
Denigrins H–L: Sulfated Derivatives of Denigrins D and E from a New Zealand Dictyodendrilla c.f. dendyi Marine Sponge

Abstract
:1. Introduction
2. Results and Discussions
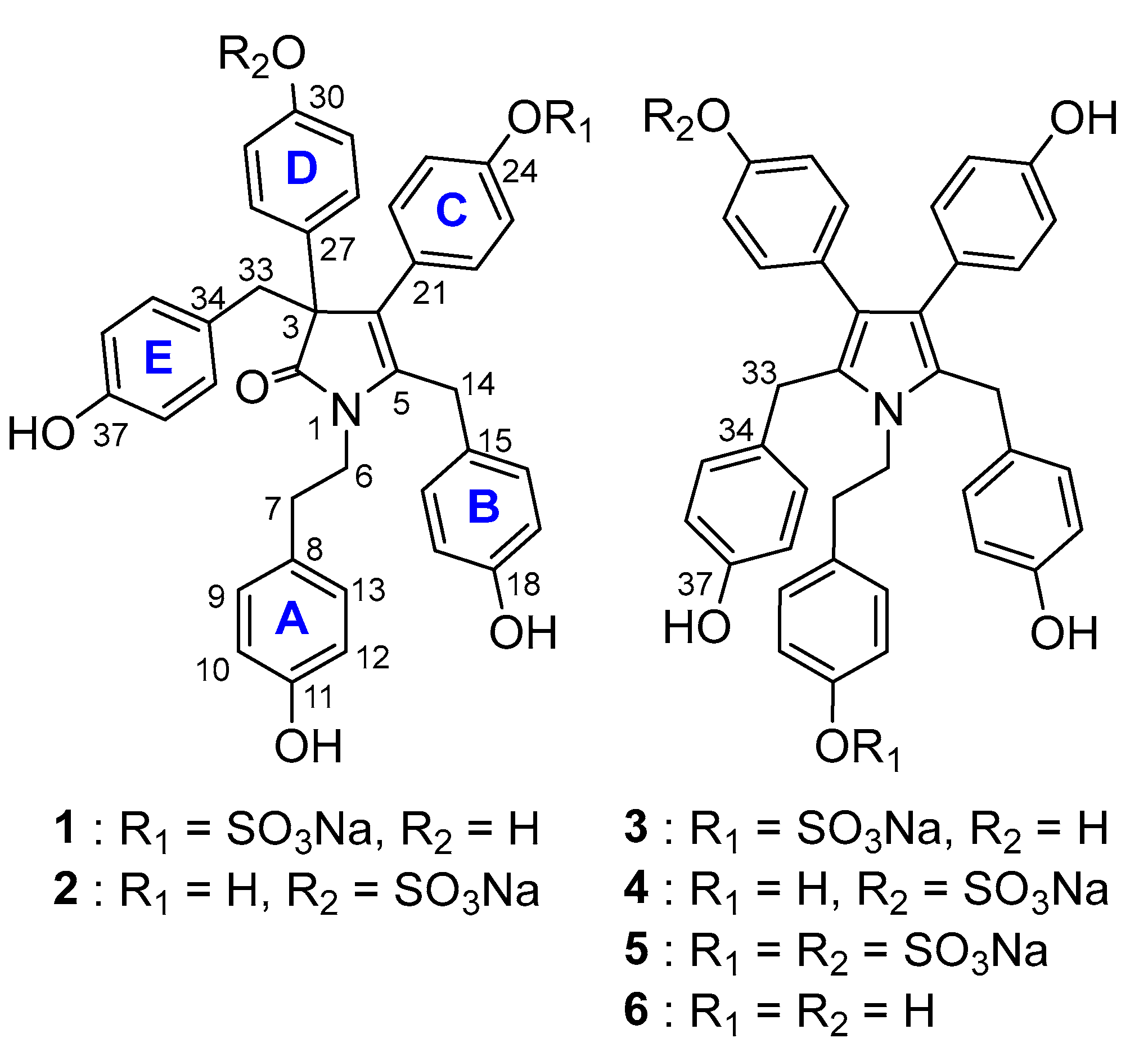
2.1. Structure Elucidation
2.2. Cytotoxicity Assay
2.3. Oxidative Rearrangement of Denigrin E Derivatives
2.4. Occurrence of the Sulfate Moiety in Sponge Metabolites
3. Materials and Methods
3.1. General Experimental Procedures
3.2. Animal Material
3.3. Cell Line and Cell Culture
3.4. MTT Cytotoxicity Assay
3.5. Extraction and Isolation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hong, L.-L.; Ding, Y.-F.; Zhang, W.; Lin, H.-W. Chemical and biological diversity of new natural products from marine sponges: A review (2009–2018). Mar. Life Sci. Technol. 2022, 4, 356–372. [Google Scholar] [CrossRef] [PubMed]
- Carroll, A.R.; Copp, B.R.; Davis, R.A.; Keyzers, R.A.; Prinsep, M.R. Marine natural products. Nat. Prod. Rep. 2023, 40, 275–325. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.M.K.; Devilal Naik, J.; Satyavathi, K.; Ramana, H.; Raghuveer Varma, P.; Purna Nagasree, K.; Smitha, D.; Venkata Rao, D. Denigrins A–C: New antitubercular 3,4-diarylpyrrole alkaloids from Dendrilla nigra. Nat. Prod. Res. 2014, 28, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Karak, M.; Oishi, T.; Torikai, K. Synthesis of anti-tubercular marine alkaloids denigrins A and B. Tetrahedron Lett. 2018, 59, 2800–2803. [Google Scholar] [CrossRef]
- Kang, U.; Cartner, L.K.; Wang, D.; Kim, C.-K.; Thomas, C.L.; Woldemichael, G.M.; Gryder, B.E.; Shern, J.F.; Khan, J.; Castello-Branco, C.; et al. Denigrins and Dactylpyrroles, Arylpyrrole Alkaloids from a Dactylia sp. Marine Sponge. J. Nat. Prod. 2020, 83, 3464–3470. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lan, P.; Banwell, M.G. Total Synthesis of Denigrin E and Its Oxidative Conversion into Co-occurring Metabolite Denigrin D. Org. Lett. 2022, 24, 2931–2934. [Google Scholar] [CrossRef] [PubMed]
- Mc Cormack, S.P.; Kelly, M.; Battershill, C.N. Description of two new species of Dysidea (Porifera, Demospongiae, Dictyoceratida, Dysideidae) from Tauranga Harbour, Bay of Plenty, New Zealand. Zootaxa 2020, 4780, 523–542. [Google Scholar] [CrossRef] [PubMed]
