


Comparative Analysis of Intestinal Microbiota Between Tetrodotoxin-Containing and Tetrodotoxin-Free Takifugu rubripes
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. TTX Concentration in Wild and Captive T. rubripes
2.2. OTU Characterization
2.3. Comparative Assessment of the Microbiota Diversity and Composition Between Captive and Wild T. rubripes
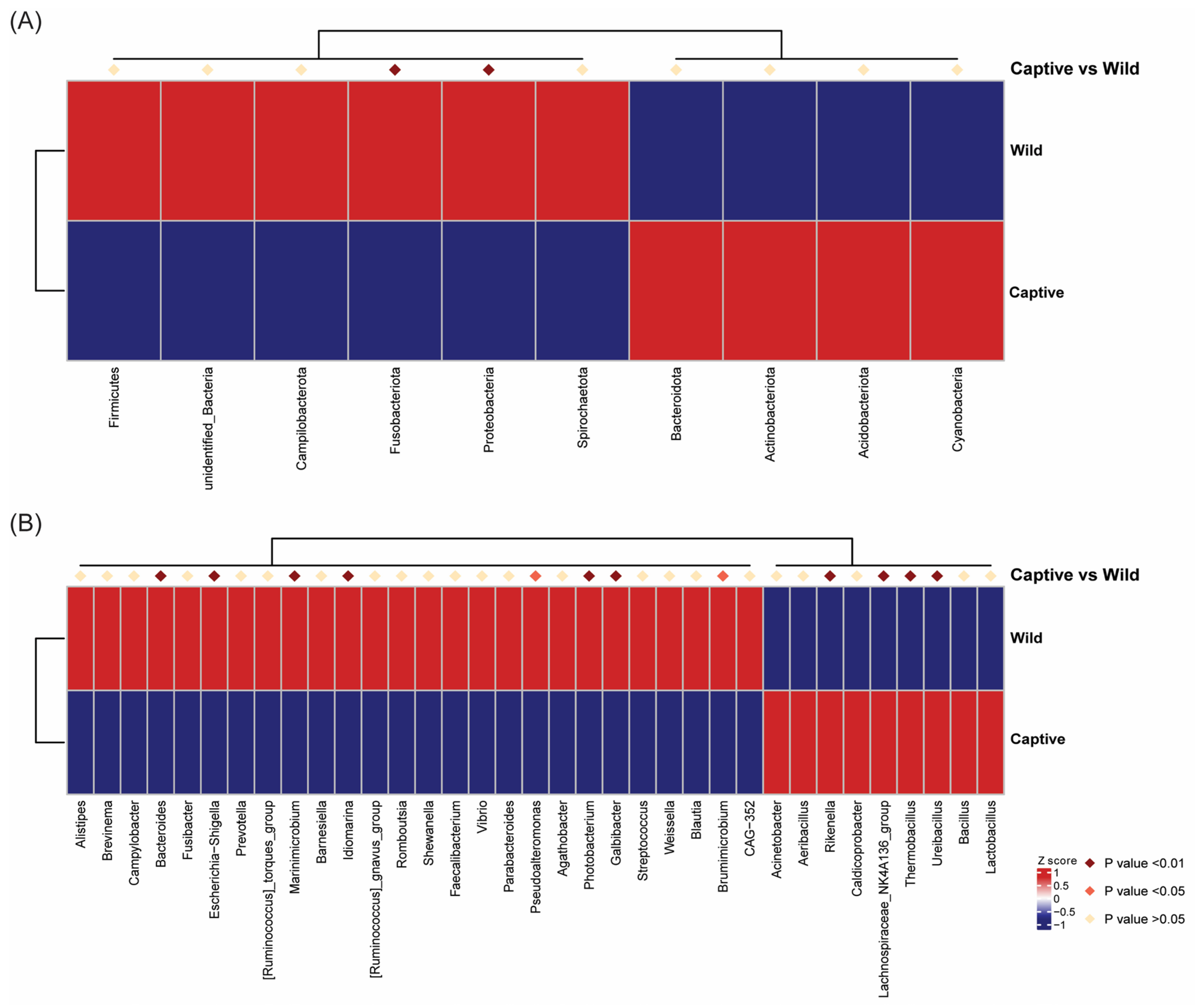
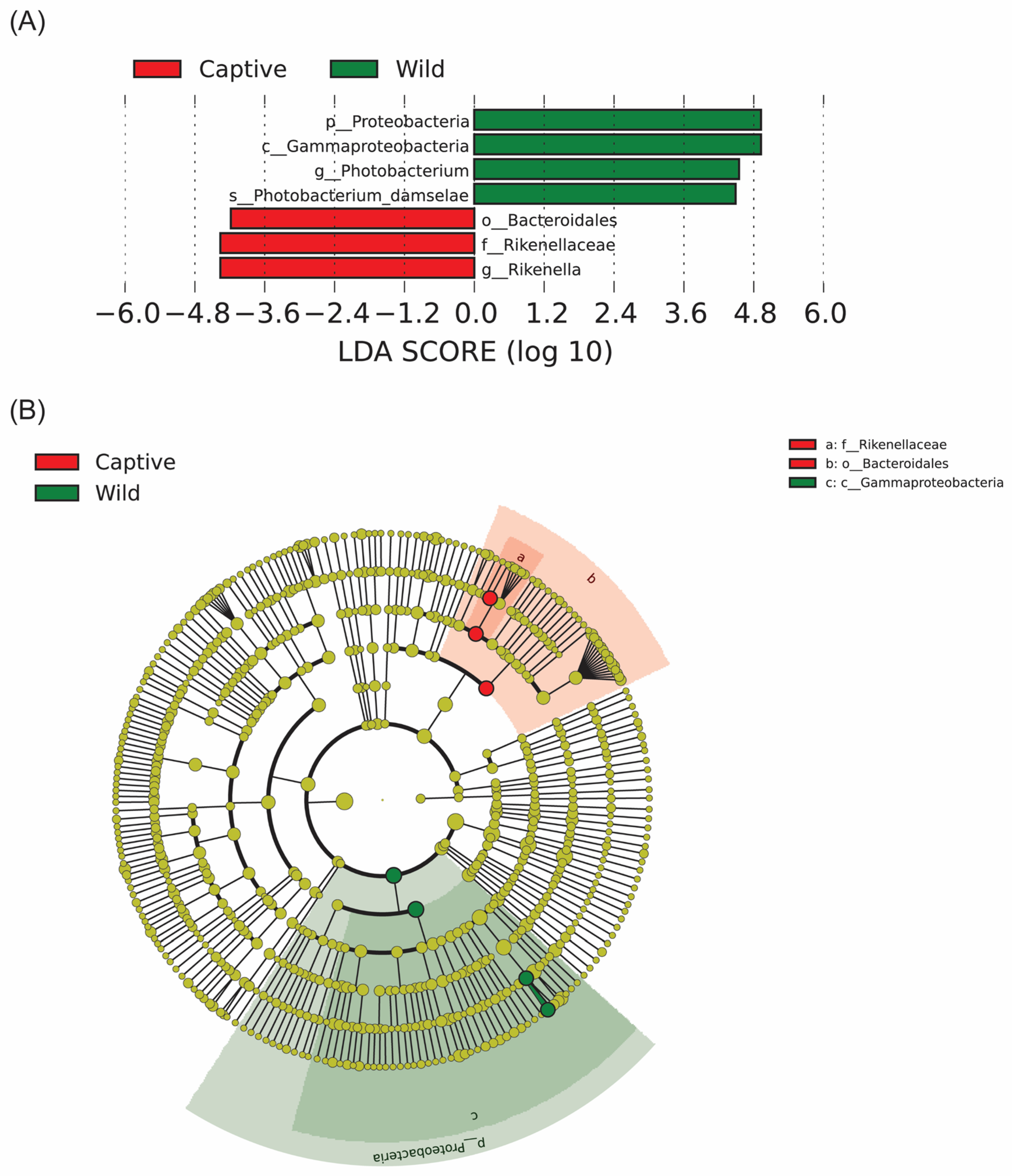
2.4. Taxonomic Classification of Microbiota
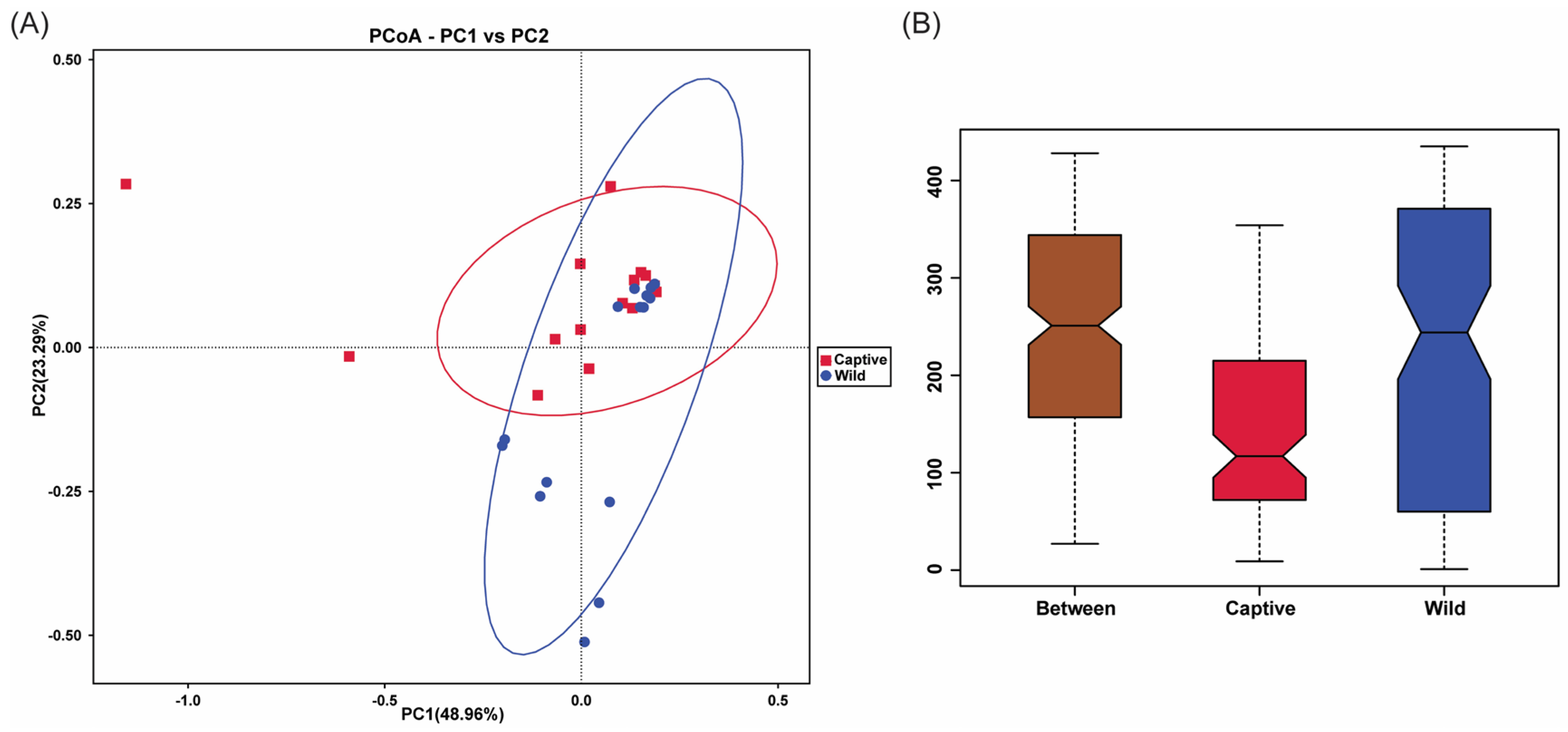
2.5. Beta Diversity Analysis
2.6. Function Prediction
3. Discussion
4. Materials and Methods
4.1. Sample Collection and TTX Detection
4.2. DNA Extraction, Amplification and Sequencing
4.3. Bioinformatics and Statistical Analysis
4.4. Functional Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lago, J.; Rodríguez, L.P.; Blanco, L.; Vieites, J.M.; Cabado, A.G. Tetrodotoxin, an Extremely Potent Marine Neurotoxin: Distribution, Toxicity, Origin and Therapeutical Uses. Mar. Drugs 2015, 13, 6384–6406. [Google Scholar] [CrossRef] [PubMed]
- Noguch, T.; Arakawa, O. Tetrodotoxin—Distribution and Accumulation in Aquatic Organisms, and Cases of Human Intoxication. Mar. Drugs 2008, 6, 220–242. [Google Scholar] [CrossRef]
- Katikou, P.; Gokbulut, C.; Kosker, A.R.; Campàs, M.; Ozogul, F. An Updated Review of Tetrodotoxin and Its Peculiarities. Mar. Drugs 2022, 20, 47. [Google Scholar] [CrossRef]
- Zhang, X.; Qiao, K.; Cui, R.; Xu, M.; Cai, S.; Huang, Q.; Liu, Z. Tetrodotoxin: The State-of-the-Art Progress in Characterization, Detection, Biosynthesis, and Transport Enrichment. Mar. Drugs 2024, 22, 531. [Google Scholar] [CrossRef]
- Matsumura, K. A monoclonal antibody against tetrodotoxin that reacts to the active group for the toxicity. Eur. J. Pharmacol. 1995, 293, 41–45. [Google Scholar]
- Shen, H.; Li, Z.; Jiang, Y.; Pan, X.; Wu, J.; Cristofori-Armstrong, B.; Smith, J.J.; Chin, Y.K.Y.; Lei, J.; Zhou, Q.; et al. Structural basis for the modulation of voltage-gated sodium channels by animal toxins. Science 2018, 362, eaau2596. [Google Scholar]
