
Chemical Investigation of the Calcareous Marine Sponge Pericharax heteroraphis, Clathridine-A Related Derivatives Isolation, Synthesis and Osteogenic Activity

 , ,
, ,  , ,
, ,  , , and
, , and
Abstract
:1. Introduction
2. Results and Discussion
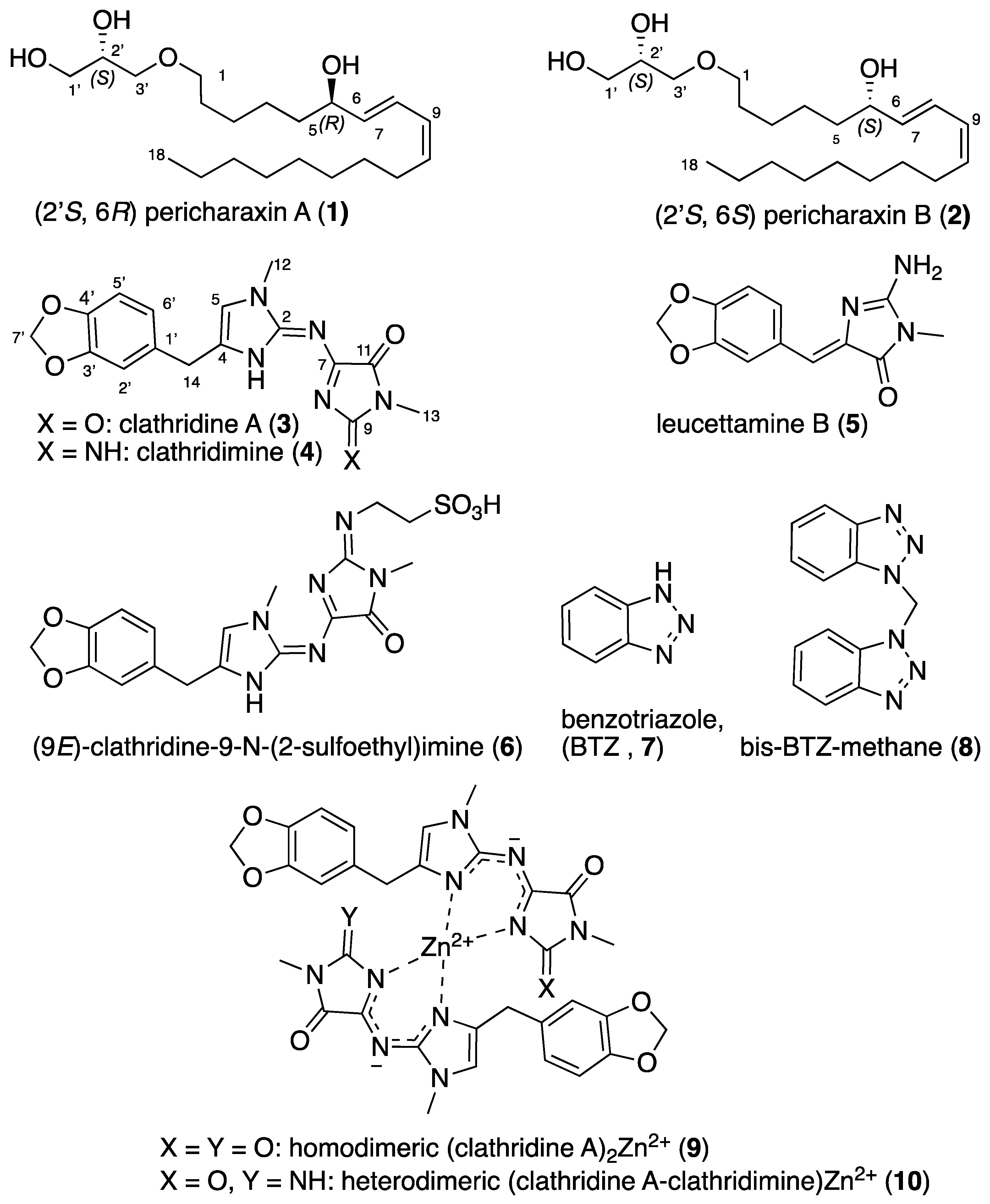
2.1. Isolation and Struture Elucidation
2.2. Synthesis of Clathridine A (3), Clathridimine (4) and Leucettamine B (5)
2.3. Complexation of Clathridine A (3) and Clathridimine (4)
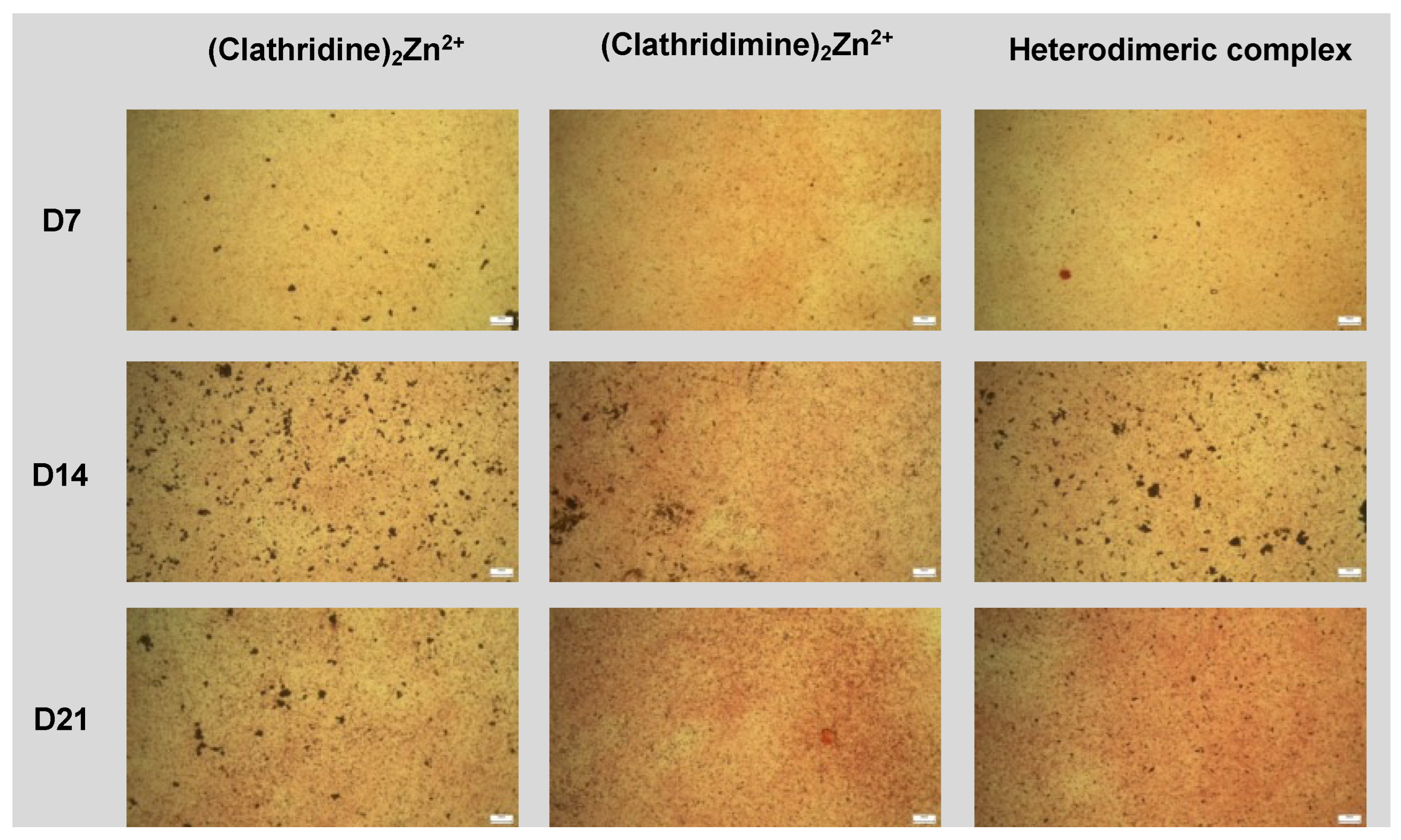
2.4. Biological Screening
3. Conclusions
4. Materials and Methods
4.1. General Procedures
4.2. Animal Material
4.3. HPLC-QTOF-ESI-MS/MS Analysis
4.4. Network Metabolomic Analyses
4.5. Extraction and Isolation of Compounds 3, 5, 6 and 9
4.6. Total Synthesis of Clathridine A (3) and Clathridimine (4)
4.7. Complexation of Clathridine A (3) and Clathridimine (4)
4.8. Total Synthesis of Leucettamine B (5)
4.9. Biological Screening
4.10. ATDC5 Micromass Model
4.11. DFT Calculations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- About Osteoporosis | International Osteoporosis Foundation. Available online: https://www.osteoporosis.foundation/patients/about-osteoporosis (accessed on 5 July 2023).
