

RNA Editing Analysis Reveals Methyl Jasmonic Acid Regulation of Fucoxanthin and Fatty Acid Metabolism in Phaeodactylum tricornutum

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. General Characteristics of RNA Editing Events in Control and MeJA-Treated P. tricornutum Samples
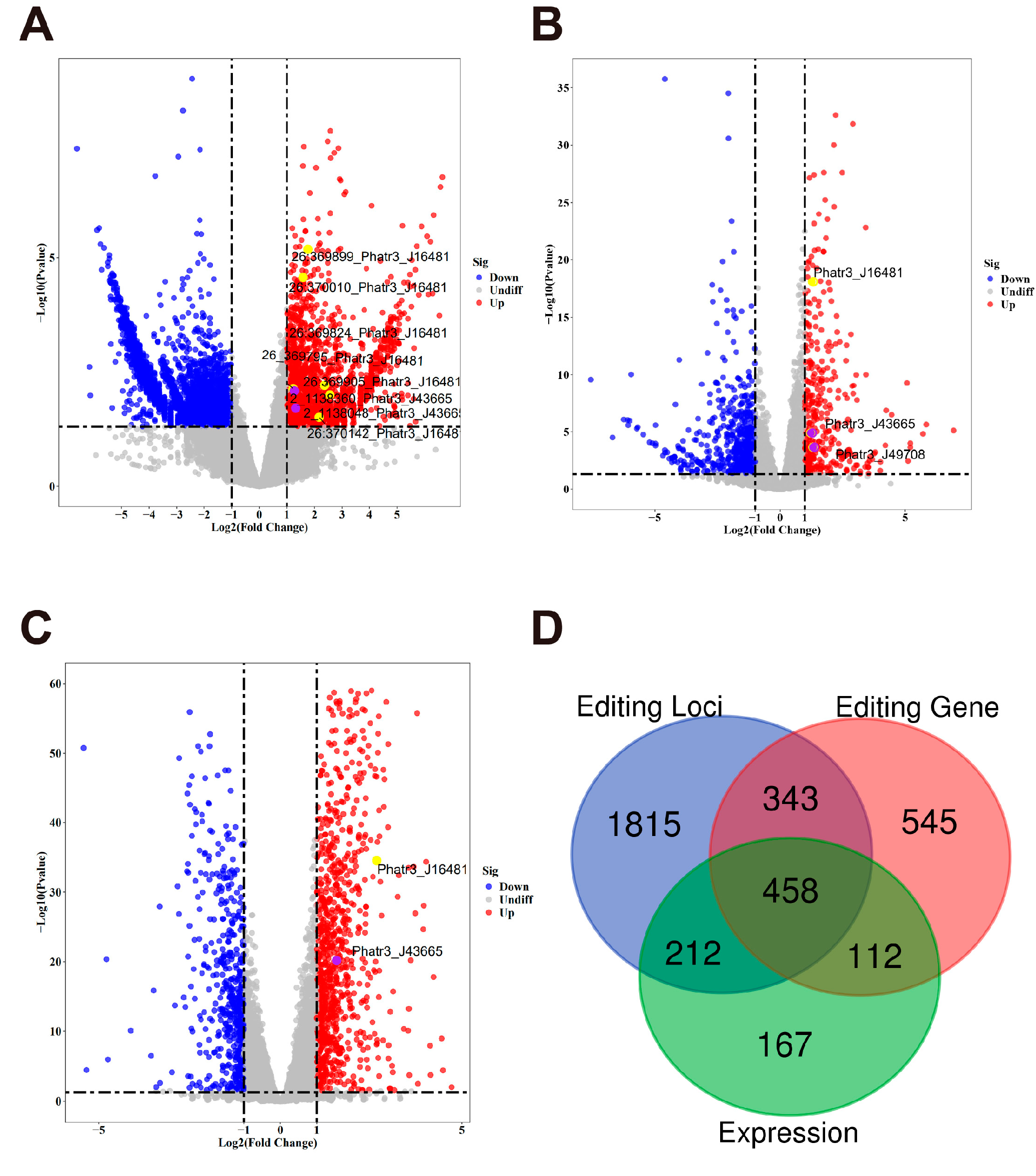
2.2. Differential Analysis of RNA Editing and Gene Expression
2.3. Functional Enrichment Analysis of Differential Gene Sets
2.4. Integrated Analysis of RNA Editing and RNA Expression
3. Discussion
4. Materials and Methods
4.1. RNA-Seq Dataset Source
4.2. Data Pre-Processing
4.3. Identification and Quantification of RNA Editing Events
4.4. Differential Editing Analysis and Functional Enrichment Analysis of Differential Editing Events
4.5. RNA Expression Quantification and Analysis
4.6. Correlation Analysis Between RNA Editing and Gene Expression
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Raja, R.; Hemaiswarya, S.; Kumar, N.A.; Sridhar, S.; Rengasamy, R. A Perspective on the Biotechnological Potential of Microalgae. Crit. Rev. Microbiol. 2008, 34, 77–88. [Google Scholar] [CrossRef] [PubMed]
- Pulz, O.; Gross, W. Valuable Products from Biotechnology of Microalgae. Appl. Microbiol. Biotechnol. 2004, 65, 635–648. [Google Scholar] [CrossRef] [PubMed]
- Rizwan, M.; Mujtaba, G.; Memon, S.A.; Lee, K.; Rashid, N. Exploring the Potential of Microalgae for New Biotechnology Applications and Beyond: A Review. Renew. Sustain. Energy Rev. 2018, 92, 394–404. [Google Scholar] [CrossRef]
- Ampofo, J.; Abbey, L. Microalgae: Bioactive Composition, Health Benefits, Safety and Prospects as Potential High-Value Ingredients for the Functional Food Industry. Foods 2022, 11, 1744. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.I.; Shin, J.H.; Kim, J.D. The Promising Future of Microalgae: Current Status, Challenges, and Optimization of a Sustainable and Renewable Industry for Biofuels, Feed, and Other Products. Microb. Cell Fact. 2018, 17, 36. [Google Scholar] [CrossRef]
- Siddiki, S.Y.A.; Mofijur, M.; Kumar, P.S.; Ahmed, S.F.; Inayat, A.; Kusumo, F.; Badruddin, I.A.; Khan, T.M.Y.; Nghiem, L.D.; Ong, H.C. Microalgae Biomass as a Sustainable Source for Biofuel, Biochemical and Biobased Value-Added Products: An Integrated Biorefinery Concept. Fuel 2022, 307, 121782. [Google Scholar] [CrossRef]
- Dolganyuk, V.; Belova, D.; Babich, O.; Prosekov, A.; Ivanova, S.; Katserov, D.; Patyukov, N.; Sukhikh, S. Microalgae: A Promising Source of Valuable Bioproducts. Biomolecules 2020, 10, 1153. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Li, K.; Duan, X.; Hill, D.; Barrow, C.; Dunshea, F.; Martin, G.; Suleria, H. Bioactive Compounds in Microalgae and Their Potential Health Benefits. Food Biosci. 2022, 49, 101932. [Google Scholar] [CrossRef]