- Mattei, C. Tetrodotoxin, a Candidate Drug for Nav1.1-Induced Mechanical Pain? Mar. Drugs 2018, 16, 72. [Google Scholar] [CrossRef]
- Nieto, F.R.; Cobos, E.J.; Tejada, M.Á.; Sánchez-Fernández, C.; González-Cano, R.; Cendán, C.M. Tetrodotoxin (TTX) as a Therapeutic Agent for Pain. Mar. Drugs 2012, 10, 281–305. [Google Scholar] [CrossRef]
- Berde, C.B.; Athiraman, U.; Yahalom, B.; Zurakowski, D.; Corfas, G.; Bognet, C. Tetrodotoxin-Bupivacaine-Epinephrine Combinations for Prolonged Local Anesthesia. Mar. Drugs 2011, 9, 2717–2728. [Google Scholar] [CrossRef]
- Huerta, M.Á.; de la Nava, J.; Artacho-Cordón, A.; Nieto, F.R. Efficacy and Security of Tetrodotoxin in the Treatment of Cancer-Related Pain: Systematic Review and Meta-Analysis. Mar. Drugs 2023, 21, 316. [Google Scholar] [CrossRef]
- Park, J.; Proux, C.; Ehanno, W.; Réthoré, L.; Vessières, E.; Bourreau, J.; Favre, J.; Kauffenstein, G.; Mattei, C.; Tricoire-Leignel, H.; et al. Tetrodotoxin Decreases the Contractility of Mesenteric Arteries, Revealing the Contribution of Voltage-Gated Na+ Channels in Vascular Tone Regulation. Mar. Drugs 2023, 21, 196. [Google Scholar] [CrossRef] [PubMed]
- Hong, B.; He, J.; Le, Q.; Bai, K.; Chen, Y.; Huang, W. Combination Formulation of Tetrodotoxin and Lidocaine as a Potential Therapy for Severe Arrhythmias. Mar. Drugs 2019, 17, 685. [Google Scholar] [CrossRef] [PubMed]
- Yotsu-Yamashita, M.; Abe, Y.; Kudo, Y.; Ritson-Williams, R.; Paul, V.J.; Konoki, K.; Cho, Y.; Adachi, M.; Imazu, T.; Nishikawa, T.; et al. First Identification of 5,11-Dideoxytetrodotoxin in Marine Animals, and Characterization of Major Fragment Ions of Tetrodotoxin and Its Analogs by High Resolution ESI-MS/MS. Mar. Drugs 2013, 11, 2799–2813. [Google Scholar] [CrossRef] [PubMed]
- Bane, V.; Lehane, M.; Dikshit, M.; O’Riordan, A.; Furey, A. Tetrodotoxin: Chemistry, Toxicity, Source, Distribution and Detection. Toxins 2014, 6, 693–755. [Google Scholar] [CrossRef]
- Rossignoli, A.E.; Mariño, C.; Martín, H.; Blanco, J. First Report of Two Gymnodimines and Two Tetrodotoxin Analogues in Invertebrates from the North Atlantic Coast of Spain. Mar. Drugs 2023, 21, 232. [Google Scholar] [CrossRef]