- Epidemiology | International Osteoporosis Foundation. Available online: https://www.osteoporosis.foundation/health-professionals/fragility-fractures/epidemiology (accessed on 20 January 2022).
- Raisz, L.G. Pathogenesis of Osteoporosis: Concepts, Conflicts, and Prospects. J. Clin. Investig. 2005, 115, 3318–3325. [Google Scholar] [CrossRef]
- Sigwart, J.D.; Blasiak, R.; Jaspars, M.; Jouffray, J.-B.; Tasdemir, D. Unlocking the Potential of Marine Biodiscovery. Nat. Prod. Rep. 2021, 38, 1235–1242. [Google Scholar] [CrossRef]
- Voigt, O.; Adamska, M.; Adamski, M.; Kittelmann, A.; Wencker, L.; Wörheide, G. Spicule Formation in Calcareous Sponges: Coordinated Expression of Biomineralization Genes and Spicule-Type Specific Genes. Sci. Rep. 2017, 7, 45658. [Google Scholar] [CrossRef]
- Voigt, O.; Fradusco, B.; Gut, C.; Kevrekidis, C.; Vargas, S.; Wörheide, G. Carbonic Anhydrases: An Ancient Tool in Calcareous Sponge Biomineralization. Front. Genet. 2021, 12, 624533. [Google Scholar] [CrossRef]
- Murshed, M. Mechanism of Bone Mineralization. Cold Spring Harb. Perspect Med. 2018, 8, a031229. [Google Scholar] [CrossRef]
- Wang, X.; Schröder, H.C.; Müller, W.E.G. Biocalcite, a Multifunctional Inorganic Polymer: Building Block for Calcareous Sponge Spicules and Bioseed for the Synthesis of Calcium Phosphate-Based Bone. Beilstein J. Nanotechnol. 2014, 5, 610–621. [Google Scholar] [CrossRef]
- Zhang, G.; Brion, A.; Willemin, A.-S.; Piet, M.-H.; Moby, V.; Bianchi, A.; Mainard, D.; Galois, L.; Gillet, P.; Rousseau, M. Nacre, a Natural, Multi-Use, and Timely Biomaterial for Bone Graft Substitution. J. Biomed. Mater. Res. Part A 2017, 105, 662–671. [Google Scholar] [CrossRef]
- Koswatta, P.B.; Lovely, C.J. Structure and Synthesis of 2-Aminoimidazole Alkaloids from Leucetta and Clathrina Sponges. Nat. Prod. Rep. 2011, 28, 511–528. [Google Scholar] [CrossRef]
- Roué, M.; Quévrain, E.; Domart-Coulon, I.; Bourguet-Kondracki, M.-L. Assessing Calcareous Sponges and Their Associated Bacteria for the Discovery of New Bioactive Natural Products. Nat. Prod. Rep. 2012, 29, 739–751. [Google Scholar] [CrossRef]
- Jourdain de Muizon, C.; Moriou, C.; Petek, S.; Ekins, M.; Rousseau, M.; Al Mourabit, A. Isolation, Synthesis and Absolute Configuration of the Pericharaxins A and B, Epimeric Hydroxy-Polyene Glycerol Ethers from the Calcarean Sponge Pericharax Heteroraphis. Mar. Drugs 2022, 20, 635. [Google Scholar] [CrossRef]
- Gong, K.-K.; Tang, X.-L.; Liu, Y.-S.; Li, P.-L.; Li, G.-Q. Imidazole Alkaloids from the South China Sea Sponge Pericharax Heteroraphis and Their Cytotoxic and Antiviral Activities. Molecules 2016, 21, 150. [Google Scholar] [CrossRef]