- Russo, M.T.; Rogato, A.; Jaubert, M.; Karas, B.J.; Falciatore, A. Phaeodactylum Tricornutum: An Established Model Species for Diatom Molecular Research and an Emerging Chassis for Algal Synthetic Biology. J. Phycol. 2023, 59, 1114–1122. [Google Scholar] [CrossRef] [PubMed]
- Celi, C.; Fino, D.; Savorani, F. Phaeodactylum Tricornutum as a Source of Value-Added Products: A Review on Recent Developments in Cultivation and Extraction Technologies. Bioresour. Technol. Rep. 2022, 19, 101122. [Google Scholar] [CrossRef]
- Scala, S.; Carels, N.; Falciatore, A.; Chiusano, M.L.; Bowler, C. Genome Properties of the Diatom Phaeodactylum Tricornutum. Plant Physiol. 2002, 129, 993–1002. [Google Scholar] [CrossRef]
- Bowler, C.; Allen, A.E.; Badger, J.H.; Grimwood, J.; Jabbari, K.; Kuo, A.; Maheswari, U.; Martens, C.; Maumus, F.; Otillar, R.P.; et al. The Phaeodactylum Genome Reveals the Evolutionary History of Diatom Genomes. Nature 2008, 456, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Butler, T.; Kapoore, R.V.; Vaidyanathan, S. Phaeodactylum Tricornutum: A Diatom Cell Factory. Trends Biotechnol. 2020, 38, 606–622. [Google Scholar] [CrossRef] [PubMed]
- De Martino, A.; Bartual, A.; Willis, A.; Meichenin, A.; Villazán, B.; Maheswari, U.; Bowler, C. Physiological and molecular evidence that environmental changes elicit morphological interconversion in the model diatom Phaeodactylum tricornutum. Protist 2011, 162, 462–481. [Google Scholar] [CrossRef]
- Kuczynska, P.; Jemiola-Rzeminska, M.; Strzalka, K. Photosynthetic Pigments in Diatoms. Mar. Drugs 2015, 13, 5847–5881. [Google Scholar] [CrossRef]
- Guan, B.; Chen, K.; Tong, Z.; Chen, L.; Chen, Q.; Su, J. Advances in Fucoxanthin Research for the Prevention and Treatment of Inflammation-Related Diseases. Nutrients 2022, 14, 4768. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.R.; Hosokawa, M.; Miyashita, K. Fucoxanthin: A Marine Carotenoid Exerting Anti-Cancer Effects by Affecting Multiple Mechanisms. Mar. Drugs 2013, 11, 5130–5147. [Google Scholar] [CrossRef] [PubMed]
- Gammone, M.A.; D’Orazio, N. Anti-Obesity Activity of the Marine Carotenoid Fucoxanthin. Mar. Drugs 2015, 13, 2196–2214. [Google Scholar] [CrossRef] [PubMed]
- Kawee-ai, A.; Kuntiya, A.; Kim, S.M. Anticholinesterase and Antioxidant Activities of Fucoxanthin Purified from the Microalga Phaeodactylum Tricornutum. Nat. Prod. Commun. 2013, 8, 1934578X1300801010. [Google Scholar] [CrossRef]