- Kudo, Y.; Yasumoto, T.; Konoki, K.; Cho, Y.; Yotsu-Yamashita, M. Isolation and Structural Determination of the First 8-epi-type Tetrodotoxin Analogs from the Newt, Cynops ensicauda popei, and Comparison of Tetrodotoxin Analogs Profiles of This Newt and the Puffer Fish, Fugu poecilonotus. Mar. Drugs 2012, 10, 655–667. [Google Scholar] [CrossRef]
- Yu, V.C.-H.; Yu, P.H.-F.; Ho, K.-C.; Lee, F.W.-F. Isolation and Identification of a New Tetrodotoxin-Producing Bacterial Species, Raoultella terrigena, from Hong Kong Marine Puffer Fish Takifugu niphobles. Mar. Drugs 2011, 9, 2384–2396. [Google Scholar] [CrossRef]
- Arakawa, O.; Takatani, T.; Taniyama, S.; Tatsuno, R. Toxins of pufferfish—Distribution, accumulation mechanism, and physiologic functions. Aqua-BioScience Monogr. 2017, 10, 41–80. [Google Scholar] [CrossRef]
- Mosher, H.S.; Fuhrman, F.A.; Buchwald, H.D.; Fischer, H.G. Tarichatoxin-tetrodotoxin, a potent neurotoxin. Science 1964, 144, 1100–1110. [Google Scholar] [CrossRef]
- Ueda, H.; Itoi, S.; Sugita, H. TTX-Bearing Planocerid Flatworm (Platyhelminthes: Acotylea) in the Ryukyu Islands, Japan. Mar. Drugs 2018, 16, 37. [Google Scholar] [CrossRef]
- Jal, S.; Khora, S.S. An overview on the origin and production of tetrodotoxin, a potent neurotoxin. J. Appl. Microbiol. 2015, 119, 907–916. [Google Scholar] [PubMed]
- Noguchi, T.; Arakawa, O.; Takatani, T. TTX accumulation in pufferfish. Comp. Biochem. Physiol. Part D Genom. Proteom. 2006, 1, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, P.; Wu, B.; Hou, J.; Ren, J.; Zhu, Y.; Xu, J.; Si, F.; Sun, Z.; Liu, X. Transcriptomic analysis reveals the genes involved in tetrodotoxin (TTX) accumulation, translocation, and detoxification in the pufferfish Takifugu rubripes. Chemosphere 2022, 303, 134962. [Google Scholar] [CrossRef] [PubMed]
- Yotsu-Yamashita, M.; Nagaoka, Y.; Muramoto, K.; Cho, Y.; Konoki, K. Pufferfish Saxitoxin and Tetrodotoxin Binding Protein (PSTBP) Analogues in the Blood Plasma of the Pufferfish Arothron nigropunctatus, A. hispidus, A. manilensis, and Chelonodon patoca. Mar. Drugs 2018, 16, 224. [Google Scholar] [CrossRef]
