
Chrysomycin A Reshapes Metabolism and Increases Oxidative Stress to Hinder Glioblastoma Progression

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
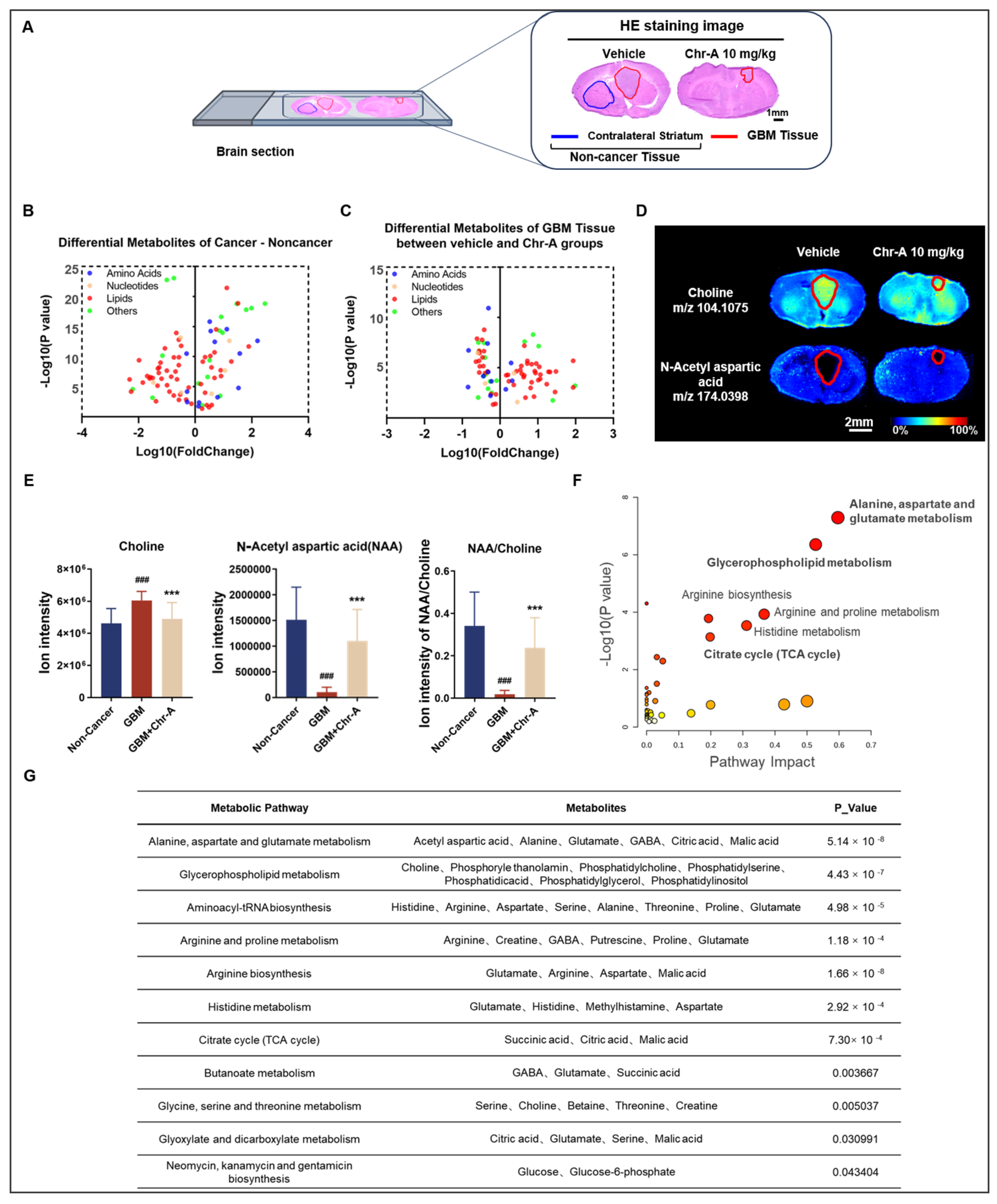
2.1. Chr-A Suppressed Orthotopic GBM Tumor Growth In Vivo
2.2. AFADESI-MSI Revealed That Chr-A Reshapes Various Metabolism Preventing GBM Progression
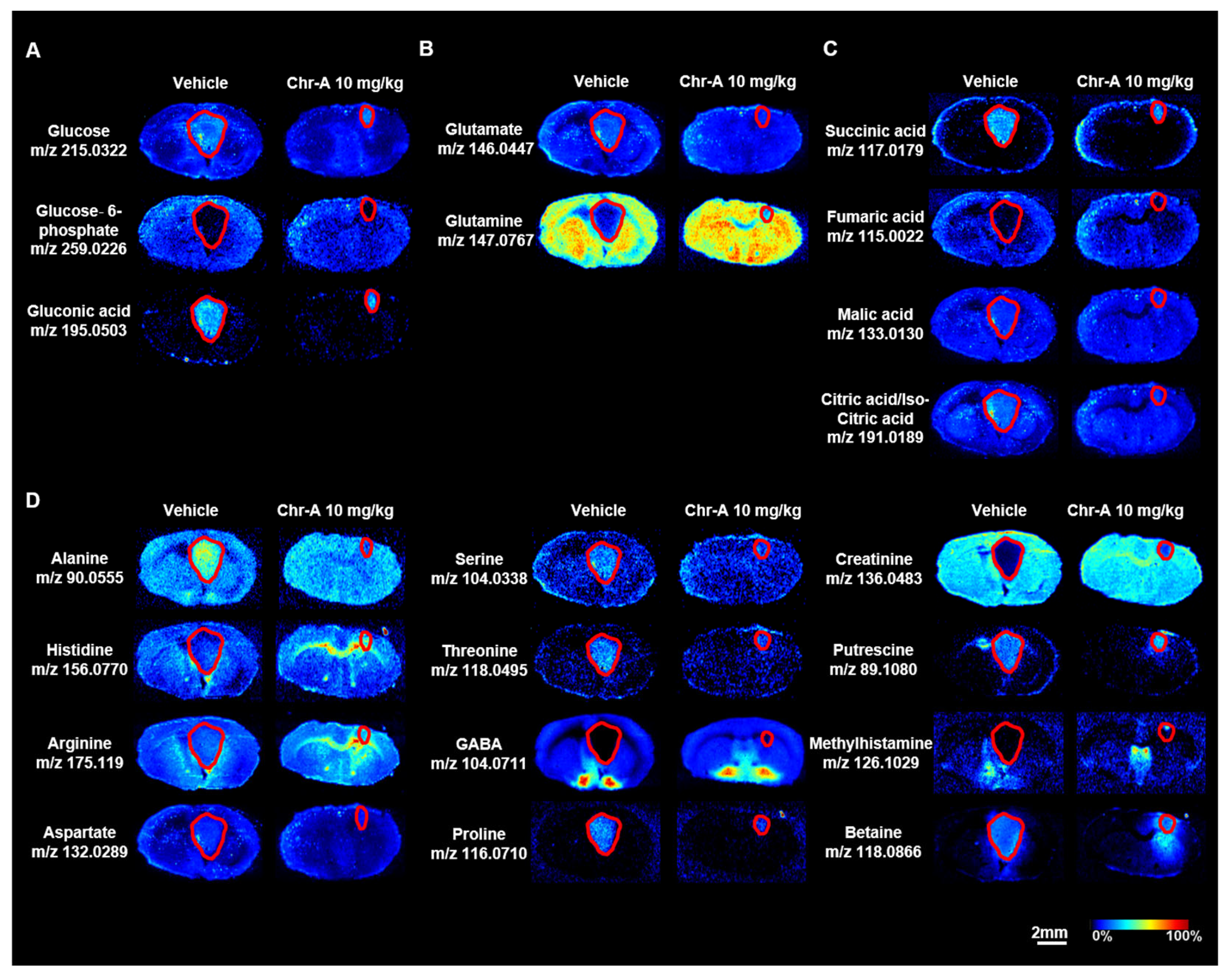
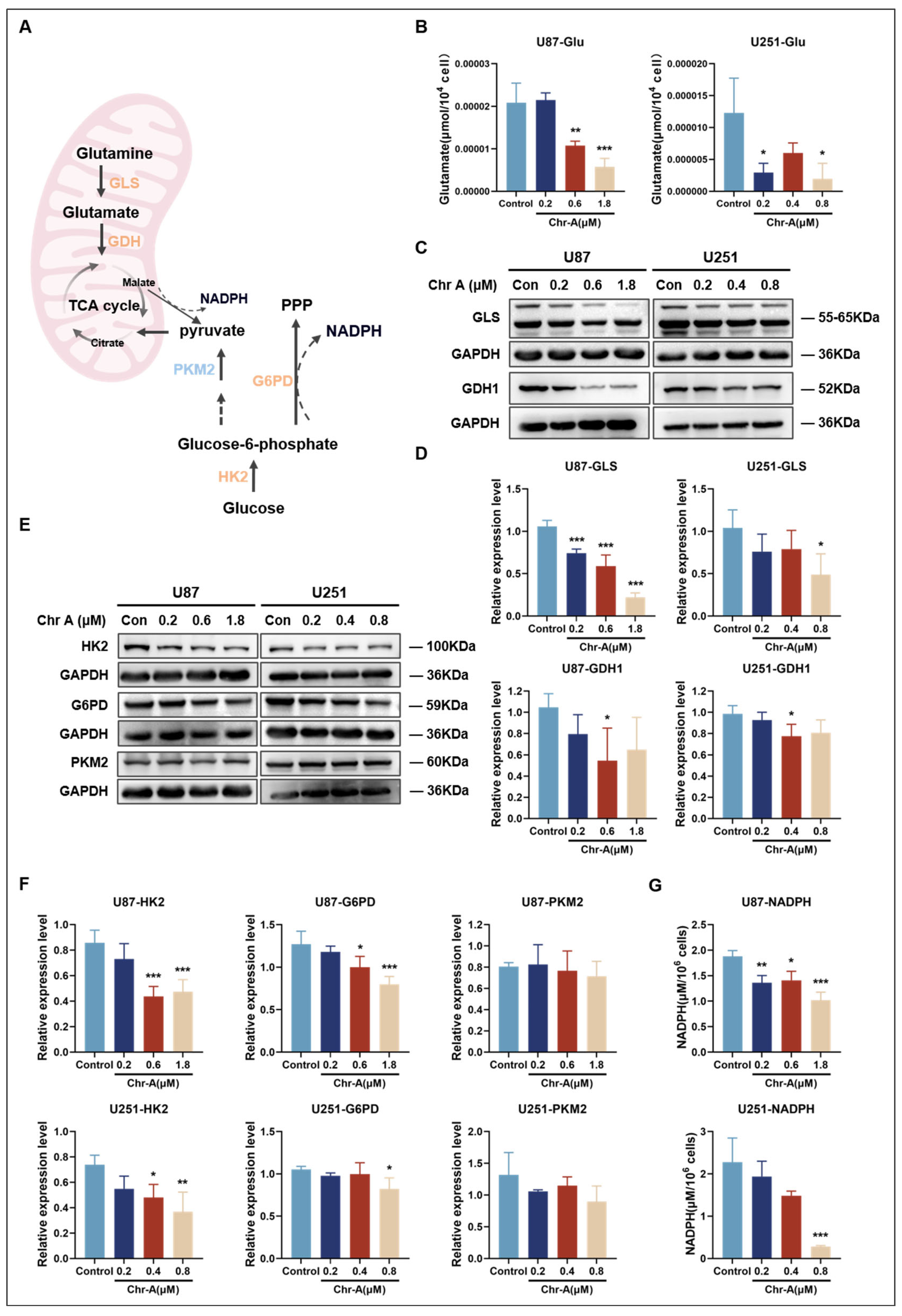
2.3. Chr-A Reduced the Abnormal Accumulation of Glucose and Glutamate and Activation of the TCA Cycle
2.4. Chr-A Corrected Phospholipid and Fatty Acid Metabolism in Glioblastoma Xenografts
2.5. Validation of Crucial Metabolites and Metabolic Enzymes in the Chr-A-Regulated Metabolic Pathways in Glioma In Vitro
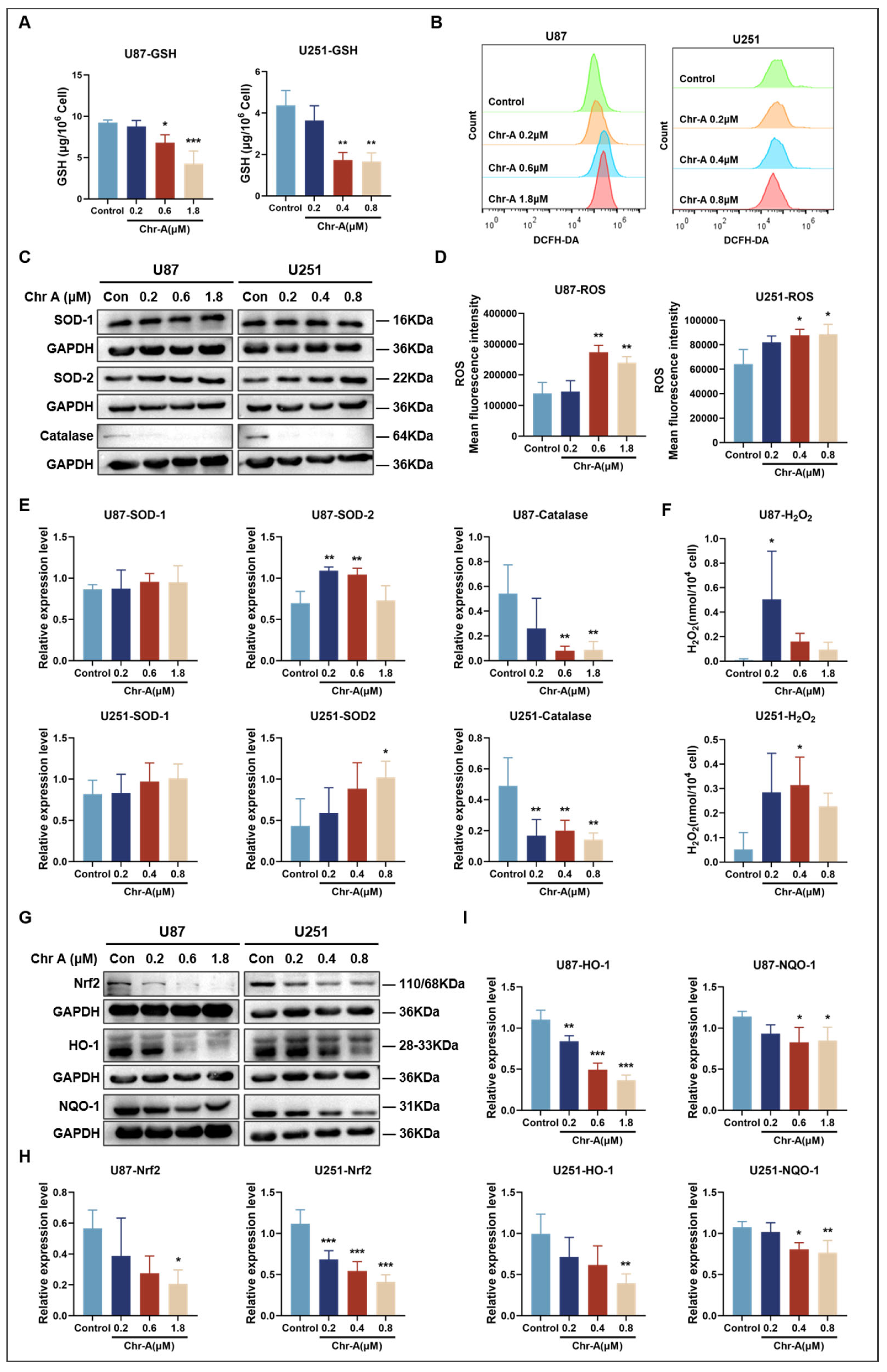
2.6. Chr-A Increased Oxidative Stress in Glioma Cells
3. Discussion
4. Materials and Methods
4.1. Chemical Reagents
4.2. U87 and U251 Cell Culture
4.3. U87 Glioma Orthotopic Model in Nude Mice and Drug Administration
4.4. MRI Analysis
4.5. AFADESI-MSI Analysis
4.6. Metabolic Pathway Enrichment Analysis
4.7. Intracellular Metabolite Measurements
4.8. ROS Flowcytometry
4.9. Western Blotting Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Schaff, L.R.; Mellinghoff, I.K. Glioblastoma and Other Primary Brain Malignancies in Adults: A Review. JAMA 2023, 329, 574–587. [Google Scholar] [CrossRef]
- Ma, R.; Taphoorn, M.J.B.; Plaha, P. Advances in the management of glioblastoma. J. Neurol. Neurosurg. Psychiatry 2021, 92, 1103–1111. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, R.; Allega, M.F.; Tardito, S. A map of the altered glioma metabolism. Trends Mol. Med. 2021, 27, 1045–1059. [Google Scholar] [CrossRef] [PubMed]
- Bernhard, C.; Reita, D.; Martin, S.; Entz-Werle, N.; Dontenwill, M. Glioblastoma Metabolism: Insights and Therapeutic Strategies. Int. J. Mol. Sci. 2023, 24, 9137. [Google Scholar] [CrossRef]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- de la Cruz-Lopez, K.G.; Castro-Munoz, L.J.; Reyes-Hernandez, D.O.; Garcia-Carranca, A.; Manzo-Merino, J. Lactate in the Regulation of Tumor Microenvironment and Therapeutic Approaches. Front. Oncol. 2019, 9, 1143. [Google Scholar] [CrossRef]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef]
- Broadfield, L.A.; Pane, A.A.; Talebi, A.; Swinnen, J.V.; Fendt, S.M. Lipid metabolism in cancer: New perspectives and emerging mechanisms. Dev. Cell 2021, 56, 1363–1393. [Google Scholar] [CrossRef]
- Yang, W.H.; Qiu, Y.; Stamatatos, O.; Janowitz, T.; Lukey, M.J. Enhancing the Efficacy of Glutamine Metabolism Inhibitors in Cancer Therapy. Trends Cancer 2021, 7, 790–804. [Google Scholar] [CrossRef]
- Stine, Z.E.; Schug, Z.T.; Salvino, J.M.; Dang, C.V. Targeting cancer metabolism in the era of precision oncology. Nat. Rev. Drug Discov. 2022, 21, 141–162. [Google Scholar] [CrossRef] [PubMed]