- Ali, A.A.; Hassanean, H.A.; Elkhayat, E.S.; Edrada, R.A.; Ebel, R.; Proksch, P. Imidazole Alkaloids from the Indopacific Sponge Pericharax Heteroraphis. Bull. Pharm. Sci. 2007, 30, 149–158. [Google Scholar] [CrossRef]
- Aghajanian, P.; Mohan, S. The Art of Building Bone: Emerging Role of Chondrocyte-to-Osteoblast Transdifferentiation in Endochondral Ossification. Bone Res. 2018, 6, 19. [Google Scholar] [CrossRef]
- Newton, P.T.; Staines, K.A.; Spevak, L.; Boskey, A.L.; Teixeira, C.C.; Macrae, V.E.; Canfield, A.E.; Farquharson, C. Chondrogenic ATDC5 Cells: An Optimised Model for Rapid and Physiological Matrix Mineralisation. Int. J. Mol. Med. 2012, 30, 1187–1193. [Google Scholar] [CrossRef]
- Shen, G. The Role of Type X Collagen in Facilitating and Regulating Endochondral Ossification of Articular Cartilage. Orthod. Craniofac. Res. 2005, 8, 11–17. [Google Scholar] [CrossRef]
- Tare, R.S.; Howard, D.; Pound, J.C.; Roach, H.I.; Oreffo, R.O.C. Tissue Engineering Strategies for Cartilage Generation—Micromass and Three Dimensional Cultures Using Human Chondrocytes and a Continuous Cell Line. Biochem. Biophys. Res. Commun. 2005, 333, 609–621. [Google Scholar] [CrossRef]
- Petek, S. WALLIS 2018 Cruise, Alis R/V. Fr. Oceanogr. Cruises 2018. [Google Scholar] [CrossRef]
- Olivon, F.; Elie, N.; Grelier, G.; Roussi, F.; Litaudon, M.; Touboul, D. MetGem Software for the Generation of Molecular Networks Based on the T-SNE Algorithm. Anal. Chem. 2018, 90, 13900–13908. [Google Scholar] [CrossRef]
- Elie, N.; Santerre, C.; Touboul, D. Generation of a Molecular Network from Electron Ionization Mass Spectrometry Data by Combining MZmine2 and MetGem Software. Anal. Chem. 2019, 91, 11489–11492. [Google Scholar] [CrossRef]
- Ciminiello, P.; Fattorusso, E.; Magno, S.; Mangoni, A. Clathridine and Its Zinc Complex, Novel Metabolites from the Marine Sponge Clathrina Clathrus. Tetrahedron 1989, 45, 3873–3878. [Google Scholar] [CrossRef]
- Roué, M.; Domart-Coulon, I.; Ereskovsky, A.; Djediat, C.; Perez, T.; Bourguet-Kondracki, M.-L. Cellular Localization of Clathridimine, an Antimicrobial 2-Aminoimidazole Alkaloid Produced by the Mediterranean Calcareous Sponge Clathrina Clathrus. J. Nat. Prod. 2010, 73, 1277–1282. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.W.; Mong, S.; Hemling, M.E.; Freyer, A.J.; Offen, P.H.; DeBrosse, C.W.; Sarau, H.M.; Westley, J.W. New Leukotriene B 4 Receptor Antagonist: Leucettamine A and Related Imidazole Alkaloids from the Marine Sponge Leucetta Microraphis. J. Nat. Prod. 1993, 56, 116–121. [Google Scholar] [CrossRef] [PubMed]