- Chau, R.; Kalaitzis, J.A.; Wood, S.A.; Neilan, B.A. Diversity and Biosynthetic Potential of Culturable Microbes Associated with Toxic Marine Animals. Mar. Drugs 2013, 11, 2695–2712. [Google Scholar] [CrossRef]
- Noguchi, T.; Hwang, D.F.; Arakawa, O.; Sugita, H.; Deguchi, Y.; Shida, Y.; Hashimoto, K. Vibrio alginolyticus, a tetrodotoxin-producing bacterium, in the intestines of the fish Fugu vermicularis vermicularis. Mar. Biol. 1987, 94, 625–630. [Google Scholar]
- Cheng, C.A.; Hwang, D.F.; Tsai, Y.H.; Chen, H.C.; Jeng, S.S.; Noguchi, T.; Ohwada, K.; Hasimoto, K. Microflora and tetrodotoxin-producing bacteria in a gastropod, Niotha clathrata. Food Chem. Toxicol. 1995, 33, 929–934. [Google Scholar]
- Wu, Z.; Xie, L.; Xia, G.; Zhang, J.; Nie, Y.; Hu, J.; Wang, S.; Zhang, R. A new tetrodotoxin-producing actinomycete, Nocardiopsis dassonvillei, isolated from the ovaries of puffer fish Fugu rubripes. Toxicon 2005, 45, 851–859. [Google Scholar]
- Ritchie, K.B.; Nagelkerken, I.; James, S.; Smith, G.W. A tetrodotoxin-producing marine pathogen. Nature 2000, 404, 354. [Google Scholar] [CrossRef]
- Wang, X.J.; Yu, R.C.; Luo, X.; Zhou, M.J.; Lin, X.T. Toxin-screening and identification of bacteria isolated from highly toxic marine gastropod Nassarius semiplicatus. Toxicon 2008, 52, 55–61. [Google Scholar] [CrossRef]
- Miyazawa, K.; Noguchi, T. Distribution and origin of tetrodotoxin. J. Toxicol. Toxin Rev. 2001, 20, 11–33. [Google Scholar]
- Demain, A.L. Induction of microbial secondary metabolism. Int. Microbiol. 1998, 1, 259–264. [Google Scholar] [PubMed]
- Strand, M.; Hedström, M.; Seth, H.; McEvoy, E.G.; Jacobsson, E.; Göransson, U.; Andersson, H.S.; Sundberg, P. The Bacterial (Vibrio alginolyticus) Production of Tetrodotoxin in the Ribbon Worm Lineus longissimus—Just a False Positive? Mar. Drugs 2016, 14, 63. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, K. Reexamination of tetrodotoxin production by bacteria. Appl. Environ. Microbiol. 1995, 61, 3468–3470. [Google Scholar]
- Johnson, J.S.; Spakowicz, D.J.; Hong, B.Y.; Petersen, L.M.; Demkowicz, P.; Chen, L.; Leopold, S.R.; Hanson, B.M.; Agresta, H.O.; Gerstein, M.; et al. Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun. 2019, 10, 5029. [Google Scholar]