- Kaddurah-Daouk, R.; Weinshilboum, R.M. Pharmacometabolomics: Implications for Clinical Pharmacology and Systems Pharmacology. Clin. Pharmacol. Ther. 2013, 95, 154–167. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Tang, F.; Luo, Z.; Chen, Y.; Xu, J.; Zhang, R.; Wang, X.; Abliz, Z. Air flow assisted ionization for remote sampling of ambient mass spectrometry and its application. Rapid Commun. Mass Spectrom. 2011, 25, 843–850. [Google Scholar] [CrossRef]
- He, J.; Luo, Z.; Huang, L.; He, J.; Chen, Y.; Rong, X.; Jia, S.; Tang, F.; Wang, X.; Zhang, R.; et al. Ambient Mass Spectrometry Imaging Metabolomics Method Provides Novel Insights into the Action Mechanism of Drug Candidates. Anal. Chem. 2015, 87, 5372–5379. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, T.; Song, X.; Huang, L.; Zang, Q.; Xu, J.; Bi, N.; Jiao, G.; Hao, Y.; Chen, Y.; et al. Spatially resolved metabolomics to discover tumor-associated metabolic alterations. Proc. Natl. Acad. Sci. USA 2019, 116, 52–57. [Google Scholar] [CrossRef]
- Huang, L.; Mao, X.; Sun, C.; Li, T.; Song, X.; Li, J.; Gao, S.; Zhang, R.; Chen, J.; He, J.; et al. Molecular Pathological Diagnosis of Thyroid Tumors Using Spatially Resolved Metabolomics. Molecules 2022, 27, 1390. [Google Scholar] [CrossRef]
- Li, T.; He, J.; Mao, X.; Bi, Y.; Luo, Z.; Guo, C.; Tang, F.; Xu, X.; Wang, X.; Wang, M.; et al. In situ biomarker discovery and label-free molecular histopathological diagnosis of lung cancer by ambient mass spectrometry imaging. Sci. Rep. 2015, 5, srep14089. [Google Scholar] [CrossRef]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Forman, H.J.; Zhang, H. Targeting oxidative stress in disease: Promise and limitations of antioxidant therapy. Nat. Rev. Drug Discov. 2021, 20, 689–709. [Google Scholar] [CrossRef]
- Cheng, X.; Geng, F.; Pan, M.; Wu, X.; Zhong, Y.; Wang, C.; Tian, Z.; Cheng, C.; Zhang, R.; Puduvalli, V.; et al. Targeting DGAT1 Ameliorates Glioblastoma by Increasing Fat Catabolism and Oxidative Stress. Cell Metab. 2020, 32, 229–242.e8. [Google Scholar] [CrossRef]
- Alves, A.; Costa, P.; Pinto, M.; Ferreira, D.; Correia-da-Silva, M. Small Molecules of Marine Origin as Potential Anti-Glioma Agents. Molecules 2021, 26, 2707. [Google Scholar] [CrossRef]
- Strelitz, F.; Flon, H.; Asheshov, I.N. Chrysomycin: A new antibiotic substance for bacterial viruses. J. Bacteriol. 1955, 69, 280–283. [Google Scholar] [CrossRef] [PubMed]
- Weiss, U.; Yoshihira, K.; Highet, R.J.; White, R.J.; Wei, T.T. The chemistry of the antibiotics chrysomycin A and B. Antitumor activity of chrysomycin A. J. Antibiot. 1982, 35, 1194–1201. [Google Scholar] [CrossRef] [PubMed]
