to 1:1, v/v) and subsequently eluted with a gradient of CH2Cl2/MeOH (10:1 to 1:1, v/v) to obtain sixteen fractions (Frs.1–16). Each fraction was detected by TLC. Fr.2 was subjected to silica gel vacuum column chromatography and eluted with petroleum ether/acetone, from 100:1 to 1:1, v/v, to give three subfractions Sfrs.2.1–2.3. Sfr.2.2 was purified by reversed-phase HPLC (OD-H, MeOH/H2O = 80/20, flow rate = 1.5 mL/min, I = 210 nm) to afford 5 (28.8 mg, tR = 36 min). Fr.4 was subjected to silica gel vacuum column chromatography and eluted with petroleum ether/acetone, from 80:1 to 1:1, v/v, to give seven subfractions Sfrs.4.1–4.7. Sfr.4.5 was purified by reversed-phase HPLC (OD-H, MeOH/H2O = 70/30, flow rate = 1.5 mL/min, I = 210 nm) to afford mixtures of 3 and 6 (24.6 mg, tR = 42 min). The mixture of 3 was purified with a chiral column (Daicel Chiral pack IC, n-hexane/isopropanol = 60:40, flow rate = 1.0 mL/min, I = 210 nm) to give 3 (11.9 mg, tR = 36 min). Fr.7 was subjected to silica gel vacuum column chromatography and eluted with petroleum ether/acetone, from 50:1 to 1:1, v/v, to give five subfractions Sfrs.7.1–7.5. Sfr.7.2 was purified by reversed-phase HPLC (OD-H, MeCN/H2O = 50/50, flow rate = 1.5 mL/min, I = 320 nm) to afford 1 (7.1 mg, tR = 72 min). Fr.9 was subjected to silica gel vacuum column chromatography and eluted with petroleum ether/acetone, from 30:1 to 1:1, v/v, to give four subfractions Sfrs.9.1–9.4. Sfr.9.4 was purified by reversed-phase HPLC (OD-H, MeOH/H2O = 60/40, flow rate = 1.5 mL/min, I = 210 nm) to afford 7 (19.7 mg, tR = 45 min). Fr.11 was subjected to silica gel vacuum column chromatography and eluted with petroleum ether/acetone, from 20:1 to 1:1, v/v, to give six subfractions Sfrs.11.1–11.6. Sfr.11.1 was purified by reversed-phase HPLC (OD-H, MeCN/H2O = 30/70, flow rate = 1.5 mL/min, I = 210 nm) to afford 4 (20.7 mg, tR = 57 min). Sfr.11.2 was purified by reversed-phase HPLC (OD-H, MeCN/H2O = 50/50, flow rate = 1.5 mL/min, I = 320 nm) to afford 2 (7.3 mg, tR = 45 min).
Lemneolemnane A (
1): colourless crystals, mp 157.9–159.5 °C; [
α]
25D +57.8 (
c 1.0, MeOH); CD (
c 1.0, MeOH) = Δ
ε197 + 51.50, Δ
ε226–50.33, Δ
ε260 + 17.29; UV (MeOH)
λmax (log
ε) = 196 (1.45) nm,
λmax (log
ε) = 213(0.91) nm,
λmax (log
ε) = 230(1.42) nm; IR (KBr)
νmax = 3415, 2808, 1728, 1600 cm
−1; HRESIMS
m/z 307.1902 [M + H]
+ (calcd for C
18H
27O
4, 307.1904). For
1H NMR and
13C NMR data, see
Table 1.
Lemneolemnane B (
2): colourless crystals, mp 186.5–188.3 °C; [
α]
25D +1.6 (
c 1.0, MeOH); CD (
c 0.5, MeOH) = Δ
ε199 + 70.30, Δ
ε225–83.68, Δ
ε255 + 17.48; UV (MeOH)
λmax (log
ε) = 196 (1.62) nm,
λmax (log
ε) = 212(1.16) nm,
λmax (log
ε) = 226(1.66) nm; IR (KBr)
νmax = 3431, 2831, 1726, 1599 cm
−1; HRESIMS
m/z 307.1898 [M + H]
+ (calcd for C
18H
27O
4, 307.1904). For
1H NMR and
13C NMR data, see
Table 1.
Lemneolemnane C (
3): colourless crystals, mp 135.6–137.9 °C; [
α]
25D −10.8 (
c 1.0, MeOH); CD (
c 0.5, MeOH) = Δ
ε201 + 18.60; UV (MeOH) λ
max (log
ε) = 195 (1.99) nm,
λmax (log
ε) = 193(0.96) nm; IR (KBr)
νmax = 1641 cm
−1; HRESIMS
m/z 323.1854 [M + H]
+ (calcd for C
18H
27O
5, 323.1853) and 345.1670 [M + Na]
+ (calcd. for C
18H
26O
5Na, 345.1672). For
1H NMR and
13C NMR data, see
Table 2.
Lemneolemnane D (
4): colourless crystals, mp 176.3–177.8 °C; [
α]
25D +181.9 (
c 1.0, MeOH); CD (
c 0.5, MeOH) = Δ
ε198 + 43.31, Δ
ε231–35.02, Δ
ε309 + 72.61; UV (MeOH) λ
max (log
ε) = 193 (1.15) nm, λ
max (log
ε) = 208 (0.78) nm,
λmax (log
ε) = 218 (0.80) nm; IR (KBr)
νmax = 3415, 1716 cm
−1; HRESIMS
m/z 249.1485 [M + H]
+ (calcd for C
15H
21O
3, 249.1485) and 266.1751 [M + NH
4]
+ (calcd. for C
15H
24O
3 N, 266.1751). For
1H NMR and
13C NMR data, see
Table 3.
3.4. Anti-Alzheimer’s Disease Activity
The transgenic Caenorhabditis elegans strain CL4176 with genotype smg-1ts (myo-3/Aβ1-42 long 3′-untranslated region (UTR)) was obtained from the Caenorhabditis Genetics Center (CGC) (University of Minnesota, Minneapolis, MN, USA). Worms were routinely cultured on nematode growth medium (NGM) plates for 72 h to grow to the L3 stage at 16 °C, using Escherichia coli OP50 as the standard food resource. Throughout the bioactivity assays, 60~80 worm eggs were placed on NGM containing 100 μM/L of the test compounds, 100 μM/L memantine as a positive control, and 0.1% dimethyl sulfoxide solvent as a negative control. When the worms grew into L3 larvae, they were then transferred into another incubator which was set at 25 °C for another 32 h. Paralysed worms were observed and counted under a dissecting microscope every 2 h until all worms were paralysed. The anti-AD activity of the tested compound was indicated by the percentage of individual paralyzed worms in the tested worm population throughout the whole experiment in contrast with the negative control group. For the anti-AD activity assay, a log-rank survival test was used to compare the significance among treatments. p values at 0.05 or lower were considered statistically significant.
3.5. X-ray Crystallographic Analysis
X-ray crystallographic data were collected on a Bruker APEX-II CCD diffractometer. The crystal was kept at 150.0 K or 293.0 K during data collection. Using Olex2, the structures were solved with the SHELXT structure solution program using intrinsic phasing and refined with the SHELXL refinement package using least squares minimization.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}