Characterizing the Anti-HIV Activity of Papuamide A

Abstract
: Papuamide A is representative of a class of marine derived cyclic depsipeptides, reported to have cytoprotective activity against HIV-1 in vitro. We show here that papuamide A acts as an entry inhibitor, preventing human immunodeficiency virus infection of host cells and that this inhibition is not specific to R5 or X4 tropic virus. This inhibition of viral entry was determined to not be due to papuamide A binding to CD4 or HIV gp120, the two proteins involved in the cell-virus recognition and binding. Furthermore, papuamide A was able to inhibit HIV pseudotype viruses expressing envelope glycoproteins from vesicular stomatitis virus or amphotropic murine leukemia virus indicating the mechanism of viral entry inhibition is not HIV-1 envelope glycoprotein specific. Time delayed addition studies with the pseudotyped viruses show that papuamide A inhibits viral infection only at the initial stage of the viral life cycle. Additionally, pretreatment studies revealed that the virus, and not the cell, is the target of papuamide A’s action. Together, these results suggest a direct virucidal mechanism of HIV-1 inhibition by papuamide A. We also demonstrate here that the other papuamides (B-D) are able to inhibit viral entry indicating that the free amino moiety of 2,3-diaminobutanoic acid residue is not required for the virucidal activity.
1. Introduction
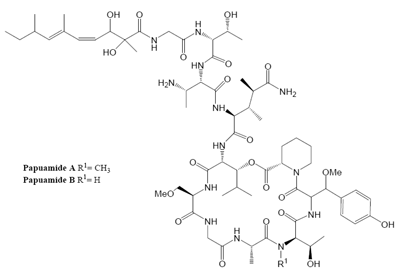
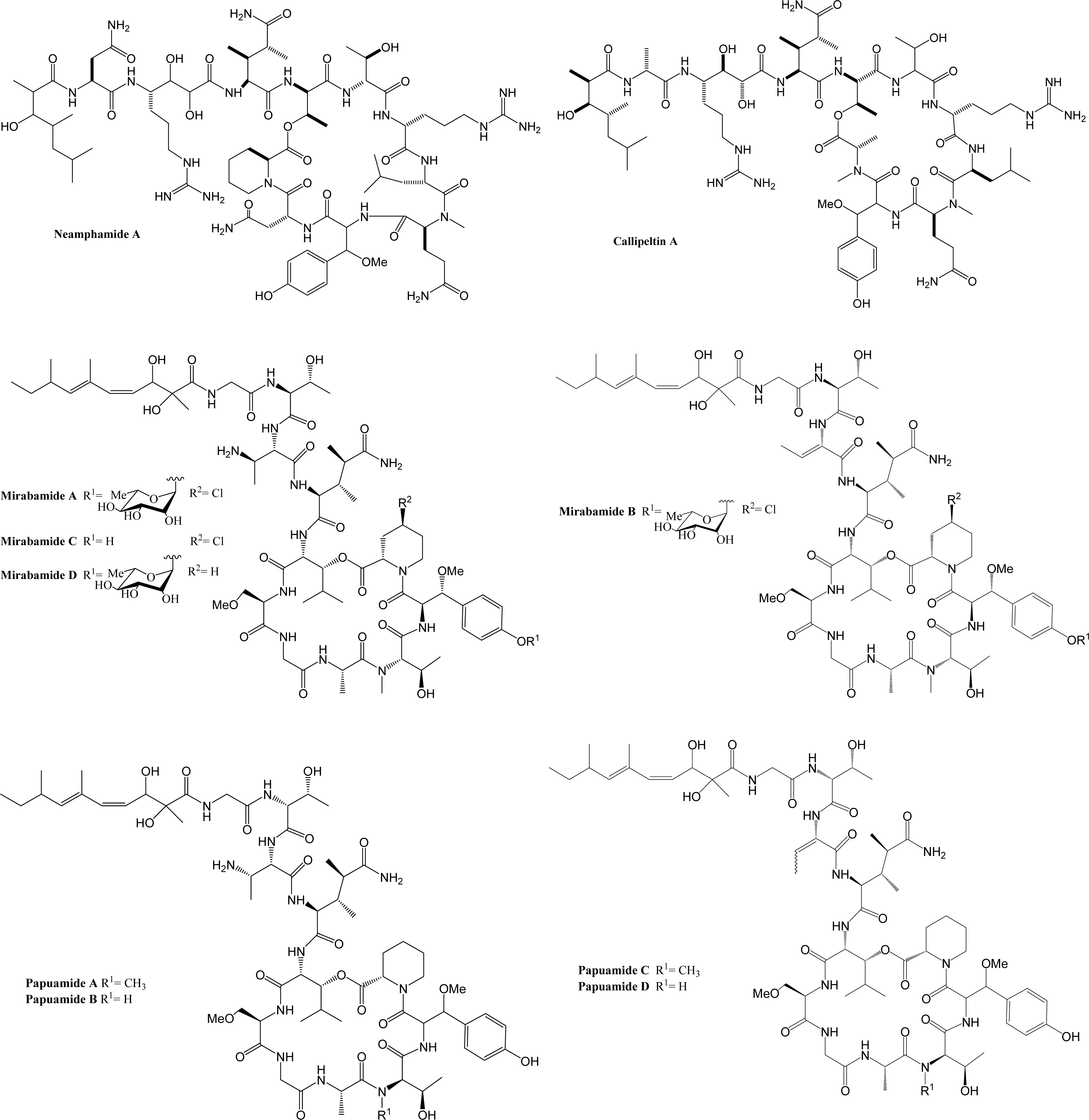
Marine organisms have proven to be an excellent source of biologically active compounds against human immunodeficiency virus (HIV) [1, 2]. One class of molecules with potent cytoprotective activity consists of structurally similar cyclic depsipeptides isolated from sponges. These marine metabolites exhibit potent inhibition of HIV induced T cell death in vitro with selectivity indexes (SI, effective dose divided by the cytotoxic dose) ranging from 3 to 29. Included in this group are: neamphamide A (SI=9.29) [3, 4], callipeltin A (SI=29) [4, 5], mirabamides A–D (SI ranging from 3–13) [6] and papuamide A and B (SI=21 for both) [4, 7]. (Chemical structures provided in Fig. 1.) The mirabamides are the most recently discovered of this group and have been reported to inhibit HIV-1 envelope mediated cell-cell fusion in a recombinant vaccinia virus fusion assay [6].
Problems associated with current therapeutic treatments and the emergence of drug resistant HIV strains [8] establishes the need for new therapeutic treatments. Agents which inhibit viral entry are particularly sought after because they can prevent infection. HIV entry is often described as a three stage process: attachment of the virus to the target cell, co-receptor binding, and fusion of the virus and cell membranes. Virus attachment is generally through HIV gp120 recognition and binding to the host cell receptor, CD4 [9, 10]. Once the virus is attached to the cell, gp120 will undergo conformational changes exposing a new epitope which then interacts with a cellular chemokine co-receptor [11–13], typically either CCR5 or CXCR4. After this dual receptor interaction has taken place, gp120 and gp41 will undergo further conformational changes exposing the extracellular region of gp41[12–16]. The N-terminal region of gp41 contains what has been called a fusion peptide that inserts itself into the cell membrane [17]. Two helical regions of gp41 fold upon each other, bringing the virus and cellular membranes into close proximity. The resultant free energy change from this structural rearrangement is thought to be sufficient for lipid mixing and fusion of the two membranes to occur, thus allowing the viral core to enter the host cell [18–20]. Compounds that inhibit any stage of this entry process may not only be useful for the treatment of HIV/AIDS infected individuals, but also have the potential to be used as microbicides in the absence of a vaccine.