- Ni, H.J.; Lv, S.Y.; Sheng, Y.T.; Wang, H.; Chu, X.H.; Zhang, H.W. Optimization of fermentation conditions and medium compositions for the production of chrysomycin a by a marine-derived strain Streptomyces sp. 891. Prep. Biochem. Biotechnol. 2021, 51, 998–1003. [Google Scholar] [CrossRef] [PubMed]
- Matson, J.A.; Rose, W.C.; Bush, J.A.; Myllymaki, R.; Bradner, W.T.; Doyle, T.W. Antitumor activity of chrysomycins M and V. J. Antibiot. 1989, 42, 1446–1448. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Chu, Y.; Gong, Y.; Hong, Y.; Song, F.; Wang, H.; Zhang, H.; Sun, X. Enhanced Transdermal Peptide-Modified Flexible Liposomes for Efficient Percutaneous Delivery of Chrysomycin A to Treat Subcutaneous Melanoma and Intradermal MRSA Infection. Adv. Health Mater. 2023, 12, e2300881. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, P.; Chen, J.; Yao, D.; Liu, Q.; Zhang, J.; Zhang, H.W.; Leung, E.L.; Yao, X.J.; Liu, L. Upgrade of chrysomycin A as a novel topoisomerase II inhibitor to curb KRAS-mutant lung adenocarcinoma progression. Pharmacol. Res. 2023, 187, 106565. [Google Scholar] [CrossRef]
- Liu, D.N.; Liu, M.; Zhang, S.S.; Shang, Y.F.; Zhang, W.F.; Song, F.H.; Zhang, H.W.; Du, G.H.; Wang, Y.H. Chrysomycin A Regulates Proliferation and Apoptosis of Neuroglioma Cells via the Akt/GSK-3beta Signaling Pathway In Vivo and In Vitro. Mar. Drugs 2023, 21, 329. [Google Scholar] [CrossRef]
- Liu, D.N.; Liu, M.; Zhang, S.S.; Shang, Y.F.; Song, F.H.; Zhang, H.W.; Du, G.H.; Wang, Y.H. Chrysomycin A Inhibits the Proliferation, Migration and Invasion of U251 and U87-MG Glioblastoma Cells to Exert Its Anti-Cancer Effects. Molecules 2022, 27, 6148. [Google Scholar] [CrossRef]
- Farahzadi, R.; Hejazi, M.S.; Molavi, O.; Pishgahzadeh, E.; Montazersaheb, S.; Jafari, S. Clinical Significance of Carnitine in the Treatment of Cancer: From Traffic to the Regulation. Oxid. Med. Cell Longev. 2023, 2023, 9328344. [Google Scholar] [CrossRef]
- Fink, M.A.; Paland, H.; Herzog, S.; Grube, M.; Vogelgesang, S.; Weitmann, K.; Bialke, A.; Hoffmann, W.; Rauch, B.H.; Schroeder, H.W.S.; et al. L-Carnitine-Mediated Tumor Cell Protection and Poor Patient Survival Associated with OCTN2 Overexpression in Glioblastoma Multiforme. Clin. Cancer Res. 2019, 25, 2874–2886. [Google Scholar] [CrossRef]
- Vance, J.E.; Steenbergen, R. Metabolism and functions of phosphatidylserine. Prog. Lipid Res. 2005, 44, 207–234. [Google Scholar] [CrossRef] [PubMed]
- Ran, S.; Thorpe, P.E. Phosphatidylserine is a marker of tumor vasculature and a potential target for cancer imaging and therapy. Int. J. Radiat. Oncol. Biol. Phys. 2002, 54, 1479–1484. [Google Scholar] [CrossRef]
- da Silva, A.M.B.; Silva-Goncalves, L.C.; Oliveira, F.A.; Arcisio-Miranda, M. Pro-necrotic Activity of Cationic Mastoparan Peptides in Human Glioblastoma Multiforme Cells Via Membranolytic Action. Mol. Neurobiol. 2018, 55, 5490–5504. [Google Scholar] [CrossRef] [PubMed]