- Bergquist, P.R. Demospongiae. Part 5. Dendroceratida and Halisarca. In The Marine Fauna of New Zealand: Porifera; New Zealand Oceanographic Institute: Wellington, New Zealand, 1996; Volume 107, pp. 1–53. [Google Scholar]
- Gris, L.; Battershill, C.N.; Prinsep, M.R. Investigation of the Dietary Preferences of Two Dorid Nudibranchs by Feeding-Choice Experiments and Chemical Analysis. J. Chem. Ecol. 2023, 49, 599–610. [Google Scholar] [CrossRef] [PubMed]
- Warabi, K.; Matsunanga, S.; Van Soest, R.W.M.; Fusetani, N. Dictyodendrins A−E, the First Telomerase-Inhibitory Marine Natural Products from the Sponge Dictyodendrilla verongiformis. J. Org. Chem. 2003, 68, 2765–2770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Conte, M.M.; Khalil, Z.; Huang, X.-C.; Capon, R.J. New dictyodendrins as BACE inhibitors from a southern Australian marine sponge, Ianthella sp. RSC Adv. 2012, 2, 4209–4214. [Google Scholar] [CrossRef]
- Zhang, H.; Conte, M.M.; Huang, X.-C.; Khalil, Z.; Capon, R.J. A search for BACE inhibitors reveals new biosynthetically related pyrrolidones, furanones and pyrroles from a southern Australian marine sponge, Ianthella sp. Org. Biomol. Chem. 2012, 10, 2656–2663. [Google Scholar] [CrossRef] [PubMed]
- Gardini, G.P. The Oxidation of Monocyclic Pyrroles. In Advances in Heterocyclic Chemistry; Katritzky, A.R., Boulton, A.J., Eds.; Academic Press: Cambridge, MA, USA, 1973; Volume 15, pp. 67–98. [Google Scholar]
- Ji Ram, V.; Sethi, A.; Nath, M.; Pratap, R. Chapter 5—Five-Membered Heterocycles. In The Chemistry of Heterocycles; Ji Ram, V., Sethi, A., Nath, M., Pratap, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2019; pp. 149–478. [Google Scholar]
- Kotoučová, H.; Strnadová, I.; Kovandová, M.; Chudoba, J.; Dvořáková, H.; Cibulka, R. Biomimetic aerobic oxidative hydroxylation of arylboronic acids to phenols catalysed by a flavin derivative. Org. Biomol. Chem. 2014, 12, 2137–2142. [Google Scholar] [CrossRef] [PubMed]
- Göthel, Q.; Köck, M. New sesquiterpene hydroquinones from the Caribbean sponge Aka coralliphagum. Beilstein J. Org. Chem. 2014, 10, 613–621. [Google Scholar] [CrossRef] [PubMed]
- Available online: http://pubs.rsc.org/marinlit (accessed on 11 May 2024).