- Yotsu-Yamashita, M.; Mebs, D.; Flachsenberger, W. Distribution of tetrodotoxin in the body of the blue-ringed octopus (Hapalochlaena maculosa). Toxicon 2007, 49, 410–412. [Google Scholar]
- Tsai, Y.H.; Hwang, D.F.; Chai, T.J.; Jeng, S.S. Occurrence of tetrodotoxin and paralytic shellfish poison in the Taiwanese crab Lophozozymus pictor. Toxicon 1995, 33, 1669–1673. [Google Scholar]
- Yotsu-Yamashita, M.; Mebs, D. The levels of tetrodotoxin and its analogue 6-epitetrodotoxin in the red-spotted newt, Notophthalmus viridescens. Toxicon 2001, 39, 1261–1263. [Google Scholar]
- Kosker, A.R.; Özogul, F.; Ayas, D.; Durmus, M.; Ucar, Y.; Regenstein, J.M.; Özogul, Y. Tetrodotoxin levels of three pufferfish species (Lagocephalus sp.) caught in the North-Eastern Mediterranean sea. Chemosphere 2019, 219, 95–99. [Google Scholar]
- Kosker, A.R.; Özogul, F.; Durmus, M.; Ucar, Y.; Ayas, D.; Šimat, V.; Özogul, Y. First report on TTX levels of the yellow spotted pufferfish (Torquigener flavimaculosus) in the Mediterranean Sea. Toxicon 2018, 148, 101–106. [Google Scholar]
- Roeselers, G.; Mittge, E.K.; Stephens, W.Z.; Parichy, D.M.; Cavanaugh, C.M.; Guillemin, K.; Rawls, J.F. Evidence for a core gut microbiota in the zebrafish. ISME J. 2011, 5, 1595–1608. [Google Scholar] [PubMed]
- Llewellyn, M.S.; Boutin, S.; Hoseinifar, S.H.; Derome, N. Teleost microbiomes: The state of the art in their characterization, manipulation and importance in aquaculture and fisheries. Front. Microbiol. 2014, 5, 207. [Google Scholar]
- Ruff, W.E.; Greiling, T.M.; Kriegel, M.A. Host-microbiota interactions in immune-mediated diseases. Nat. Rev. Microbiol. 2020, 18, 521–538. [Google Scholar]
- Kiron, V. Fish immune system and its nutritional modulation for preventive health care. Anim. Feed Sci. Technol. 2012, 173, 111–133. [Google Scholar]
- Kogut, M.H.; Lee, A.; Santin, E. Microbiome and pathogen interaction with the immune system. Poult. Sci. 2020, 99, 1906–1913. [Google Scholar]
- Kurilshikov, A.; Wijmenga, C.; Fu, J.; Zhernakova, A. Host Genetics and Gut Microbiome: Challenges and Perspectives. Trends Immunol. 2017, 38, 633–647. [Google Scholar]