- He, H.Y.; Faulkner, D.J.; Lee, A.Y.; Clardy, J. A New Imidazole Alkaloid from the Marine Sponge Leucetta Microrhaphis. J. Org. Chem. 1992, 57, 2176–2178. [Google Scholar] [CrossRef]
- Finšgar, M.; Milošev, I. Inhibition of Copper Corrosion by 1,2,3-Benzotriazole: A Review. Corros. Sci. 2010, 52, 2737–2749. [Google Scholar] [CrossRef]
- Alieva G., K.; Kadirova, S.A.; Nuralieva, G.A.; Ashurov, J.M. Synthesis and Study of Complex Compounds with Benzotriazole Products of 3D-Metals. Int. J. Mater. Chem. 2021, 11, 1–9. [Google Scholar] [CrossRef]
- Koswatta, P.B.; Kasiri, S.; Das, J.K.; Bhan, A.; Lima, H.M.; Garcia-Barboza, B.; Khatibi, N.N.; Yousufuddin, M.; Mandal, S.S.; Lovely, C.J. Total Synthesis and Cytotoxicity of Leucetta Alkaloids. Bioorganic Med. Chem. 2017, 25, 1608–1621. [Google Scholar] [CrossRef]
- Cossfo, F.P.; Aizpurua, J.M.; Palomo, C. Synthetic Applications of Chromium(VI) Reagents in Combination with Chlorotrimethylsilane. Can. J. Chem. 1986, 64, 225–231. [Google Scholar] [CrossRef]
- Yamamoto, H.; Pfleiderer, W. 2-Methylamino-4,5-Imidazoledione. A Revised Structure for Creatone, Methylparabanic Acid Imide, and (3-Methylguanidino)Glyoxylic Acid. BCSJ 1982, 55, 1912–1914. [Google Scholar] [CrossRef]
- Yamamoto, H.; Ohira, C.; Aso, T.; Pfleiderer, W. 2-Amino-1-Methyl-1 H -Imidazole-4,5-Dione: Synthesis and the Dimroth Type Rearrangement to Creatone (2-Methyl-Amino-1 H -Imidazole-4,5-Dione). BCSJ 1987, 60, 4115–4120. [Google Scholar] [CrossRef]
- Alvi, K.A.; Peters, B.M.; Hunter, L.M.; Crews’, P. 2-Aminoimidazoles and Their Zinc Complexes from Indo-Pacific Leucetta Sponges and Nofodoris Nudibranchs. Tetrahedron 1993, 49, 329–336. [Google Scholar] [CrossRef]
- Tian, H.; Ermolenko, L.; Gabant, M.; Vergne, C.; Moriou, C.; Retailleau, P.; Al-Mourabit, A. Pyrrole-Assisted and Easy Oxidation of Cyclic α-Amino Acid- Derived Diketopiperazines under Mild Conditions. Adv. Synth. Catal. 2011, 353, 1525–1533. [Google Scholar] [CrossRef]
- Vergne, C.; Appenzeller, J.; Ratinaud, C.; Martin, M.-T.; Debitus, C.; Zaparucha, A.; Al-Mourabit, A. Debromodispacamides B and D: Isolation from the Marine Sponge Agelas Mauritiana and Stereoselective Synthesis Using a Biomimetic Proline Route. Org. Lett. 2008, 10, 493–496. [Google Scholar] [CrossRef] [PubMed]
- Ermolenko, L.; Zhaoyu, H.; Lejeune, C.; Vergne, C.; Ratinaud, C.; Nguyen, T.B.; Al-Mourabit, A. Concise Synthesis of Didebromohamacanthin A and Demethylaplysinopsine: Addition of Ethylenediamine and Guanidine Derivatives to the Pyrrole-Amino Acid Diketopiperazines in Oxidative Conditions. Org. Lett. 2014, 16, 872–875. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.L.; Berkowitz, D.B. Formal. Alpha.-Vinylation of Amino Acids. Use of a New Benzeneselenolate Equivalent. J. Org. Chem. 1993, 58, 6966–6975. [Google Scholar] [CrossRef]