The structural uniqueness of natural product metabolites often leads to the identification of new targets in the treatment of diseases and novel inhibitors of viral entry are a much desired class of drugs. Therefore, we investigated the mechanism by which papuamide A protects against HIV induced cytopathicity. In the present study, papuamide A’s ability to inhibit HIV entry is demonstrated. Papuamide A is shown to target the virus and this virucidal mechanism is investigated. In addition, papuamides B, C and D are shown to also inhibit HIV entry.
2. Results
2.2 Papuamide A does not show significant binding to either sCD4 or gp120.
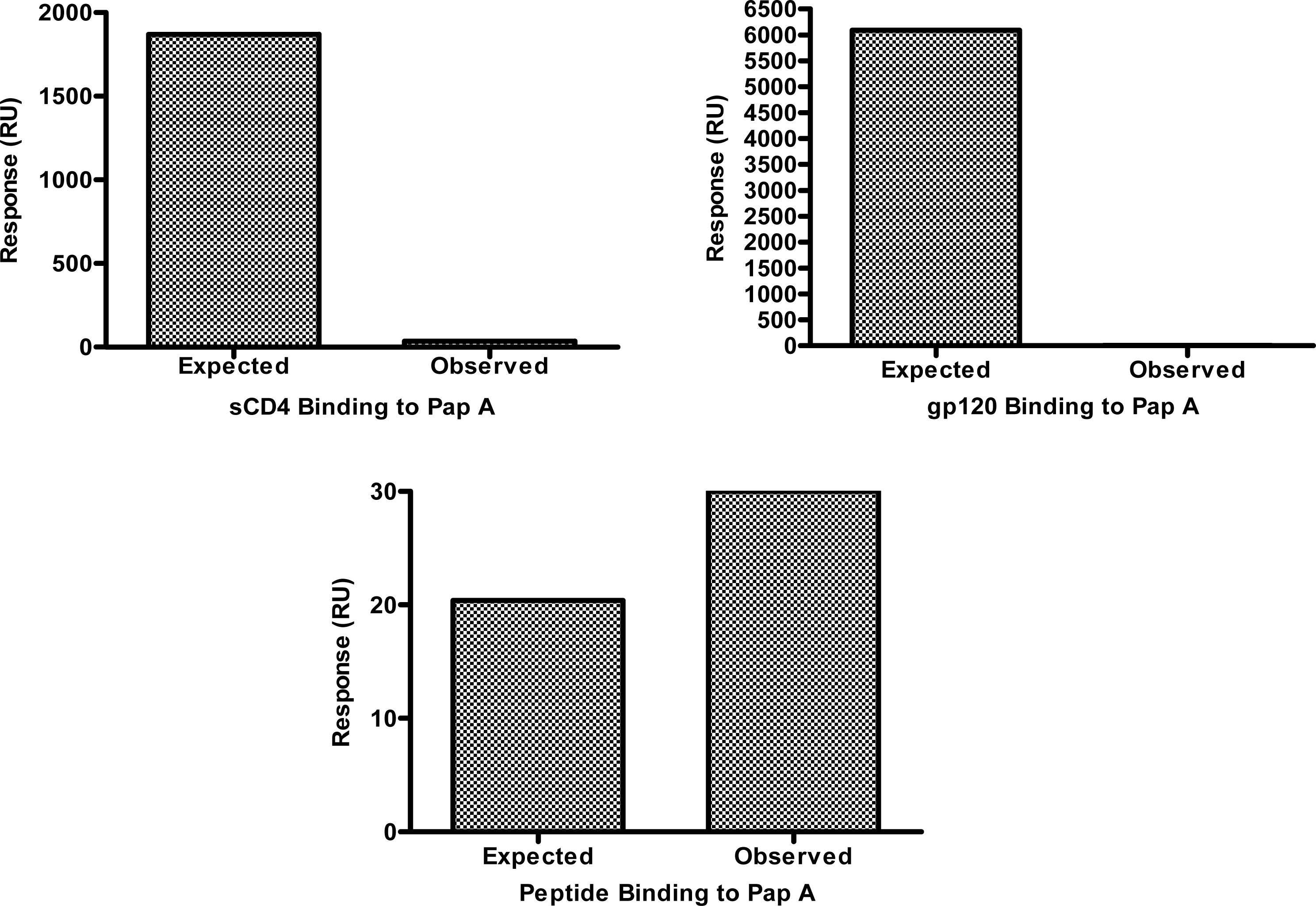
The lack of tropism specificity demonstrated above suggested that the chemokine co-receptors (CXCR4 and CCR5) and the co-receptor binding domain of gp120 are not the targets of papuamide A inhibition. However, the initial step of HIV entry, binding of gp120 to CD4, is thought to be similar regardless of viral tropism, therefore, the ability of papuamide A to interact with CD4 or gp120 was assessed. Successful biotinylation of papuamide A without a significant loss of activity was achieved (data not shown) and the biotinylated papuamide A was utilized to quantify binding of sCD4 or gp120 by surface plasmon resonance (SPR). Fig. 3 shows the observed binding response (response in RU measured by the instrument) versus the expected binding response (see formula and explanation below) for sCD4 and gp120 with papuamide A. SPR binding responses for both proteins were determined not to be significant because the observed response was 600 times lower than the calculated expected response for sCD4 at the maximum concentration tested, and the observed response for gp120 was 54 times lower than the calculated expected response at the maximum concentration tested. Papuamide A was shown to interact with a positive control binding peptide, as expected in this system.
Values used to determine the expected response are in Table 1. The expected response at saturation is determined by the following formula: , (LCR=Ligand capture response, amount of papuamide A captured onto the biosensor chip; MWL=molecular weight of ligand, papuamide A; and MWA=molecular weight of analyte, the protein or peptide.)[23–26] The observed response, determined by the BIAcore instrument detector in response units (RU), can be lower than the expected response if the concentration of analyte used is below the dissociation constant (KD) concentration value, KD values were calculated by the software from the binding responses obtained with multiple concentrations of analyte. When this is the case, the fold difference in concentration values (KD/concentration of analyte used) is approximately proportionate to the fold difference in responses (expected response/observe response). Therefore, to correct for the expected response at the maximum concentration tested, the expected response at saturation was divided by the fold difference in concentration values to determine the expected response at the maximum value tested.