- Zhou, Y.L.; He, G.L.; Jin, T.; Chen, Y.J.; Dai, F.Y.; Luo, L.; Lin, S.M. High dietary starch impairs intestinal health and microbiota of largemouth bass, Micropterus salmoides. Aquaculture 2021, 534, 736261. [Google Scholar]
- Joe, J.T.X.; Chiou, P.P.; Kuo, C.Y.; Lin, J.H.J.; Wu, J.L.; Lu, M.W. The microbiota profile and transcriptome analysis of immune response during metamorphosis stages in orange spotted grouper (Epinephelus coioides). Fish Shellfish Immunol. 2019, 90, 141–149. [Google Scholar]
- Wassel, M.A.; Makabe-Kobayashi, Y.; Iqbal, M.M.; Takatani, T.; Sakakura, Y.; Hamasaki, K. The impact of tetrodotoxin (TTX) on the gut microbiome in juvenile tiger pufferfish, Takifugu rubripes. Sci. Rep. 2024, 14, 16684. [Google Scholar]
- Li, Z.; Tian, J.; Lai, Y.; Lee, C.-H.; Cai, Z.; Yu, C.-F. Puffer Fish Gut Microbiota Studies Revealed Unique Bacterial Co-Occurrence Patterns and New Insights on Tetrodotoxin Producers. Mar. Drugs 2020, 18, 278. [Google Scholar] [CrossRef]
- Melnikova, D.I.; Magarlamov, T.Y. The Microbial Community of Tetrodotoxin-Bearing and Non-Tetrodotoxin-Bearing Ribbon Worms (Nemertea) from the Sea of Japan. Mar. Drugs 2020, 18, 177. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Zhang, Z.; Xing, Z.; Li, Q.; Kong, N.; Wang, L.; Song, L. The variation of intestinal autochthonous bacteria in cultured tiger pufferfish Takifugu rubripes. Front. Cell. Infect. Microbiol. 2022, 12, 1062512. [Google Scholar]
- Yu, G.; Ou, W.; Liao, Z.; Xu, H.; Liang, M.; Zhang, Y.; Mai, K. Intestinal homeostasis of juvenile tiger puffer Takifugu rubripes was sensitive to dietary arachidonic acid in terms of mucosal barrier and microbiota. Aquaculture 2019, 502, 97–106. [Google Scholar]
- Jin, X.; Chen, Z.; Shi, Y.; Gui, J.; Zhao, Z. Response of gut microbiota to feed-borne bacteria depends on fish growth rate: A snapshot survey of farmed juvenile Takifugu obscurus. Microb. Biotechnol. 2002, 15, 683–702. [Google Scholar]
- Riazati, N.; Kable, M.E.; Stephensen, C.B. Association of intestinal bacteria with immune activation in a cohort of healthy adults. Microbiol. Spectr. 2023, 11, e0102723. [Google Scholar]