- Michael, J.P.; Pattenden, G. Marine Metabolites and Metal Ion Chelation: The Facts and the Fantasies. Angew. Chem. Int. Ed. Engl. 1993, 32, 1–23. [Google Scholar] [CrossRef]
- Schilling, M.; Levasseur, M.; Barbier, M.; Oliveira-Correia, L.; Henry, C.; Touboul, D.; Farine, S.; Bertsch, C.; Gelhaye, E. Wood Degradation by Fomitiporia Mediterranea M. Fischer: Exploring Fungal Adaptation Using Metabolomic Networking. JoF 2023, 9, 536. [Google Scholar] [CrossRef] [PubMed]
- Caudal, F.; Rodrigues, S.; Dufour, A.; Artigaud, S.; Le Blay, G.; Petek, S.; Bazire, A. Extracts from Wallis Sponges Inhibit Vibrio Harveyi Biofilm Formation. Microorganisms 2023, 11, 1762. [Google Scholar] [CrossRef] [PubMed]
- Loaëc, N.; Attanasio, E.; Villiers, B.; Durieu, E.; Tahtouh, T.; Cam, M.; Davis, R.A.; Alencar, A.; Roué, M.; Bourguet-Kondracki, M.-L.; et al. Marine-Derived 2-Aminoimidazolone Alkaloids. Leucettamine B-Related Polyandrocarpamines Inhibit Mammalian and Protozoan DYRK & CLK Kinases. Mar. Drugs 2017, 15, 316. [Google Scholar] [CrossRef]
- Ohta, S.; Tsuno, N.; Maeda, K.; Nakamura, S.; Taguchi, N.; Yamashita, M.; Kawasaki, I. Total Synthesis of a Marine Imidazole Alkaloid, Clathridine A. Tetrahedron Lett. 2000, 41, 4623–4627. [Google Scholar] [CrossRef]
- Nelea, V.; Luo, L.; Demers, C.N.; Antoniou, J.; Petit, A.; Lerouge, S.; Wertheimer, M.R.; Mwale, F. Selective Inhibition of Type X Collagen Expression in Human Mesenchymal Stem Cell Differentiation on Polymer Substrates Surface-Modified by Glow Discharge Plasma. J. Biomed. Mater. Res. Part A 2005, 75A, 216–223. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Lee, Y.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Rigaku, O.D. CrysAlis PRO. Rigaku Oxford Diffraction, Yarnton, Oxfordshire, England. 2018. Available online: https://veranova.com/solid-form-particle-engineering/structureconfirmation/?gad_source=1&gclid=EAIaIQobChMI5t3rhcTchQMVlmIPAh3DBgbMEAAYASAAEgJ9nPD_BwE (accessed on 11 April 2024).
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. J. Appl. Cryst. 2009, 42, 339–341. Available online: https://www.scirp.org/reference/referencespapers?referenceid=1770039 (accessed on 11 April 2024).
- Sheldrick, G.M. Acta Cryst. 2015, A71, 3–8. Available online: https://www.scirp.org/reference/referencespapers?referenceid=1989712 (accessed on 11 April 2024).
- Spek, A.L. Acta Cryst. 2015, C71, 9–18. Available online: https://scripts.iucr.org/cgi-bin/paper?s2053229614024929 (accessed on 11 April 2024).