- Krick, R.; Thumm, M. Atg8 lipidation is coordinated in a PtdIns3P-dependent manner by the PROPPIN Atg21. Autophagy 2016, 12, 2260–2261. [Google Scholar] [CrossRef]
- Li, W.; Ren, L.; Zheng, X.; Liu, J.; Wang, J.; Ji, T.; Du, G. 3-O-Acetyl-11-keto- beta -boswellic acid ameliorated aberrant metabolic landscape and inhibited autophagy in glioblastoma. Acta Pharm. Sin. B 2020, 10, 301–312. [Google Scholar] [CrossRef] [PubMed]
- Zalba, S.; Ten Hagen, T.L. Cell membrane modulation as adjuvant in cancer therapy. Cancer Treat Rev. 2017, 52, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Kishi, Y.; Okudaira, S.; Tanaka, M.; Hama, K.; Shida, D.; Kitayama, J.; Yamori, T.; Aoki, J.; Fujimaki, T.; Arai, H. Autotaxin is overexpressed in glioblastoma multiforme and contributes to cell motility of glioblastoma by converting lysophosphatidylcholine to lysophosphatidic acid. J. Biol. Chem. 2006, 281, 17492–17500. [Google Scholar] [CrossRef]
- Mauler, J.; Lohmann, P.; Maudsley, A.A.; Sheriff, S.; Hoevels, M.; Meissner, A.K.; Hamisch, C.; Brunn, A.; Deckert, M.; Filss, C.P.; et al. Diagnostic Accuracy of MR Spectroscopic Imaging and (18)F-FET PET for Identifying Glioma: A Biopsy-Controlled Hybrid PET/MRI Study. J. Nucl. Med. 2024, 65, 16–21. [Google Scholar] [CrossRef]
- Paul, S.; Ghosh, S.; Kumar, S. Tumor glycolysis, an essential sweet tooth of tumor cells. Semin. Cancer Biol. 2022, 86 Pt 3, 1216–1230. [Google Scholar] [CrossRef]
- Fu, L.H.; Qi, C.; Hu, Y.R.; Lin, J.; Huang, P. Glucose Oxidase-Instructed Multimodal Synergistic Cancer Therapy. Adv. Mater. 2019, 31, e1808325. [Google Scholar] [CrossRef] [PubMed]
- Tardito, S.; Oudin, A.; Ahmed, S.U.; Fack, F.; Keunen, O.; Zheng, L.; Miletic, H.; Sakariassen, P.Ø.; Weinstock, A.; Wagner, A.; et al. Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat. Cell Biol. 2015, 17, 1556–1568. [Google Scholar] [CrossRef] [PubMed]
- Curthoys, N.P.; Watford, M. Regulation of glutaminase activity and glutamine metabolism. Annu. Rev. Nutr. 1995, 15, 133–159. [Google Scholar] [CrossRef]
- Eniafe, J.; Jiang, S. The functional roles of TCA cycle metabolites in cancer. Oncogene 2021, 40, 3351–3363. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Cheng, S.Y. Targeting phospholipid metabolism for glioblastoma therapy. Neuro Oncol. 2021, 23, 343–344. [Google Scholar] [CrossRef]
- Young, R.S.E.; Bowman, A.P.; Tousignant, K.D.; Poad, B.L.J.; Gunter, J.H.; Philp, L.K.; Nelson, C.C.; Ellis, S.R.; Heeren, R.M.A.; Sadowski, M.C.; et al. Isomeric lipid signatures reveal compartmentalized fatty acid metabolism in cancer. J. Lipid Res. 2022, 63, 100223. [Google Scholar] [CrossRef]