- Yang, Y.-H.; Du, L.; Hosokawa, M.; Miyashita, K.; Kokubun, Y.; Arai, H.; Taroda, H. Fatty Acid and Lipid Class Composition of the Microalga Phaeodactylum Tricornutum. J. Oleo Sci. 2017, 66, 363–368. [Google Scholar] [CrossRef]
- Neumann, U.; Louis, S.; Gille, A.; Derwenskus, F.; Schmid-Staiger, U.; Briviba, K.; Bischoff, S.C. Anti-Inflammatory Effects of Phaeodactylum Tricornutum Extracts on Human Blood Mononuclear Cells and Murine Macrophages. J. Appl. Phycol. 2018, 30, 2837–2846. [Google Scholar] [CrossRef]
- Pereira, H.; Sá, M.; Maia, I.; Rodrigues, A.; Teles, I.; Wijffels, R.H.; Navalho, J.; Barbosa, M. Fucoxanthin production from Tisochrysis lutea and Phaeodactylum tricornutum at industrial scale. Algal Res. 2021, 56, 102322. [Google Scholar] [CrossRef]
- Kim, S.M.; Jung, Y.-J.; Kwon, O.-N.; Cha, K.H.; Um, B.-H.; Chung, D.; Pan, C.-H. A Potential Commercial Source of Fucoxanthin Extracted from the Microalga Phaeodactylum tricornutum. Appl. Biochem. Biotechnol. 2012, 166, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Lourenço-Lopes, C.; Garcia-Oliveira, P.; Carpena, M.; Fraga-Corral, M.; Jimenez-Lopez, C.; Pereira, A.G.; Prieto, M.A.; Simal-Gandara, J. Scientific Approaches on Extraction, Purification and Stability for the Commercialization of Fucoxanthin Recovered from Brown Algae. Foods 2020, 9, 1113. [Google Scholar] [CrossRef] [PubMed]
- McClure, D.D.; Luiz, A.; Gerber, B.; Barton, G.W.; Kavanagh, J.M. An investigation into the effect of culture conditions on fucoxanthin production using the marine microalgae Phaeodactylum tricornutum. Algal Res. 2018, 29, 41–48. [Google Scholar] [CrossRef]
- Yang, R.; Wei, D. Improving Fucoxanthin Production in Mixotrophic Culture of Marine Diatom Phaeodactylum tricornutum by LED Light Shift and Nitrogen Supplementation. Front. Bioeng. Biotechnol. 2020, 8, 820. [Google Scholar] [CrossRef] [PubMed]
- Afonso, C.; Bragança, A.R.; Rebelo, B.A.; Serra, T.S.; Abranches, R. Optimal Nitrate Supplementation in Phaeodactylum tricornutum Culture Medium Increases Biomass and Fucoxanthin Production. Foods 2022, 11, 568. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, W.; Zhang, Y.; Zhang, X.; Lang, D.; Zhang, X. The Roles of Methyl Jasmonate to Stress in Plants. Funct. Plant Biol. 2018, 46, 197–212. [Google Scholar] [CrossRef]
- Ho, T.-T.; Murthy, H.N.; Park, S.-Y. Methyl Jasmonate Induced Oxidative Stress and Accumulation of Secondary Metabolites in Plant Cell and Organ Cultures. Int. J. Mol. Sci. 2020, 21, 716. [Google Scholar] [CrossRef] [PubMed]
- Khan, V.; Jha, A.; Seth, T.; Iqbal, N.; Umar, S. Exploring the Role of Jasmonic Acid in Boosting the Production of Secondary Metabolites in Medicinal Plants: Pathway for Future Research. Ind. Crops Prod. 2024, 220, 119227. [Google Scholar] [CrossRef]