2.3 Papuamide A does not exhibit virus envelope specificity.
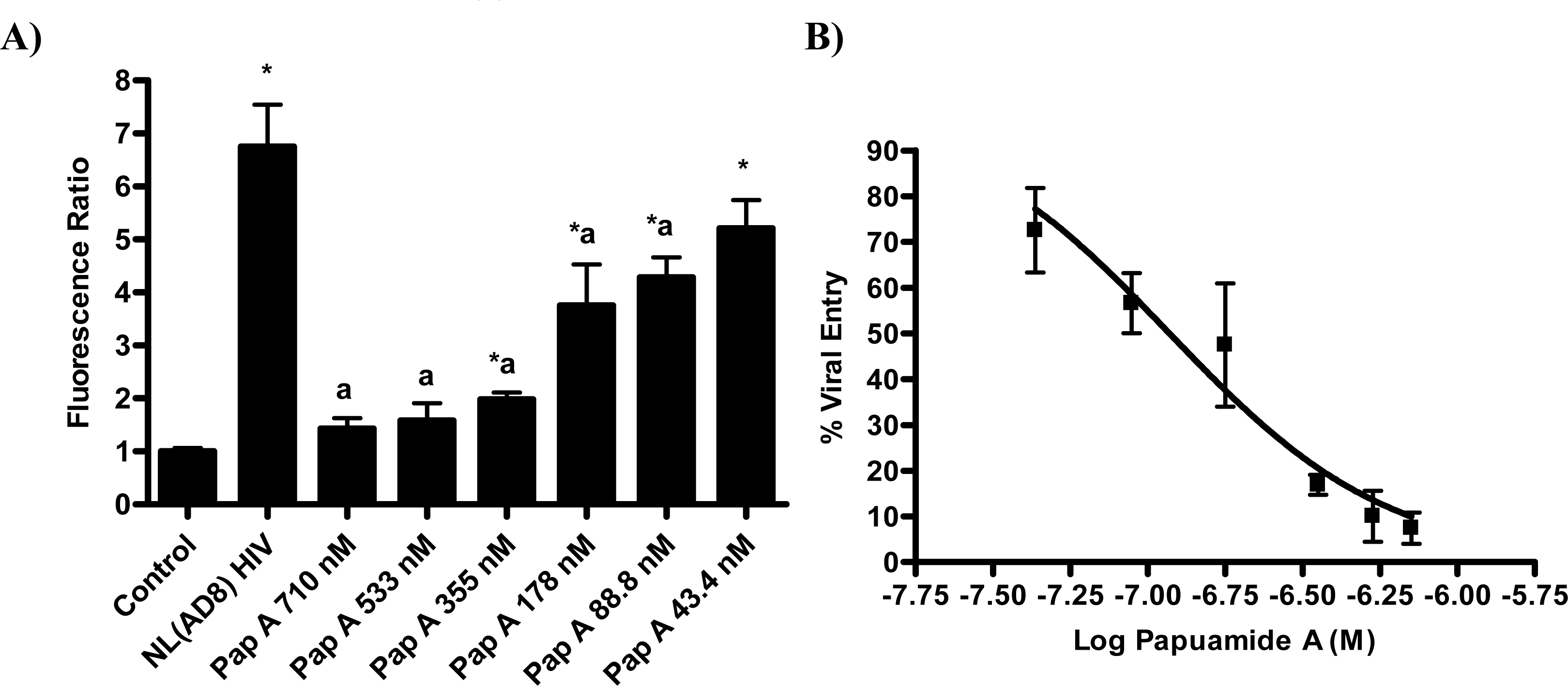
The proteins known to be critical for the initial stage of virus attachment to the cell do not appear to be the targets of papuamide A. This finding lead to the investigation of a more general mechanism of viral entry inhibition. Inhibition of infection was evaluated using multiple pseudotype viruses (engineered HIV virions bearing various envelope glycoproteins). CEM-SS, a human T4 lymphoblastoid cell line, was used for all infections except for those using pseudotype virus expressing JRFL, which requires the presence of CCR5. For the JRFL pseudotype virus experiments, CEM.NKR-CCR5, a CEM derived cell line expressing CCR5, was used. Pseudotype viruses were produced by co-transfection of envelope glycoprotein plasmid and an env-defective HIV vector, DHIV-3-GFP, [27] which contains a green fluorescence protein (GFP) reporter gene in place of nef (this allows for quantification of HIV infection by flow cytometry). Papuamide A was tested at a concentration of 174nM which was previously determined to inhibit approximately 50% of infection in this system (data not shown).
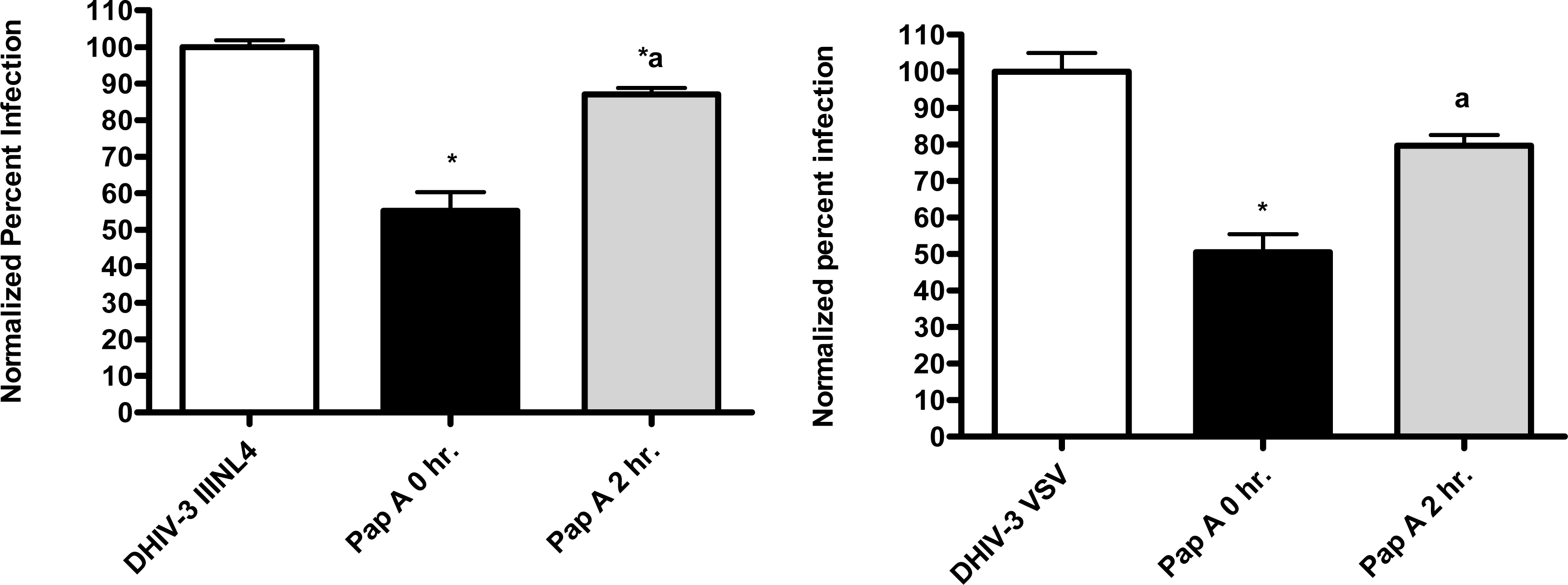
First, the ability of papuamide A to inhibit infection in this system was confirmed using viruses expressing LAI (X4 tropic) or JRFL (R5 tropic) envelope proteins (Fig. 4). This lack of viral tropism specificity was consistent with that observed in the virus entry inhibition studies above. Next, the defective HIV-GFP virus was pseudotyped with envelope glycoproteins from either vesicular stomatitis virus (VSV) or amphotropic murine leukemia virus (aMLV), and the sensitivity to papuamide A inhibition was tested. Papuamide A effectively inhibited infection by these pseudotype viruses (Fig. 4). These results confirm that papuamide A does not act by inhibiting the recognition and attachment of the virus to the cell and also suggests that papuamide A’s activity in not selective for gp41 mediated fusion. VSV and aMLV envelopes result in the virus entering through a receptor mediated endocytic pathway [27–30] and through the PIT-2 sodium phosphate co-transporter mediated pathway [31, 32], respectively.