- Melone, M.A.B.; Valentino, A.; Margarucci, S.; Galderisi, U.; Giordano, A.; Peluso, G. The carnitine system and cancer metabolic plasticity. Cell Death Dis. 2018, 9, 228. [Google Scholar] [CrossRef]
- Qin, L.; An, N.; Yuan, B.; Zhu, Q.; Feng, Y. The Metabolomic Characteristics and Dysregulation of Fatty Acid Esters of Hydroxy Fatty Acids in Breast Cancer. Metabolites 2023, 13, 1108. [Google Scholar] [CrossRef]
- Benlebna, M.; Balas, L.; Gaillet, S.; Durand, T.; Coudray, C.; Casas, F.; Feillet-Coudray, C. Potential physio-pathological effects of branched fatty acid esters of hydroxy fatty acids. Biochimie 2021, 182, 13–22. [Google Scholar] [CrossRef]
- Cluntun, A.A.; Lukey, M.J.; Cerione, R.A.; Locasale, J.W. Glutamine Metabolism in Cancer: Understanding the Heterogeneity. Trends Cancer 2017, 3, 169–180. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Q.; Huang, Z.; Li, B.; Nice, E.C.; Huang, C.; Wei, L.; Zou, B. Targeting Glucose Metabolism Enzymes in Cancer Treatment: Current and Emerging Strategies. Cancers 2022, 14, 4568. [Google Scholar] [CrossRef]
- Mele, L.; Paino, F.; Papaccio, F.; Regad, T.; Boocock, D.; Stiuso, P.; Lombardi, A.; Liccardo, D.; Aquino, G.; Barbieri, A.; et al. A new inhibitor of glucose-6-phosphate dehydrogenase blocks pentose phosphate pathway and suppresses malignant proliferation and metastasis in vivo. Cell Death Dis. 2018, 9, 572. [Google Scholar] [CrossRef] [PubMed]
- Patra, K.C.; Hay, N. The pentose phosphate pathway and cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qi, H.; Liu, Y.; Duan, C.; Liu, X.; Xia, T.; Chen, D.; Piao, H.L.; Liu, H.X. The double-edged roles of ROS in cancer prevention and therapy. Theranostics 2021, 11, 4839–4857. [Google Scholar] [CrossRef]
- Ying, M.; You, D.; Zhu, X.; Cai, L.; Zeng, S.; Hu, X. Lactate and glutamine support NADPH generation in cancer cells under glucose deprived conditions. Redox Biol. 2021, 46, 102065. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, F.; Hamanaka, R.; Wheaton, W.W.; Weinberg, S.; Joseph, J.; Lopez, M.; Kalyanaraman, B.; Mutlu, G.M.; Budinger, G.R.; Chandel, N.S. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc. Natl. Acad. Sci. USA 2010, 107, 8788–8793. [Google Scholar] [CrossRef] [PubMed]
- Ali, S.S.; Ahsan, H.; Zia, M.K.; Siddiqui, T.; Khan, F.H. Understanding oxidants and antioxidants: Classical team with new players. J. Food Biochem. 2020, 44, e13145. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Putnam, C.D.; Arvai, A.S.; Bourne, Y.; Tainer, J.A. Active and inhibited human catalase structures: Ligand and NADPH binding and catalytic mechanism. J. Mol. Biol. 2000, 296, 295–309. [Google Scholar] [CrossRef]
- Basak, P.; Sadhukhan, P.; Sarkar, P.; Sil, P.C. Perspectives of the Nrf-2 signaling pathway in cancer progression and therapy. Toxicol. Rep. 2017, 4, 306–318. [Google Scholar] [CrossRef]