- Prinsep, M.R.; Blunt, J.W.; Munro, M.H.G. A New Sterol Sulfate from the Marine Sponge Stylopus australis. J. Nat. Prod. 1989, 52, 657–659. [Google Scholar] [CrossRef]
- Fan, G.; Li, Z.; Shen, S.; Zeng, Y.; Yang, Y.; Xu, M.; Bruhn, T.; Bruhn, H.; Morschhäuser, J.; Bringmann, G.; et al. Baculiferins A–O, O-sulfated pyrrole alkaloids with anti-HIV-1 activity, from the Chinese marine sponge Iotrochota baculifera. Bioorg. Med. Chem. 2010, 18, 5466–5474. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Chang, L.C. Novel Sulfated Sesterterpene Alkaloids from the Marine Sponge Fasciospongia sp. Org. Lett. 2007, 9, 3037–3040. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Kondratyuk, T.P.; Tan, G.T.; Pezzuto, J.M.; Chang, L.C. Bioactive Sulfated Sesterterpene Alkaloids and Sesterterpene Sulfates from the Marine Sponge Fasciospongia sp. J. Nat. Prod. 2009, 72, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Khalil, Z.; Conte, M.M.; Plisson, F.; Capon, R.J. A search for kinase inhibitors and antibacterial agents: Bromopyrrolo-2-aminoimidazoles from a deep-water Great Australian Bight sponge, Axinella sp. Tetrahedron Lett. 2012, 53, 3784–3787. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| No. | δH, Mult (J in Hz) | δH, Mult (J in Hz) | δH, Mult (J in Hz) | δH, Mult (J in Hz) | δH, Mult (J in Hz) |
| 6 | 3.41, m 3.11 b, m | 3.41, m 3.08 b, m | 3.59, m | 3.58, m | 3.61, m |
| 7 | 2.70, m 2.19, ddd (13.2, 8.9, 4.7) | 2.69, m 2.18, ddd (13.1, 8.7, 4.8) | 2.45, m | 2.45, m | 2.45, m |
| 9/13 | 6.712 c, d (8.5) | 6.706 c, d (8.4) | 6.78, d (8.5) | 6.66, d (9.0) | 6.80, d (8.6) |
| 10/12 | 6.65, d (8.5) | 6.65, d (8.6) | 7.13, d (8.4) | 6.63, d (8.3) | 7.14 d, d (8.6) |
| 14 | 3.77, d (17.0) 3.28 e, m | 3.79, d (16.9) 3.29 e, m | 3.85, s | 3.80, s | 3.94, s |
| 16/20 | 6.34, d (8.4) | 6.34, d (8.3) | 6.93, d (8.6) | 6.89 f, d (8.3) | 6.95 g, d (8.6) |
| 17/19 | 6.705 c, d (8.6) | 6.714 c, d (8.4) | 6.69, d (8.5) | 6.70 h, d (9.0) | 6.710 c, i, d (8.6) |
| 22/26 | 6.79, d (8.9) | 6.66, d (8.9) | 6.92, d (8.6) | 6.92, d (9.0) | 6.95 g, d (8.6) |
| 23/25 | 7.07, d (8.9) | 6.56, d (8.7) | 6.61, d (8.7) | 6.62, d (8.3) | 6.63, d (8.7) |
| 28/32 | 7.25, d (8.8) | 7.42, d (9.1) | 6.92, d (8.6) | 7.06, d (8.6) | 7.09, d (8.8) |
| 29/31 | 6.87. d (8.8) | 7.39, d (9.0) | 6.61, d (8.7) | 7.12, d (8.3) | 7.14 d, d (8.6) |
| 33 | 3.64, d (12.9) 3.09 b, d (12.7) | 3.68, d (12.9) 3.09 b, d (12.7) | 3.85, s | 3.80, s | 3.87, s |
| 35/39 | 6.72, d (8.7) | 6.68, d (8.6) | 6.93, d (8.6) | 6.91 f, d (8.3) | 6.95 g, d (8.6) |
| 36/38 | 6.64, d (8.6) | 6.63, d (8.7) | 6.69, d (8.5) | 6.69 h, d 8.3) | 6.708 c, i, d (8.6) |
| 1 | 2 | 3 | 4 | 5 | 6 | |
|---|---|---|---|---|---|---|
| No. | δC, Type | δC, Type | δC, Type | δC, Type | δC, Type | δC, Type |
| 2 | 183.6, C | 182.9, C | 128.3, C | 128.73 b, c, C | 128.6 d, C | 128.3, C |
| 3 | 62.3, C | 62.4, C | 123.4, C | 122.9, C | 123.0, C | 123.3, C |
| 4 | 124.8, C | 124.9, C | 123.4, C | 123.4, C | 123.5, C | 123.3, C |
| 5 | 139.9, C | 138.7, C | 128.3, C | 128.68 b, c, C | 128.7 d, C | 128.3, C |