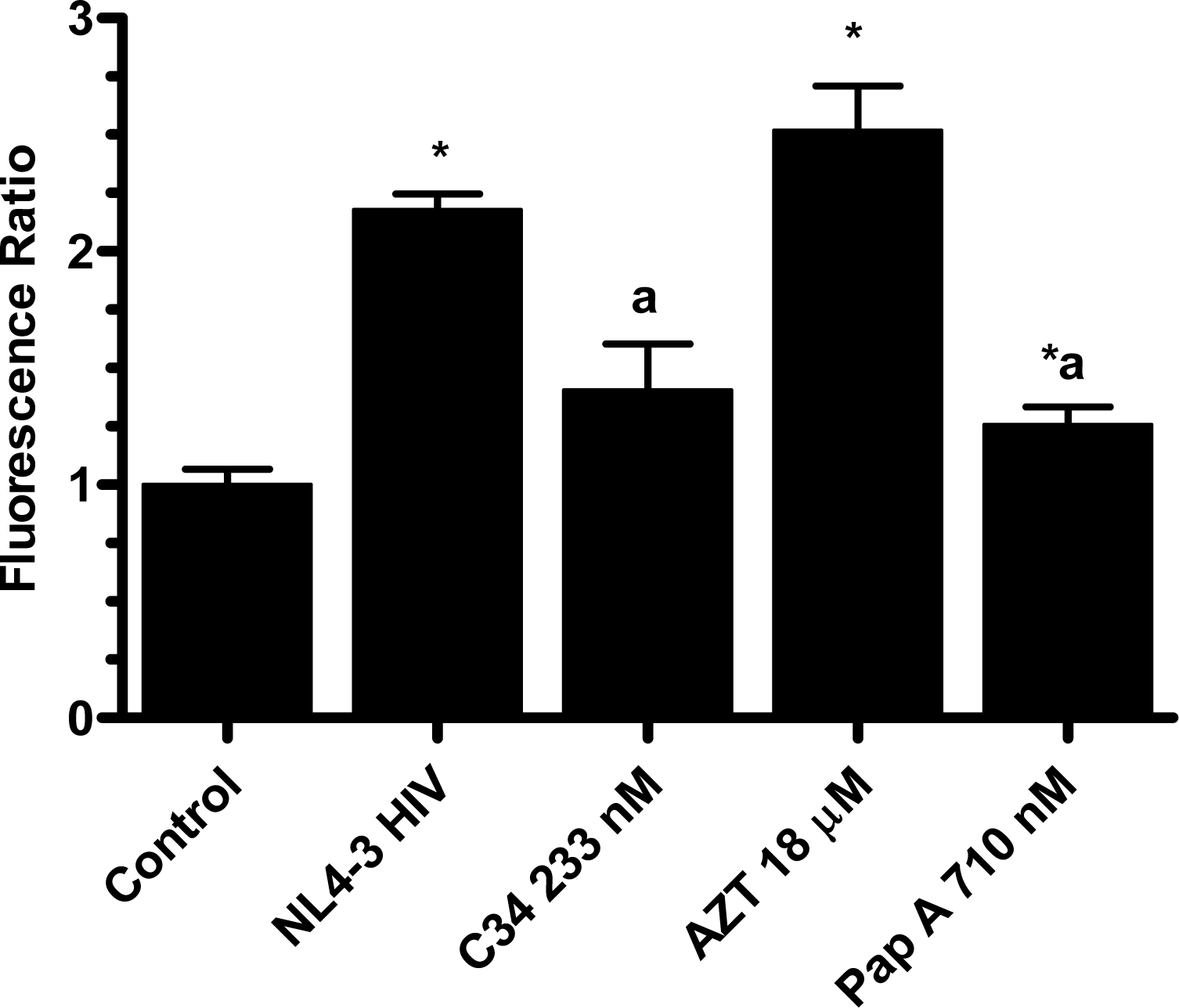
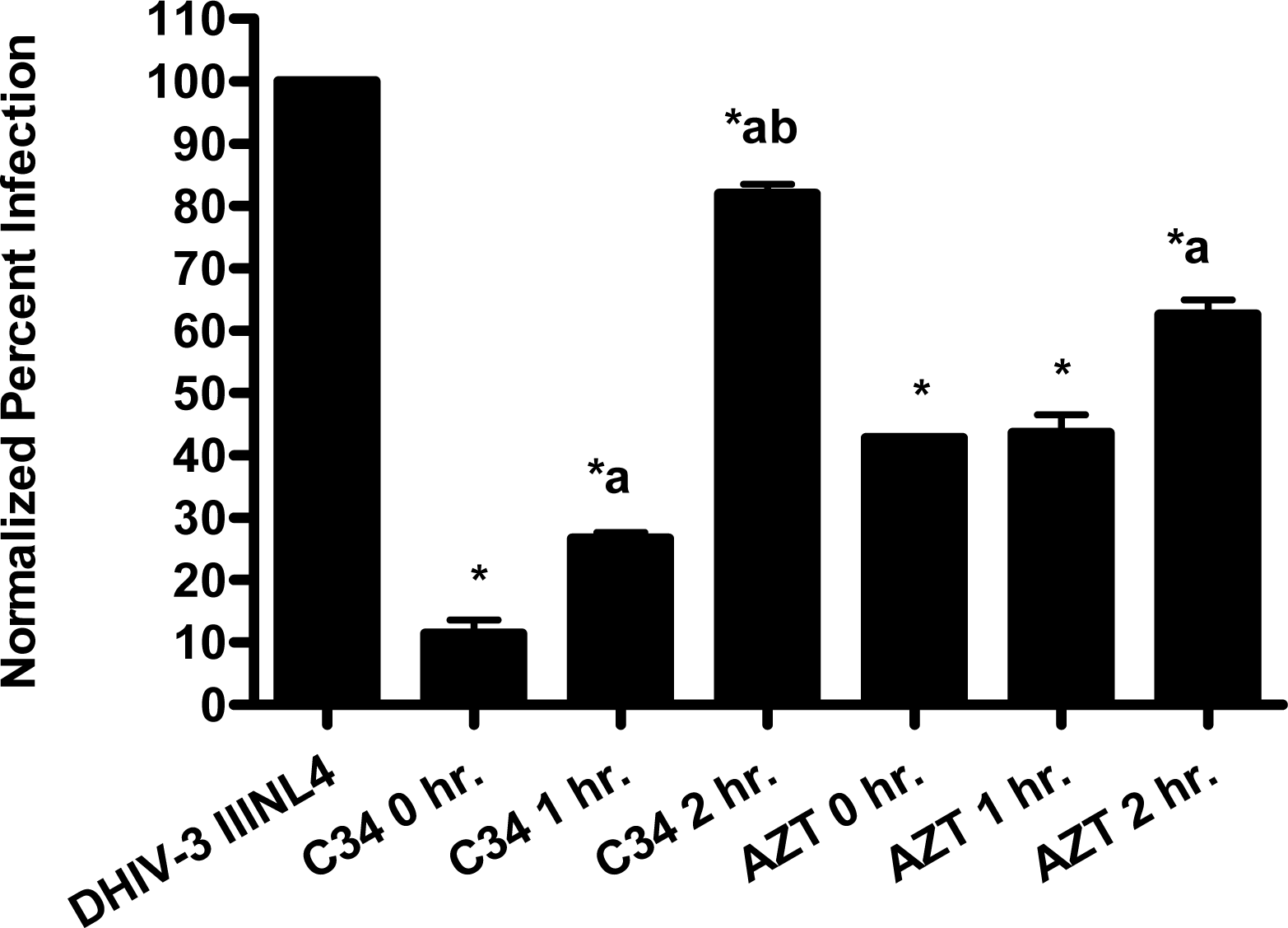
The pseudotype virus assay described above is a single round infectivity assay that can detect inhibition of early viral life cycle events. To confirm the activity of papuamide A in the pseudotyped virus system was due to inhibition of the initial viral entry steps, time delayed addition studies were performed. Compounds which are active during the entry process of the viral life cycle show a strong time dependency in their effectiveness. This is illustrated in Fig. 5a with the controls C34, an inhibitor of fusion, and AZT, a reverse transcriptase inhibitor. C34 showed a significant decrease in effectiveness when addition was delayed by 1 hour, with an even greater loss of activity when addition was delayed 2 hours. The profile of AZT showed no difference in activity when added 1 hour after infection, and only a minor loss of activity when added 2 hours after infection. Papuamide A’s profile (Fig. 5b) was similar to that of C34, indicating the observed activity is at the initial stages of the viral life cycle. A similar loss of inhibition due to delayed addition was also observed for pseudotype virus expressing VSV envelope glycoprotein when compared to pseudotype virus expressing an HIV envelope glycoprotein (Fig. 5c).