- Jain, S.K.; Pathania, A.S.; Parshad, R.; Raina, C.; Ali, A.; Gupta, A.P.; Kushwaha, M.; Aravinda, S.; Bhushan, S.; Bharate, S.B.; et al. Correction: Chrysomycins A-C, antileukemic naphthocoumarins from Streptomyces sporoverrucosus. RSC Adv. 2022, 12, 23680–23682. [Google Scholar] [CrossRef] [PubMed]
- He, M.J.; Pu, W.; Wang, X.; Zhang, W.; Tang, D.; Dai, Y. Comparing DESI-MSI and MALDI-MSI Mediated Spatial Metabolomics and Their Applications in Cancer Studies. Front. Oncol. 2022, 12, 891018. [Google Scholar] [CrossRef] [PubMed]
- Kampa, J.M.; Kellner, U.; Marsching, C.; Ramallo Guevara, C.; Knappe, U.J.; Sahin, M.; Giampà, M.; Niehaus, K.; Bednarz, H. Glioblastoma multiforme: Metabolic differences to peritumoral tissue and IDH-mutated gliomas revealed by mass spectrometry imaging. Neuropathology 2020, 40, 546–558. [Google Scholar] [CrossRef]
- Wang, H.-Y.J.; Huang, C.-Y.; Wei, K.-C.; Hung, K.-C. A mass spectrometry imaging and lipidomic investigation reveals aberrant lipid metabolism in the orthotopic mouse glioma. J. Lipid Res. 2022, 63, 100304. [Google Scholar] [CrossRef]
- Randall, E.C.; Lopez, B.G.C.; Peng, S.; Regan, M.S.; Abdelmoula, W.M.; Basu, S.S.; Santagata, S.; Yoon, H.; Haigis, M.C.; Agar, J.N.; et al. Localized Metabolomic Gradients in Patient-Derived Xenograft Models of Glioblastoma. Cancer Res. 2020, 80, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Moffett, J.; Ross, B.; Arun, P.; Madhavarao, C.; Namboodiri, A. N-Acetylaspartate in the CNS: From neurodiagnostics to neurobiology. Prog. Neurobiol. 2007, 81, 89–131. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Updates 2018, 38, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef]
- Stoica, C.; Ferreira, A.K.; Hannan, K.; Bakovic, M. Bilayer Forming Phospholipids as Targets for Cancer Therapy. Int. J. Mol. Sci. 2022, 23, 5266. [Google Scholar] [CrossRef]
- Yang, S.; Zhao, J.; Cui, X.; Zhan, Q.; Yi, K.; Wang, Q.; Xiao, M.; Tan, Y.; Hong, B.; Fang, C.; et al. TCA-phospholipid-glycolysis targeted triple therapy effectively suppresses ATP production and tumor growth in glioblastoma. Theranostics 2022, 12, 7032–7050. [Google Scholar] [CrossRef]
- Ackerstaff, E.; Glunde, K.; Bhujwalla, Z.M. Choline phospholipid metabolism: A target in cancer cells? J. Cell Biochem. 2003, 90, 525–533. [Google Scholar] [CrossRef]
- Kurabe, N.; Hayasaka, T.; Ogawa, M.; Masaki, N.; Ide, Y.; Waki, M.; Nakamura, T.; Kurachi, K.; Kahyo, T.; Shinmura, K.; et al. Accumulated phosphatidylcholine (16:0/16:1) in human colorectal cancer; possible involvement of LPCAT4. Cancer Sci. 2013, 104, 1295–1302. [Google Scholar] [CrossRef]
- Patel, D.; Witt, S.N. Ethanolamine and Phosphatidylethanolamine: Partners in Health and Disease. Oxid. Med. Cell Longev. 2017, 2017, 4829180. [Google Scholar] [CrossRef] [PubMed]