- Lu, Y.; Jiang, P.; Liu, S.; Gan, Q.; Cui, H.; Qin, S. Methyl Jasmonate-or Gibberellins A3-Induced Astaxanthin Accumulation Is Associated with up-Regulation of Transcription of β-Carotene Ketolase Genes (Bkts) in Microalga Haematococcus pluvialis. Bioresour. Technol. 2010, 101, 6468–6474. [Google Scholar] [CrossRef]
- Liu, H.; Chen, Y.; Wang, H.; Huang, Y.; Hu, Y.; Zhao, Y.; Gong, Y. Identification of Potential Factors for the Promotion of Fucoxanthin Synthesis by Methyl Jasmonic Acid Treatment of Phaeodactylum tricornutum. Mar. Drugs 2023, 22, 7. [Google Scholar] [CrossRef]
- Schaub, M.; Keller, W. RNA Editing by Adenosine Deaminases Generates RNA and Protein Diversity. Biochimie 2002, 84, 791–803. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.; Fei, Y.; Page, M. Biological Significance of RNA Editing in Cells. Mol. Biotechnol. 2012, 52, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.P.; Maggi, L.B.; Weber, J.D. The Role of RNA Editing in Cancer Development and Metabolic Disorders. Front. Endocrinol. 2018, 9, 762. [Google Scholar] [CrossRef]
- Maas, S.; Rich, A. Changing Genetic Information Through RNA Editing. Bioessays 2000, 22, 790–802. [Google Scholar] [CrossRef]
- Takenaka, M.; Zehrmann, A.; Verbitskiy, D.; Härtel, B.; Brennicke, A. RNA Editing in Plants and Its Evolution. Annu. Rev. Genet. 2013, 47, 335–352. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.H.; Li, Q.; Shanmugam, R.; Piskol, R.; Kohler, J.; Young, A.N.; Liu, K.I.; Zhang, R.; Ramaswami, G.; Ariyoshi, K. Dynamic Landscape and Regulation of RNA Editing in Mammals. Nature 2017, 550, 249–254. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Nie, W.; Ahmad, I.; Chen, G.; Zhu, B. The Occurrence, Characteristics, and Adaptation of A-to-I RNA Editing in Bacteria: A Review. Front. Microbiol. 2023, 14, 1143929. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-X.; Huang, A.; Li, Y.; Molloy, D.P.; Huang, C. Emerging Roles of the C-to-U RNA Editing in Plant Stress Responses. Plant Sci. 2024, 349, 112263. [Google Scholar] [CrossRef] [PubMed]
- Yablonovitch, A.L.; Deng, P.; Jacobson, D.; Li, J.B. The Evolution and Adaptation of A-to-I RNA Editing. PLoS Genet. 2017, 13, e1007064. [Google Scholar] [CrossRef] [PubMed]
- Nishikura, K. Functions and regulation of RNA editing by ADAR deaminases. Annu. Rev. Biochem. 2010, 79, 321–349. [Google Scholar] [CrossRef] [PubMed]
- Stukenberg, D.; Zauner, S.; Dell’Aquila, G.; Maier, U.G. Optimizing CRISPR/Cas9 for the Diatom Phaeodactylum tricornutum. Front. Plant Sci. 2023, 9, 74. [Google Scholar] [CrossRef]