2.4 Papuamide A interacts directly with the virus, not the target cell, to inhibit infection.
Current FDA approved inhibitors of viral entry and the majority of those under investigation target CD4, gp120, the chemokine co-receptors, or gp41 mediated fusion.[33] Papuamide A’s inhibition of viral entry has been shown to be independent of these proteins and appears to be novel in its mechanism. To further evaluate papuamide A’s mechanism, pretreatment studies were performed to determine if papuamide A was interacting directly with the virus or the target cell.
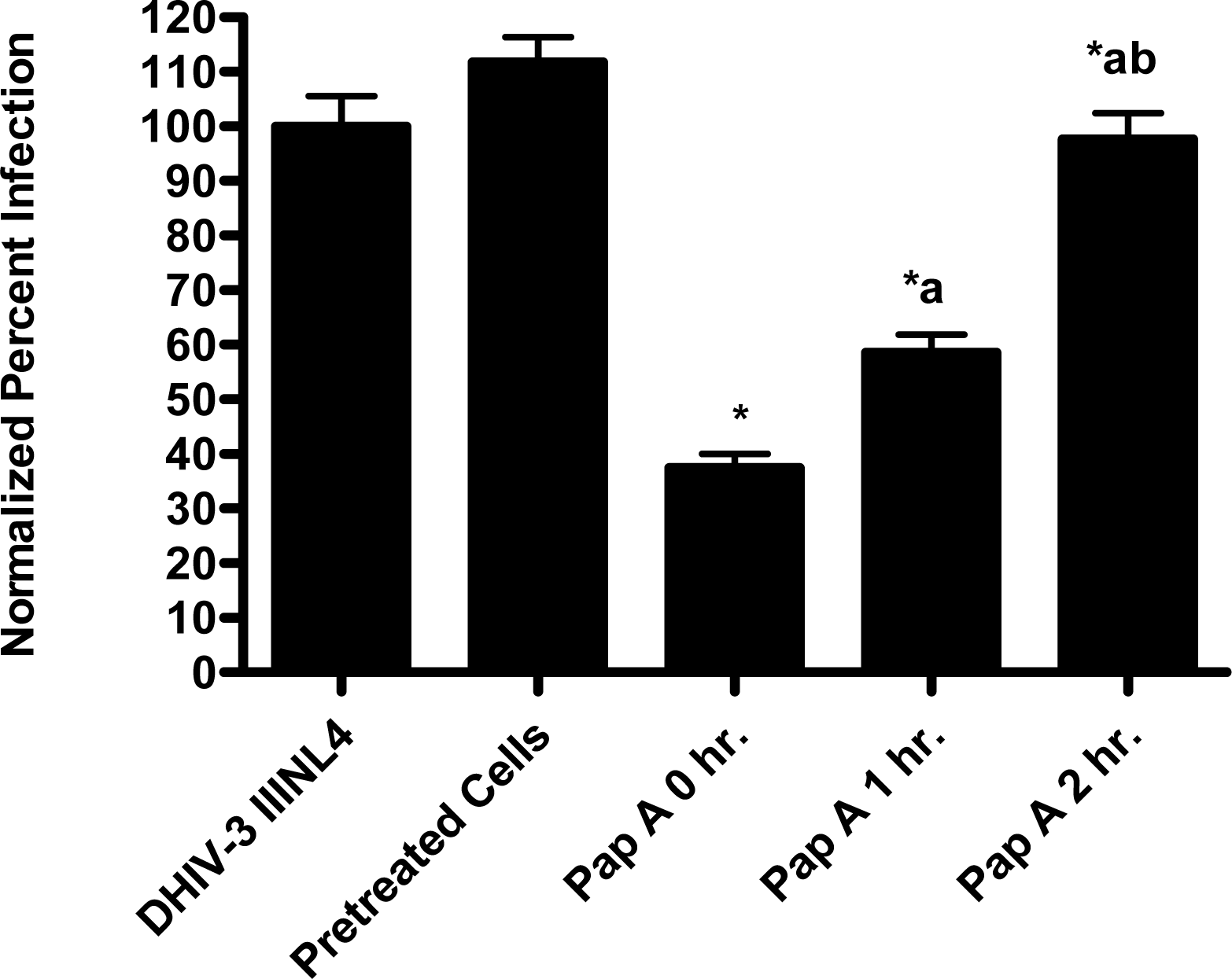
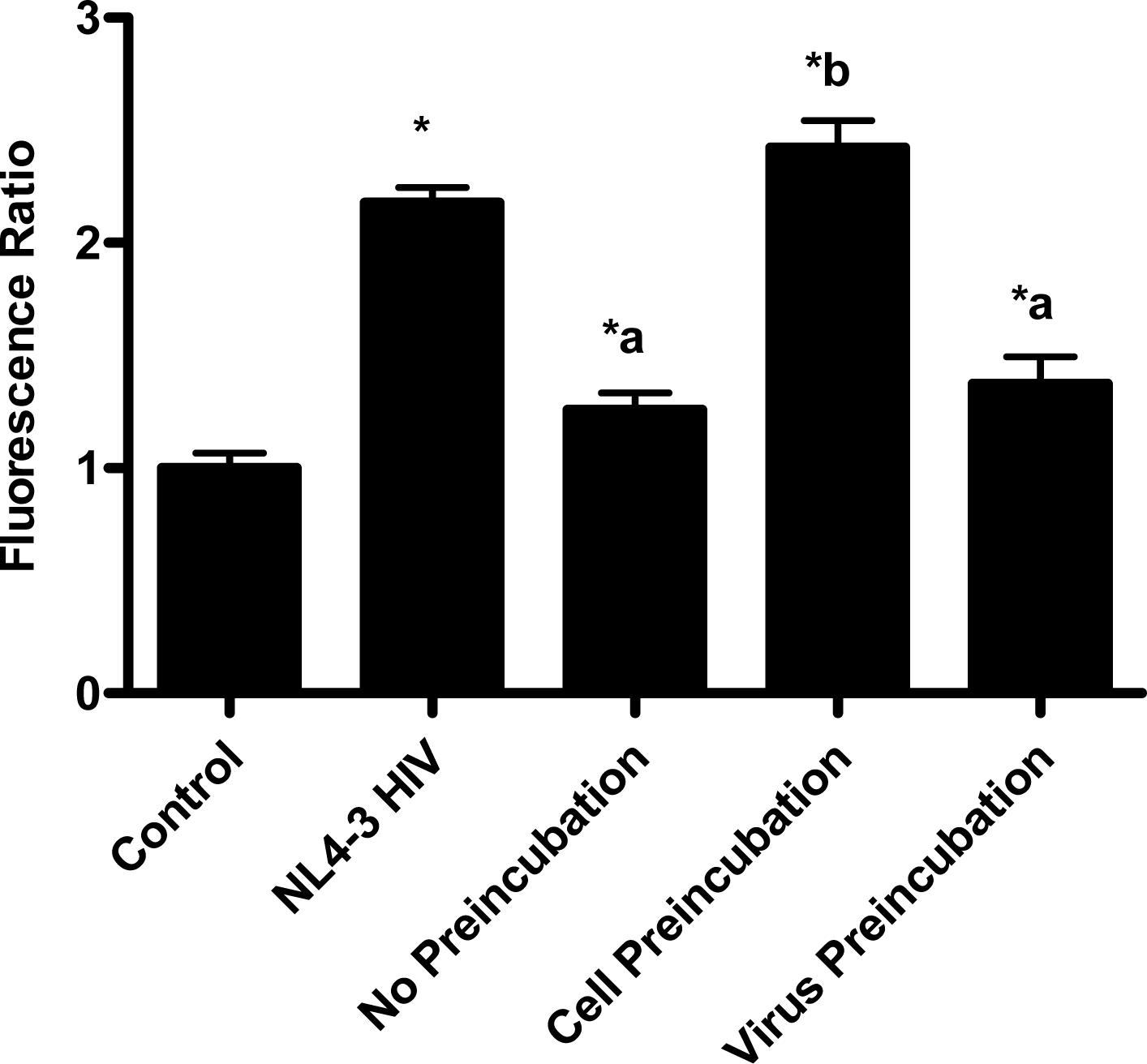
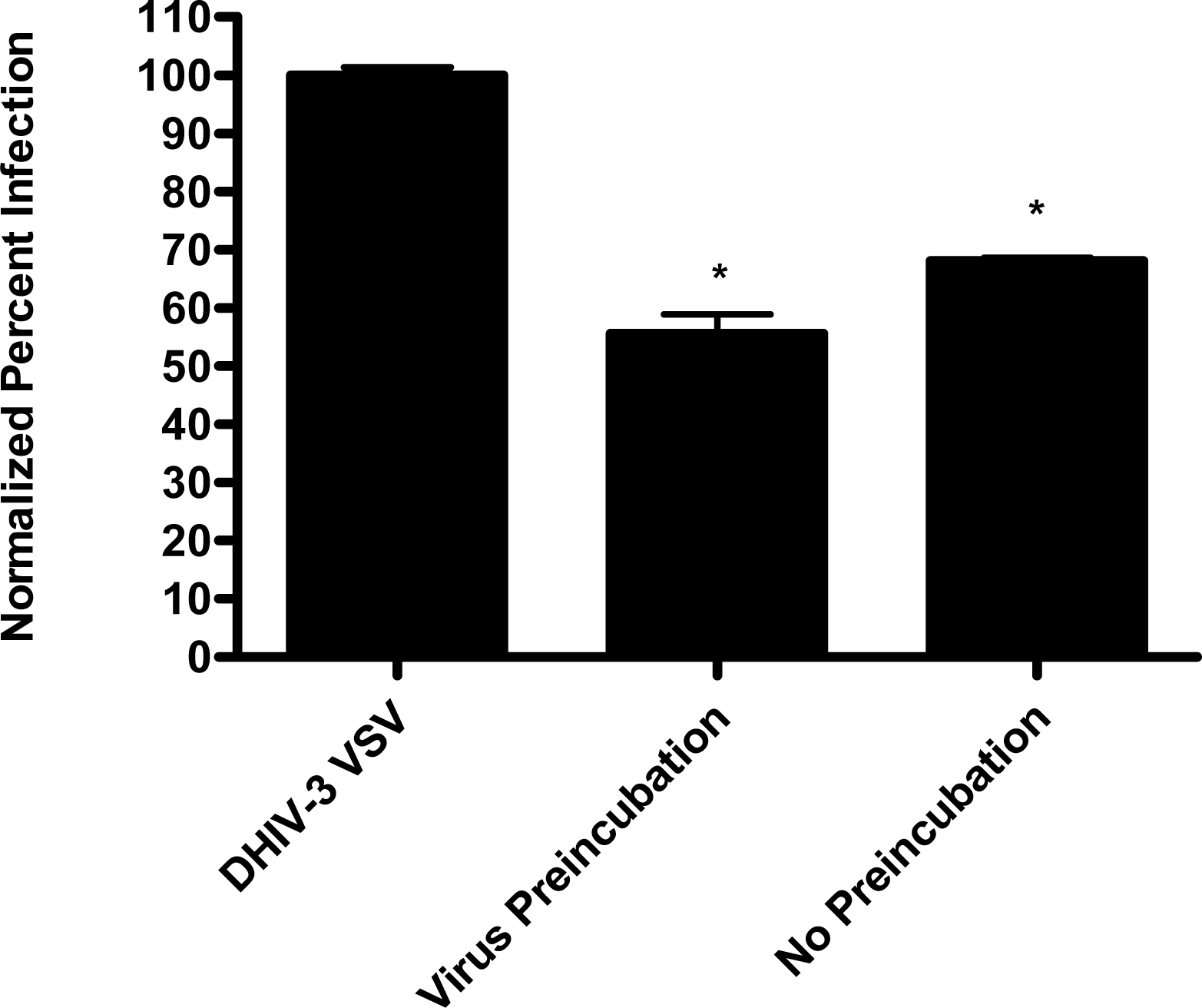
In the virion fusion assay, cells were incubated with papuamide A for 2 hours prior to infection. Papuamide A containing medium was removed followed by two washes. This pretreatment protocol did not result in inhibition of viral entry demonstrating that papuamide A does not have an irreversible effect on the cell (Fig. 6a). Preincubation of the cells with papuamide A overnight also failed to inhibit viral infection (data not shown). Modification of the fusion assay was performed so that HIV was pretreated with papuamide A for 30 minutes prior to infection. Virus pretreatment, followed by dilution of virus, resulted in a rate of inhibition similar to when papuamide A was added at the same time as virus (Fig. 6a). However, in this experiment papuamide A could only be diluted, not completely removed from the virus and medium. Effective removal of unbound papuamide A after virus incubation requires ultracentrifugation, but ultracentrifugation of HIV enveloped virus greatly decreases virus infectivity [34, 35]. To overcome this problem, VSV-G pseudotype virus was used