- Noguchi, T.; Jeon, J.K.; Arakawa, O.; Sugita, H.; Deguchi, Y.; Shida, Y.; Hashimoto, K. Occurrence of tetrodotoxin and anhydrotetrodotoxin in Vibrio sp. isolated from the intestines of a xanthid crab (Atergatis floridus). J. Biochem. 1986, 99, 311–314. [Google Scholar]
- Narita, H.; Matsubara, S.; Miwa, N.; Akahane, S.; Marukami, M.; Goto, T.; Nara, M.; Noguchi, T.; Saito, T.; Shida, Y.; et al. Vibrio alginolyticus, a TTX-producing bacterium isolated from the Starfish (Astropecten polycanthus). Nippon Suisan Gakkaishi 1987, 53, 617–621. [Google Scholar]
- Sugita, H.; Ueda, R.; Noguchi, T.; Arakawa, O.; Hashimoto, K.; Deguchi, Y. Identification of a tetrodotoxinproducing bacterium isolated from the xanthid crab (Atergatis floridus). Nippon Suisan Gakkaishi 1987, 53, 1693. [Google Scholar]
- Carroll, S.; McEvoy, E.; Gibson, R. The production of tetrodotoxin-like substances by nemertean worms in conjunction with bacteria. J. Exp. Mar. Biol. Ecol. 2003, 288, 51–63. [Google Scholar]
- Bowman, J.P. Bioactive Compound Synthetic Capacity and Ecological Significance of Marine Bacterial Genus Pseudoalteromonas. Mar. Drugs 2007, 5, 220–241. [Google Scholar] [CrossRef]
- Melnikova, D.I.; Beleneva, I.A.; Tyunin, A.P.; Magarlamov, T.Y. The taxonomic composition, characteristics, and neurotoxic activities of ribbon worm-associated bacteria from the Sea of Japan. Russ. J. Mar. Biol. 2017, 43, 383–391. [Google Scholar]
- Zhang, D.; Yu, Z.; Zeng, B.; Liu, X. Genome-wide analysis of the ABC gene family in almond and functional predictions during flower development, freezing stress, and salt stress. BMC Plant Biol. 2024, 24, 12. [Google Scholar]
- Romersi, R.F.; Nicklisch, S.C.T. Interactions of Environmental Chemicals and Natural Products with ABC and SLC Transporters in the Digestive System of Aquatic Organisms. Front. Physiol. 2022, 12, 767766. [Google Scholar]
- Wang, H.; Liu, S.; Xun, X.; Li, M.; Lou, J.; Zhang, Y.; Shi, J.; Hu, J.; Bao, Z.; Hu, X. Toxin- and species-dependent regulation of ATP-binding cassette (ABC) transporters in scallops after exposure to paralytic shellfish toxin-producing dinoflagellates. Aquat. Toxicol. 2021, 230, 105697. [Google Scholar]