- Macrae, C.F.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Shields, G.P.; Taylor, R.; Towler, M.; van de Streek, J. J. Appl. Cryst. 2006, 39, 453–457. Available online: https://scripts.iucr.org/cgi-bin/paper?s002188980600731x (accessed on 11 April 2024).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clathridine Zinc Complex (9) | Clathridimine Zinc Complex (27) | Clathridine–Clathridimine Heterodimeric Zinc Complex (10) | ||||
|---|---|---|---|---|---|---|
| Position | δC, Type | δH Mult, (J in Hz) | δC, Type | δH Mult, (J in Hz) | δC, Type | δH Mult, (J in Hz) |
| 2 | 149.0, C 1 | - | 149.5, C 1 | - | 149.0 3, 149.5 4 | - |
| 4 | 136.3, C 1 | - | 135.4, C 1 | - | 136.3 3, 135.4 4 | - |
| 5 | 117.7, CH | 6.63, s | 117.2, CH | 6.63, s | 117.7 3, 117.2 4 | 6.63, s |
| 7 | nd | - | nd | - | nd | - |
| 9 | 161.4, C 1 | - | 158.7, C 1 | - | 161.2 3, 159.0 4 | - |
| 11 | 164.9, C 1 | - | 164.0, C 1 | - | 164.9 3, 164.0 4 | - |
| 12 | 32.7, CH3 | 3.80, s | 32.7, CH3 | 3.81, s | 32.7, CH3 | 3.80, s |
| 13 | 24.7, CH3 | 3.04, s | 25.6, CH3 | 3.16, s | 24.7, CH3 | 3.14 4, 3.05 3, s 2 |
| 14a | 33.30, CH2 | 3.51, d (16.3) | 33.30, CH2 | 3.51, d (16.2) | 33.27, CH2 | 3.51, d (16.3) |
| 14b | 3.37, d (16.3) | 3.34, d (16.2) | 3.35, d (16.3) 2 | |||
| 1′ | 131.1, C 1 | - | 131.0, C 1 | - | 131.0, C 1 | - |
| 2′ | 108.6, CH | 6.25, s | 108.4, CH | 6.22, s | 108.5, CH | 6.24, s |
| 3′ | 147.7, C 1 | - | 147.8, C 1 | - | 147.7, C 1 | - |
| 4′ | 146.5, C 1 | - | 146.3, C 1 | - | 146.4, C 1 | - |
| 5′ | 107.9, CH | 6.502, d (7.9) | 107.9, CH | 6.492, d (8.3) | 107.8, CH | 6.499, d (7.9) 2 |
| 6′ | 121.7, CH | 6.26, d (7.9) | 121.2, CH | 6.23, d (8.3) | 121.2, CH | 6.24, m |
| 7′ | 101.2, CH2 | 5.88, dd (13.6, 1.1) | 101.3, CH2 | 5.90, dd (15.4, 0.9) | 101.3, CH2 | 5.89, dd (13.8, 1.3) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jourdain de Muizon, C.; Moriou, C.; Levasseur, M.; Touboul, D.; Iorga, B.I.; Nedev, H.; Van Elslande, E.; Retailleau, P.; Petek, S.; Folcher, E.; et al. Chemical Investigation of the Calcareous Marine Sponge Pericharax heteroraphis, Clathridine-A Related Derivatives Isolation, Synthesis and Osteogenic Activity. Mar. Drugs 2024, 22, 196. https://doi.org/10.3390/md22050196
Jourdain de Muizon C, Moriou C, Levasseur M, Touboul D, Iorga BI, Nedev H, Van Elslande E, Retailleau P, Petek S, Folcher E, et al. Chemical Investigation of the Calcareous Marine Sponge Pericharax heteroraphis, Clathridine-A Related Derivatives Isolation, Synthesis and Osteogenic Activity. Marine Drugs. 2024; 22(5):196. https://doi.org/10.3390/md22050196
Chicago/Turabian StyleJourdain de Muizon, Capucine, Céline Moriou, Marceau Levasseur, David Touboul, Bogdan I. Iorga, Hristo Nedev, Elsa Van Elslande, Pascal Retailleau, Sylvain Petek, Eric Folcher, and et al. 2024. "Chemical Investigation of the Calcareous Marine Sponge Pericharax heteroraphis, Clathridine-A Related Derivatives Isolation, Synthesis and Osteogenic Activity" Marine Drugs 22, no. 5: 196. https://doi.org/10.3390/md22050196
APA StyleJourdain de Muizon, C., Moriou, C., Levasseur, M., Touboul, D., Iorga, B. I., Nedev, H., Van Elslande, E., Retailleau, P., Petek, S., Folcher, E., Bianchi, A., Thomas, M., Viallon, S., Peyroche, S., Nahle, S., Rousseau, M., & Al-Mourabit, A. (2024). Chemical Investigation of the Calcareous Marine Sponge Pericharax heteroraphis, Clathridine-A Related Derivatives Isolation, Synthesis and Osteogenic Activity. Marine Drugs, 22(5), 196. https://doi.org/10.3390/md22050196