- Ali, M.; Abbasi, B.H.; Ali, G.S. Elicitation of Antioxidant Secondary Metabolites with Jasmonates and Gibberellic Acid in Cell Suspension Cultures of Artemisia absinthium L. Plant Cell Tissue Organ Cult. (PCTOC) 2015, 120, 1099–1106. [Google Scholar] [CrossRef]
- Sabater-Jara, A.; Onrubia, M.; Moyano, E.; Bonfill, M.; Palazón, J.; Pedreño, M.A.; Cusidó, R.M. Synergistic Effect of Cyclodextrins and Methyl Jasmonate on Taxane Production in Taxus x Media Cell Cultures. Plant Biotechnol. J. 2014, 12, 1075–1084. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhang, J.; Lv, J.; Li, J.; Gao, Y.; Patience, B.E.; Niu, T.; Yu, J.; Xie, J. Effect of Methyl Jasmonate Treatment on Primary and Secondary Metabolites and Antioxidant Capacity of the Substrate and Hydroponically Grown Chinese Chives. Front. Nutr. 2022, 9, 859035. [Google Scholar] [CrossRef] [PubMed]
- Mc Gee, D.; Archer, L.; Parkes, R.; Fleming, G.T.A.; Santos, H.M.; Touzet, N. The Role of Methyl Jasmonate in Enhancing Biomass Yields and Bioactive Metabolites in Stauroneis sp.(Bacillariophyceae) Revealed by Proteome and Biochemical Profiling. J. Proteom. 2021, 249, 104381. [Google Scholar] [CrossRef] [PubMed]
- Mohibbullah, M.; Haque, M.N.; Sohag, A.A.M.; Hossain, M.T.; Zahan, M.S.; Uddin, M.J.; Hannan, M.A.; Moon, I.S.; Choi, J.-S. A Systematic Review on Marine Algae-Derived Fucoxanthin: An Update of Pharmacological Insights. Mar. Drugs 2022, 20, 279. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Côme, M.; Ulmann, L.; Chini Zittelli, G.; Faraloni, C.; Nazih, H.; Ouguerram, K.; Chénais, B.; Mimouni, V. Preventive Effects of the Marine Microalga Phaeodactylum Tricornutum, Used as a Food Supplement, on Risk Factors Associated with Metabolic Syndrome in Wistar Rats. Nutrients 2019, 11, 1069. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, N.G.; Ramadan, A.M.; Amer, M.; Morsy, Y.; Mohamed, R.A.; Said, O.A.; Alnufaei, A.A.; Ibrahim, M.I.M.; Hassanein, S.E.; Eissa, H.F. RNA editing-induced structural and functional adaptations of NAD9 in Triticum aestivum under drought stress. Front. Plant Sci. 2024, 15, 1490288. [Google Scholar] [CrossRef]
- Nymark, M.; Valle, K.C.; Hancke, K.; Winge, P.; Andresen, K.; Johnsen, G.; Bones, A.M.; Brembu, T. Molecular and Photosynthetic Responses to Prolonged Darkness and Subsequent Acclimation to Re-Illumination in the Diatom Phaeodactylum tricornutum. PLoS ONE 2013, 8, e58722. [Google Scholar] [CrossRef]