- Zhu, B.; Bai, Y.; Yeo, Y.Y.; Lu, X.; Rovira-Clavé, X.; Chen, H.; Yeung, J.; Nkosi, D.; Glickman, J.; Delgado-Gonzalez, A.; et al. A multi-omics spatial framework for host-microbiome dissection within the intestinal tissue microenvironment. Nat. Commun. 2025, 16, 1230. [Google Scholar]
- GB 5009.206-2016; National Standard of the People’s Republic of China GB 5009.206-2016 Determination of Tetrodotoxin in Aquatic Products. China Food and Drug Administration, National Health and Family Planning Commission: Beijing, China, 2016.
- Zhou, F.; Liu, B.; Liu, X.; Li, Y.; Wang, L.; Huang, J.; Luo, G.; Wang, X. The Impact of Microbiome and Microbiota-Derived Sodium Butyrate on Drosophila Transcriptome and Metabolome Revealed by Multi-Omics Analysis. Metabolites 2021, 11, 298. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- White, J.R.; Nagarajan, N.; Pop, M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput. Biol. 2009, 5, e1000352. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Aßhauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, H.; Hou, J.; Zhu, Y.; Wu, B.; Ren, J.; Sun, Z.; Liu, X. Comparative Analysis of Intestinal Microbiota Between Tetrodotoxin-Containing and Tetrodotoxin-Free Takifugu rubripes. Mar. Drugs 2025, 23, 140. https://doi.org/10.3390/md23040140
Zhang H, Hou J, Zhu Y, Wu B, Ren J, Sun Z, Liu X. Comparative Analysis of Intestinal Microbiota Between Tetrodotoxin-Containing and Tetrodotoxin-Free Takifugu rubripes. Marine Drugs. 2025; 23(4):140. https://doi.org/10.3390/md23040140
Chicago/Turabian StyleZhang, Hanyuan, Jilun Hou, Youxiu Zhu, Biyin Wu, Jiangong Ren, Zhaohui Sun, and Xia Liu. 2025. "Comparative Analysis of Intestinal Microbiota Between Tetrodotoxin-Containing and Tetrodotoxin-Free Takifugu rubripes" Marine Drugs 23, no. 4: 140. https://doi.org/10.3390/md23040140
APA StyleZhang, H., Hou, J., Zhu, Y., Wu, B., Ren, J., Sun, Z., & Liu, X. (2025). Comparative Analysis of Intestinal Microbiota Between Tetrodotoxin-Containing and Tetrodotoxin-Free Takifugu rubripes. Marine Drugs, 23(4), 140. https://doi.org/10.3390/md23040140