| 6 | 44.4, CH2 | 44.3, CH2 | 47.1, CH2 | 47.3, CH2 | 47.2, CH2 | 47.3, CH2 |
| 7 | 34.9, CH2 | 34.9, CH2 | 37.7, CH2 | 37.6, CH2 | 37.6, CH2 | 37.7, CH2 |
| 8 | 130.8, C | 130.6, C | 136.6, C | 130.94 c, C | 136.5, C | 131.0, C |
| 9/13 | 130.9, CH | 131.0, CH | 130.6, CH | 130.92 c, CH | 130.4, CH | 130.9, CH |
| 10/12 | 116.2, CH | 116.2, CH | 122.5, CH | 116.1, CH | 122.5, CH | 116.1, CH |
| 11 | 157.1, C | 157.1, C | 152.6, C | 157.0, C | 152.5, C | 157.0, C |
| 14 | 31.0, CH2 | 31.1, CH2 | 30.7, CH2 | 30.63 c, e, CH2 | 30.7, CH2 | 30.7, CH2 |
| 15 | 128.2, C | 128.40 c, f, C | 132.8, C | 132.7 g, C | 132.7 h, C | 132.9, C |
| 16/20 | 130.1, CH | 130.1, CH | 130.0, CH | 129.92 c, i, CH | 130.00 c, j, CH | 129.9, CH |
| 17/19 | 116.9, CH | 116.8, CH | 116.3, CH | 116.32 c, k, CH | 116.41 c, l, CH | 116.3, CH |
| 18 | 157.3, C | 157.2, C | 156.7, C | 156.68 c, m, C | 156.83 c, n, C | 156.7, C |
| 21 | 131.7, C | 126.1, C | 129.7, C | 129.4, C | 129.3, C | 129.7, C |
| 22/26 | 130.0, CH | 130.3, CH | 132.5, CH | 132.5, CH | 132.6, CH | 132.5, CH |
| 23/25 | 122.1, CH | 116.0, CH | 115.6, CH | 115.7, CH | 115.8, CH | 115.6, CH |
| 24 | 152.8, C | 157.63 c, C | 156.1, C | 156.2, C | 156.3, C | 156.1, C |
| 27 | 132.0, C | 137.9, C | 129.7, C | 135.2, C | 135.1, C | 129.7, C |
| 28/32 | 129.0, CH | 128.8, CH | 132.5, CH | 132.0, CH | 132.0, CH | 132.5, CH |
| 29/31 | 116.8, CH | 122.8, CH | 115.6, CH | 121.9, CH | 121.9, CH | 115.6, CH |
| 30 | 158.1, C | 153.5, C | 156.1, C | 151.6, C | 151.7, C | 156.0, C |
| 33 | 38.5, CH2 | 38.6, CH2 | 30.7, CH2 | 30.57 c, e, CH2 | 30.7, CH2 | 30.7, CH2 |
| 34 | 128.4, C | 128.41c, f, C | 132.8, C | 132.8 g, C | 132.6 h, C | 132.9, C |
| 35/39 | 132.5, CH | 132.6, CH | 130.0, CH | 129.87 c, i, CH | 129.96 c, j, CH | 129.9, CH |
| 36/38 | 115.8, CH | 115.7, CH | 116.3, CH | 116.30 c, k, CH | 116.37 c, l, CH | 116.3, CH |
| 37 | 157.6, C | 157.58 c, C | 156.7, C | 156.70 c, m, C | 156.78 c, n, C | 156.7, C |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gris, L.; Prinsep, M.R.; Peters, L.M.; Battershill, C.N. Denigrins H–L: Sulfated Derivatives of Denigrins D and E from a New Zealand Dictyodendrilla c.f. dendyi Marine Sponge. Mar. Drugs 2024, 22, 231. https://doi.org/10.3390/md22050231
Gris L, Prinsep MR, Peters LM, Battershill CN. Denigrins H–L: Sulfated Derivatives of Denigrins D and E from a New Zealand Dictyodendrilla c.f. dendyi Marine Sponge. Marine Drugs. 2024; 22(5):231. https://doi.org/10.3390/md22050231
Chicago/Turabian StyleGris, Lauren, Michèle R. Prinsep, Linda M. Peters, and Christopher N. Battershill. 2024. "Denigrins H–L: Sulfated Derivatives of Denigrins D and E from a New Zealand Dictyodendrilla c.f. dendyi Marine Sponge" Marine Drugs 22, no. 5: 231. https://doi.org/10.3390/md22050231
APA StyleGris, L., Prinsep, M. R., Peters, L. M., & Battershill, C. N. (2024). Denigrins H–L: Sulfated Derivatives of Denigrins D and E from a New Zealand Dictyodendrilla c.f. dendyi Marine Sponge. Marine Drugs, 22(5), 231. https://doi.org/10.3390/md22050231