for pretreatment studies because VSV-G pseudotype virus has been shown to retain infectivity after ultracentrifugation [34]. VSV-G pseudotype virus was treated with papuamide A and incubated overnight. Virus was pelleted, papuamide A containing supernatant removed and pellet suspended in fresh medium. Virus pretreatment with papuamide A resulted in an approximate 50% decreased infectivity of the virus (Fig. 6b). This decreased infectivity was the same as that observed when papuamide A was added to cells at the same time as VSV-G pseudotype virus (control virus underwent overnight incubation with vehicle and ultracentrifugation to control for potential loss of infectivity due to this process). These results indicate that papuamide A either remains stably bound in an inhibitory association with the virions after ultracentrifugation and washing or, alternatively, papuamide A inactivates the virus.
2.5 Effect of binding phosphatidylserine
Papuamide B only differs from papuamide A by one methyl group. Papuamide B has been shown to bind to and cause leakage of phosphatidylserine (PS) containing liposomes, but not those composed solely of phosphatidylcholine [36]. PS is a phospholipid normally present on the inner leaflet of the cell membrane, but during apoptosis the regulation of PS is altered and PS is present on the outer leaflet. Callahan et al. showed PS is present in the envelopes of HIV virions and reported that binding of PS by Annexin V, a specific binder of PS, was able to inhibit monocyte and macrophage infection, but not T cell infection [37]. These results suggest that binding of PS by papuamide A may disrupt the viral membrane. Therefore, the possibility of PS being the target of papuamide A’s virucidal activity was investigated.