- Guéguen, N.; Le Moigne, D.; Amato, A.; Salvaing, J.; Maréchal, E. Lipid Droplets in Unicellular Photosynthetic stramenopiles. Front. Plant Sci. 2021, 12, 639276. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zheng, G.; Li, X.; Lin, H.; Jiang, P.; Qin, S. Cloning and Characterization of a Novel Diacylglycerol Acyltransferase from the Diatom Phaeodactylum tricornutum. J. Appl. Phycol. 2013, 25, 1509–1512. [Google Scholar] [CrossRef]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. ANNOVAR: Functional Annotation of Genetic Variants from High-Throughput Sequencing Data. Nucleic Acids Res. 2010, 38, e164. [Google Scholar] [CrossRef]
- Love, M.; Anders, S.; Huber, W. Differential Analysis of Count Data–the DESeq2 Package. Genome Biol. 2014, 15, 10–1186. [Google Scholar]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.-C.; Mendell, J.T.; Salzberg, S.L. StringTie Enables Improved Reconstruction of a Transcriptome from RNA-Seq Reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Control_1 | Control_2 | Control_3 | MeJA_1 | MeJA_2 | MeJA_3 | ||
|---|---|---|---|---|---|---|---|---|
| Editing Loci | 87,064 | 87,151 | 88,696 | 74,651 | 79,182 | 77,649 | ||
| Total | 262,911 | 231,482 | ||||||
| Specific Editing Loci | C to T | 83,450 (31.6%) | C to T | 73,534 (31.6%) | ||||
| A to G | 81,934 (31%) | A to G | 72,790 (31.4%) | |||||
| Gene | Chrom | p-Value (Gene) | Pos | Class | p-Value (Loci) | ExonicFunc |
|---|---|---|---|---|---|---|
| phatr3_j43665 | Chr:2 | 1.28 × 10−5 | 1138048 | G->T | 0.019683503 | synonymous SNV |
| 1138360 | C->G | 0.008246374 | nonsynonymous SNV |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Liu, H.; Xu, R.; Li, W.; Yang, H.; Bao, X.; Hang, Y.; Gong, Y.; Zhao, Y. RNA Editing Analysis Reveals Methyl Jasmonic Acid Regulation of Fucoxanthin and Fatty Acid Metabolism in Phaeodactylum tricornutum. Mar. Drugs 2025, 23, 66. https://doi.org/10.3390/md23020066
Huang S, Liu H, Xu R, Li W, Yang H, Bao X, Hang Y, Gong Y, Zhao Y. RNA Editing Analysis Reveals Methyl Jasmonic Acid Regulation of Fucoxanthin and Fatty Acid Metabolism in Phaeodactylum tricornutum. Marine Drugs. 2025; 23(2):66. https://doi.org/10.3390/md23020066
Chicago/Turabian StyleHuang, Sihui, Hao Liu, Ruihao Xu, Wangchang Li, Han Yang, Xinlei Bao, Yuqing Hang, Yifu Gong, and Yuxiang Zhao. 2025. "RNA Editing Analysis Reveals Methyl Jasmonic Acid Regulation of Fucoxanthin and Fatty Acid Metabolism in Phaeodactylum tricornutum" Marine Drugs 23, no. 2: 66. https://doi.org/10.3390/md23020066
APA StyleHuang, S., Liu, H., Xu, R., Li, W., Yang, H., Bao, X., Hang, Y., Gong, Y., & Zhao, Y. (2025). RNA Editing Analysis Reveals Methyl Jasmonic Acid Regulation of Fucoxanthin and Fatty Acid Metabolism in Phaeodactylum tricornutum. Marine Drugs, 23(2), 66. https://doi.org/10.3390/md23020066