- Medes, G.; Thomas, A.; Weinhouse, S. Metabolism of neoplastic tissue. IV. A study of lipid synthesis in neoplastic tissue slices in vitro. Cancer. Res. 1953, 13, 27–29. [Google Scholar] [PubMed]
- Lin, H.; Patel, S.; Affleck, V.S.; Wilson, I.; Turnbull, D.M.; Joshi, A.R.; Maxwell, R.; Stoll, E.A. Fatty acid oxidation is required for the respiration and proliferation of malignant glioma cells. Neuro-Oncol. 2017, 19, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Restall, I.J.; Cseh, O.; Richards, L.M.; Pugh, T.J.; Luchman, H.A.; Weiss, S. Brain Tumor Stem Cell Dependence on Glutaminase Reveals a Metabolic Vulnerability through the Amino Acid Deprivation Response Pathway. Cancer Res. 2020, 80, 5478–5490. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Li, X.; Wu, Y.; Zhang, G.; Liu, X.; Li, Y.; Bao, Y.; Yang, W.; Cui, H. EGFR activates GDH1 transcription to promote glutamine metabolism through MEK/ERK/ELK1 pathway in glioblastoma. Oncogene 2020, 39, 2975–2986. [Google Scholar] [CrossRef]
- Blaha, C.S.; Ramakrishnan, G.; Jeon, S.M.; Nogueira, V.; Rho, H.; Kang, S.; Bhaskar, P.; Terry, A.R.; Aissa, A.F.; Frolov, M.V.; et al. A non-catalytic scaffolding activity of hexokinase 2 contributes to EMT and metastasis. Nat. Commun. 2022, 13, 899. [Google Scholar] [CrossRef]
- Yang, H.C.; Wu, Y.H.; Yen, W.C.; Liu, H.Y.; Hwang, T.L.; Stern, A.; Chiu, D.T. The Redox Role of G6PD in Cell Growth, Cell Death, and Cancer. Cells 2019, 8, 1055. [Google Scholar] [CrossRef]
- Ye, H.; Huang, H.; Cao, F.; Chen, M.; Zheng, X.; Zhan, R. HSPB1 Enhances SIRT2-Mediated G6PD Activation and Promotes Glioma Cell Proliferation. PLoS ONE 2016, 11, e0164285. [Google Scholar] [CrossRef]
- Hosios, A.M.; Manning, B.D. Cancer Signaling Drives Cancer Metabolism: AKT and the Warburg Effect. Cancer Res. 2021, 81, 4896–4898. [Google Scholar] [CrossRef] [PubMed]
- Roberts, D.J.; Tan-Sah, V.P.; Smith, J.M.; Miyamoto, S. Akt phosphorylates HK-II at Thr-473 and increases mitochondrial HK-II association to protect cardiomyocytes. J. Biol. Chem. 2013, 288, 23798–23806. [Google Scholar] [CrossRef] [PubMed]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef]
- Lien, E.C.; Lyssiotis, C.A.; Cantley, L.C. Metabolic Reprogramming by the PI3K-Akt-mTOR Pathway in Cancer. Recent Results Cancer Res. 2016, 207, 39–72. [Google Scholar] [PubMed]
- Hoxhaj, G.; Manning, B.D. The PI3K-AKT network at the interface of oncogenic signalling and cancer metabolism. Nat. Rev. Cancer 2020, 20, 74–88. [Google Scholar] [CrossRef]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef]