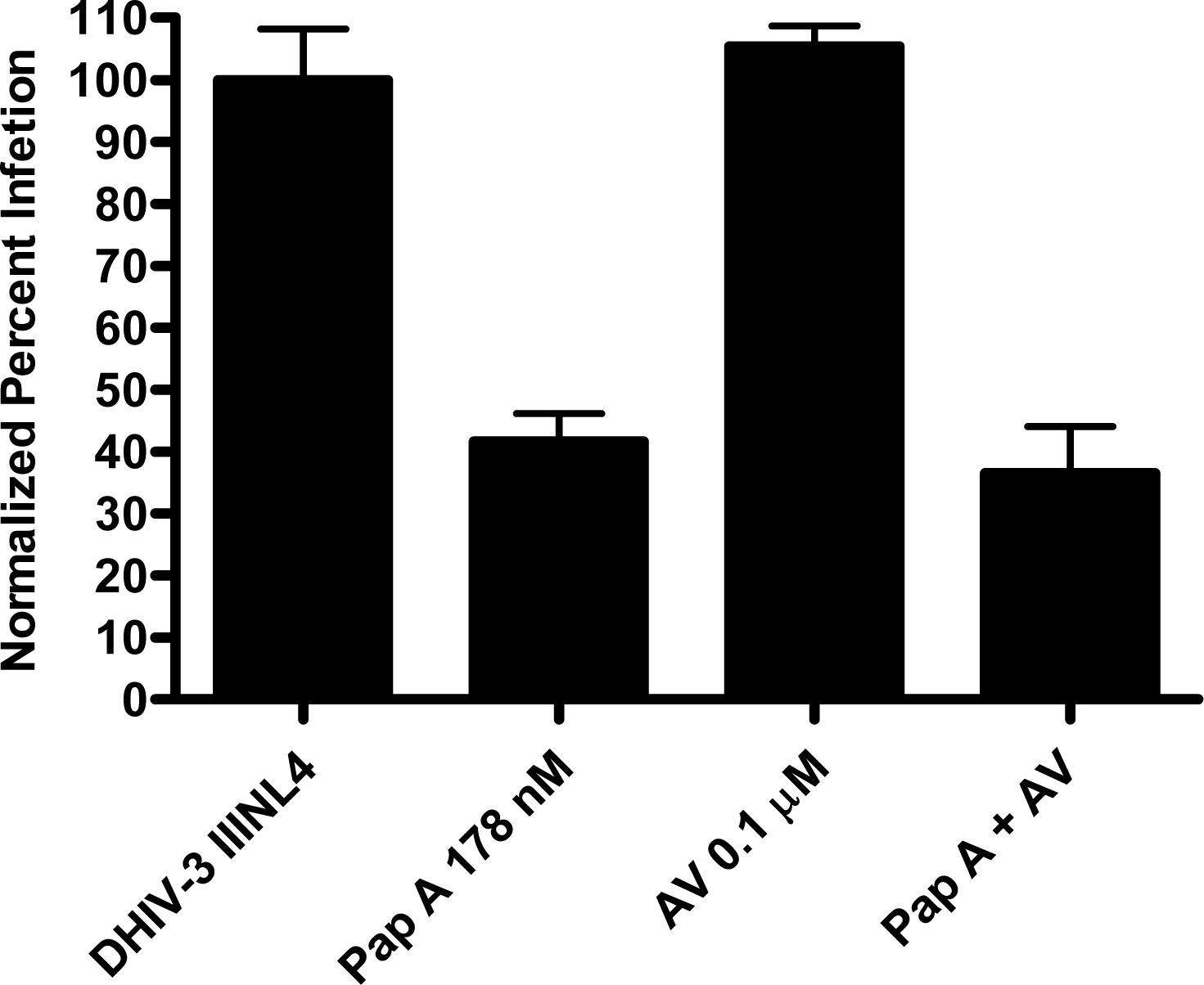
Papuamide A’s ability to bind to phosphatidylserine was observed using surface plasmon resonance. Twenty percent phosphatidylserine and 80% phosphatidylcholine (PC) vesicles were tested alongside 100% PC vesicles. The PS:PC vesicles exhibited binding to papuamide A while the 100% PC vesicles did not (data not shown). This indicates there is a preferential binding of papuamide A to phosphatidylserine. To determine if binding of PS alone was sufficient to inhibit viral entry, annexin V (AV) was tested in the virion based fusion assay. AV (tested at the concentrations similar to and higher than that shown to inhibit HIV infection) did not have any inhibitory effect on either X4 or R5 virus entry, while papuamide A remained active (Fig. 7a). In addition the effect of papuamide A was unchanged in the presence of AV when tested in the pseudotype assay (Fig. 7b). The results suggest that PS binding does not appear to be necessary for papuamide A to block entry.
3. Conclusion
In this study, we determine that the mechanism of papuamide A cytoprotection against HIV is through inhibition of virus entry. Papuamide A’s ability to inhibit both X4 and R5 tropic virus was similar to that recently published for the mirabamides, which were shown to inhibit fusion [6]. Further work shows that papuamide A inhibition does not target the key proteins involved in the viral entry process. Instead papuamide A works through a direct interaction with the virus and its virucidal activity appears to be independent of the type of envelope glycoprotein expressed. Phosphatidylserine (PS) a phospholipid present on the viral membrane has been proposed to be the target of papuamide B. Data presented here shows that while papuamide A does selectively bind PS, binding of PS alone is not sufficient to block viral entry.
Papuamide A’s activity may be representative of the group of marine depsipeptides shown to inhibit HIV induced cytopathicity. Papuamide A shares many chemical features with this group including an aliphatic tail, depsipeptide cyclization, a 3,4-dimethylglutamine residue and an available tyrosine hydroxyl (glycosylated in the mirabamides). Previously anti-HIV activity was reported for papuamides A and B. Here we show that papuamide C and D inhibit HIV entry as well, although less potently than A and B. This suggests that the free amino group of the 2,3-aminobutanoic acid residue of papuamides A and B is not required although it may contribute to the proposed virucidal activity of these compounds.
A proposed model for the mechanism of virucidal activity for these compounds can be based upon the membrane targeting mechanism proposed for a antifungal sterol dependent lipopeptide [38]. In this model, the lipopeptide has an aliphatic tail which inserts itself into the fungal membrane. The interaction is then stabilized through tyrosine binding to sterol present in the fungal membrane [38]. Cholesterol is a major component of the HIV viral membrane due to budding of the virus from cellular membrane microdomains rich in cholesterol and sphingolipids [39–41]. Common attributes of papuamide A and the other active depsipeptides include a tyrosine available to interact with cholesterol and a hydrophobic tail which could insert into the viral membrane. We hypothesize this could lead to disruption of the viral membrane resulting in a virucidal effect. Cooperative binding to PS may or may not contribute to papuamide A’s targeting of the viral membrane over the cellular membrane.
4. Experimental Section
4.1 Cells lines
The following cell lines were obtained through the AIDS Research and Reference Reagent Program: HeLa T4+, a human cervical epithelial carcinoma (HeLa) cell line rendered CD4+ by retrovirus-mediated gene transfer [42]; TZM-bl, a HeLa cell line stably expressing CD4 and CCR5 with β-galactosidase and luciferase reporter genes [43–45]; CEM-SS, a human T4 lymphoblastoid cell line [46, 47]; and CEM.NKR-CCR5, a human T4 lymphoblastoid cell expressing CCR5 [48, 49]. These cell lines were contributed by: Dr. Richard Axel; Dr. John C. Kappes, Dr. Xiaoyun Wu and Tranzyme Inc.; Dr. Peter L. Nara; and Dr. Alexandra Trkola, respectively. The HeLa T4+ and TZM-bl cell lines were maintained in Dulbecco’s modified Eagles medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and the CEM-SS and CEM.NKR-CCR5 cell lines maintained in RPMI supplemented with 10% FBS and 200 μM L-glutamine. The human embryonic kidney (HEK) cell line transformed to express the large T antigen, 293FT (Invitrogen, Carlsbad, CA), was maintained in DMEM, 10% FBS and 200 μM L-glutamine.
4.2 Plasmids
The pAdVAntage plasmid was purchased through Invitrogen (Carlsbad, CA). Dr. Eric O. Freed at the National Institutes of Health (NIH) kindly provided pNL4-3 and pIIINL4env [50]. pNL(AD8) was obtained through the AIDS Research and Reference Reagent Program, contributed by Dr. Eric O. Freed [51]. The plasmid pSCA was kindly provided by Paula M. Cannon, University of Southern California [52]. The following plasmids have been described elsewhere, pMM310 [21], pDHIV-3 [27], pHCMV-VSVG [34], pLET-JRFL [53], and pLET-LAI [53, 54].