- Porstmann, T.; Griffiths, B.; Chung, Y.L.; Delpuech, O.; Griffiths, J.R.; Downward, J.; Schulze, A. PKB/Akt induces transcription of enzymes involved in cholesterol and fatty acid biosynthesis via activation of SREBP. Oncogene 2005, 24, 6465–6481. [Google Scholar] [CrossRef]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Jia, J.; Zheng, M.; Zhang, C.; Li, B.; Lu, C.; Bai, Y.; Tong, Q.; Hang, X.; Ge, Y.; Zeng, L.; et al. Killing of Staphylococcus aureus persisters by a multitarget natural product chrysomycin A. Sci. Adv. 2023, 9, eadg5995. [Google Scholar] [CrossRef] [PubMed]
- Yoo, N.J.; Kim, H.R.; Kim, Y.R.; An, C.H.; Lee, S.H. Somatic mutations of the KEAP1 gene in common solid cancers. Histopathology 2012, 60, 943–952. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Selective modification of glutathione metabolism. Science 1983, 220, 472–477. [Google Scholar] [CrossRef]
- Mitsuishi, Y.; Taguchi, K.; Kawatani, Y.; Shibata, T.; Nukiwa, T.; Aburatani, H.; Yamamoto, M.; Motohashi, H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 2012, 22, 66–79. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Value |
|---|---|
| Spray voltage | 4500 V |
| Tube voltage | 0 V |
| Spray solvent flow | 6 μL/min |
| Spray gas pressure | 0.7 MPa |
| Extracting gas flow | 25 L/min |
| Spray angle | 60° |
| X axis move speed | 0.1 mm/s |
| Y axis step size | 0.1 mm |
| Distance from sprayer to surface | 0.7 mm |
| Distance from sprayer to tube | 3 mm |
| Distance from orifice to guide tube | 10 mm |
| Spray solution | ACN/H2O (8:2, v/v) |
| Parameter | Value |
|---|---|
| Polarity | Positive/Negative |
| Scan mode | Full scan SIM |
| Scan range | 70–1000 Da |
| Capillary temperature | 350 °C |
| Sheath gas flow rate | 0 L/min |
| Aux gas flow rate | 0 L/min |
| Sweep gas flow rate | 0 L/min |
| Aux gas heater temperature | 0 °C |
| Maximum inject time | 200 ms for Full MS |
| AGC target | 3 × 106 |
| Resolution | 70,000 |
| Scan rate | ~2.2 scans/s |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, D.-N.; Zhang, W.-F.; Feng, W.-D.; Xu, S.; Feng, D.-H.; Song, F.-H.; Zhang, H.-W.; Fang, L.-H.; Du, G.-H.; Wang, Y.-H. Chrysomycin A Reshapes Metabolism and Increases Oxidative Stress to Hinder Glioblastoma Progression. Mar. Drugs 2024, 22, 391. https://doi.org/10.3390/md22090391
Liu D-N, Zhang W-F, Feng W-D, Xu S, Feng D-H, Song F-H, Zhang H-W, Fang L-H, Du G-H, Wang Y-H. Chrysomycin A Reshapes Metabolism and Increases Oxidative Stress to Hinder Glioblastoma Progression. Marine Drugs. 2024; 22(9):391. https://doi.org/10.3390/md22090391
Chicago/Turabian StyleLiu, Dong-Ni, Wen-Fang Zhang, Wan-Di Feng, Shuang Xu, Dan-Hong Feng, Fu-Hang Song, Hua-Wei Zhang, Lian-Hua Fang, Guan-Hua Du, and Yue-Hua Wang. 2024. "Chrysomycin A Reshapes Metabolism and Increases Oxidative Stress to Hinder Glioblastoma Progression" Marine Drugs 22, no. 9: 391. https://doi.org/10.3390/md22090391