4.3 Drugs
Papuamides A-D were the generous gift of Drs. David Williams and Raymond Andersen from the University of British Columbia [7]. Papuamide A was dissolved in 50:50, H2O:MeOH at a stock concentration of 10 mg/ml. Azidothymidine (AZT) was purchased from Sigma-Aldrich (St. Louis, MO) and kept at a stock concentration of 10 mg/ml in DMSO. HIV-1 IIIB C34 Peptide (C34) was obtained through the AIDS Research and Reference Reagent Program contributed by the Division of AIDS, NIAID [55]. C34 was dissolved in H2O at a concentration of 1 mg/ml. All concentrations of drugs used in text and figures are provided as final concentration in culture.
4.4 Virion based fusion assay.
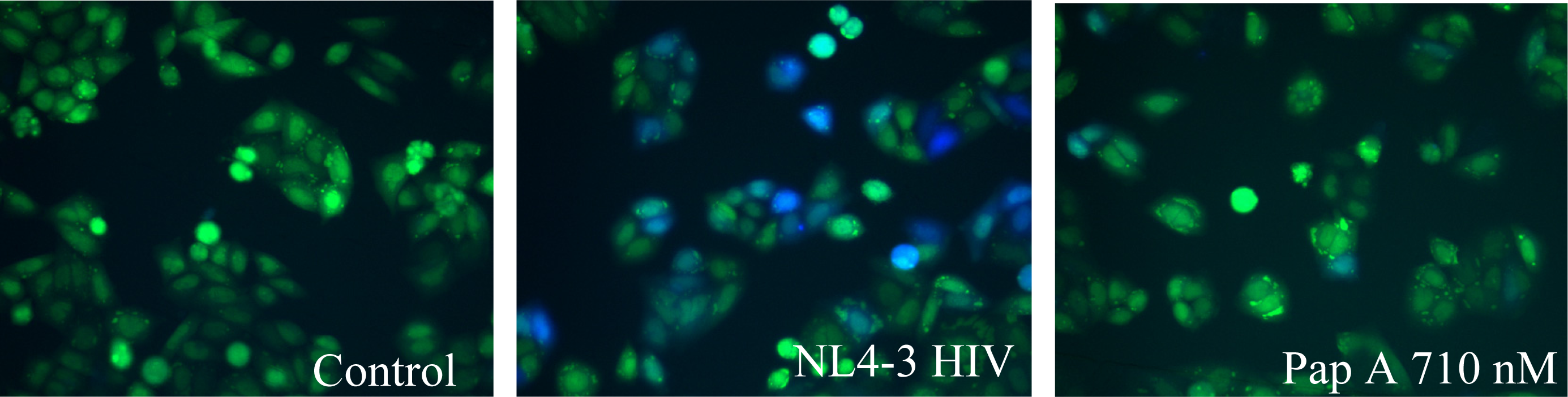
The fusion assay used was developed by Cavrois et al. [21]. Briefly, production of virus was accomplished by co-transfection of 20 μg of pMM310 (plasmid encoding β-lactamase linked to Vpr, BlaM-Vpr) [56],10 μg pAdVAntage (Invitrogen, Carlsbad, CA); and 60 μg pNL4-3(X4 tropic proviral DNA) [50] or pNL(AD8) (R5 tropic proviral DNA) [51] into 293FT cells using standard calcium phosphate transfection. Virus was collected after 48 hours and titered using the RETROtek p24 ELISA kit (ZeptoMatrix, Buffalo, NY). HeLaT4 or TZM-bl cells, 200μL at a concentration of 1 X 105 cells/mL, were plated into 96 well, black walled, flat bottom plates and placed in a 37°C, 5% CO2 humidified incubator overnight for attachment. Medium was aspirated and 200 μL of medium containing virus, at a predetermined p24 concentration of 200 ng/mL, was added with test compounds or vehicle. Cells were incubated at 37°C, 5% CO2 for 2 hours. Virus was aspirated off and cells rinsed twice with room temperature CO2 independent medium (Invitrogen, Carlsbad, CA). Cells were then loaded with CCF2-AM fluorescent substrate (Invitrogen, Carlsbad, CA) according to manufacturer’s protocol. Cells were incubated, protected from light, for 1 hour at room temperature and washed twice with room temperature CO2 independent medium. Next, 200 μL of room temperature CO2 independent medium supplemented with 10% FBS and 2.5mM probenecid (Sigma-Aldrich, St. Louis, MO) was added and the reaction was allowed to develop for 5–7 hours.
When fusion occurs, β-lactamase (BlaM) present in the virus cleaves the CCF2-AM substrate. This cleavage results in a change of the fluorescence emission from green to blue. Images of the cells were taken with a fluorescent microscope equipped with the appropriate filter set, Excitation filter 405±10 nm, Dicroic mirror 425, Emission Filter (blue) 460±20 nm, and Emission Filter (green) 530±15 nm. Using ImageJ, an image software program provided by NIH, the amount of green and blue fluorescence was determined. To quantify the observed fusion, a fluorescence ratio was calculated by dividing the amount of blue fluorescence by the amount of green fluorescence. This value was then normalized to the non-infected control by dividing the calculated values by the average control value.
4.5 Papuamide A-protein binding interactions
Binding interactions were quantified by surface plasmon resonance (SPR). Studies were performed at the University of Utah Protein Interactions facility on a BIAcore 3000 instrument. Briefly, papuamide A was biotinylated using the EZ-Link Sulfo-NHS-LC-LC-Biotin reagent (Pierce Biotechnology, Rockford, IL) with reaction products purified by reverse phase high performance liquid chromatography (HPLC). Biotinylation was confirmed by mass spec and presumed to target the free amine of the 2,3-diaminobutenoic acid. Biotinylated papuamide A was captured onto a chip with a carboxymethylated dextran matrix preimmobilized with streptavidin. Analytes were then flowed over the chip in hepes buffered saline (HBS) running buffer containing 1 mg/mL bovine serum albumin to reduce nonspecific binding. Soluble CD4 (sCD4) [57] and gp120 were obtained through the AIDS Research and Reference Reagent Program, contributed by Progenics Pharmaceuticals, Inc. and Division of AIDS, NIAID, NIH, respectively. A papuamide A positive control binding peptide was identified from a library of peptides (Catalog #6405, AIDS Research and Reference Reagent Program, NIH) contributed by Division of AIDS, NIAID, NIH. Stock concentration of 5 μM for sCD4, 8.3 μM for gp120 and 500 μM for the peptide were diluted 1/100 followed by three serial dilutions in HBS for testing. Binding responses reported as an arbitrary resonance unit (RU). Data analyzed using the SCRUBBER software, developed by the University of Utah Protein Interactions Facility.
4.6 Pseudotype virus assay
The production of pseudotype virus was accomplished by co-transfection of 30 μg of pDHIV-3 and 15 μg envelope plasmid by calcium phosphate mediated transfection of 293FT cells. pDHIV-3 [27] is a plasmid which encodes for all HIV proteins except envelope and nef and contains a green fluorescence protein (GFP) reporter gene in place of nef. Envelope glycoprotein plasmids used included: pSCA [52], amphotropic murine leukemia virus glycoprotein (aMLV); pHCMV-VSVG [34], vesicular stomatitis virus glycoprotein (VSV); pLET-JRFL [53], a R5 tropic HIV envelope glycoprotein; and, pLET-LAI [53, 54] and pIIINL4env [50], X4 tropic HIV envelope glycoproteins.
For inhibition studies, a 1.5 mL aliquot of pseudotype virus, with or without 178 nM papuamide A, was added to 2.5 × 105 CEM-SS or CEM.NKR-CCR5 cells in microfuge tube. Cells underwent a spinoculation at 1,700 × g for 2 hours at 25°C. Virus was aspirated and cells resuspended in 1mL RPMI, 10%FBS and plated into a 12 well culture plate. Cells were incubated for 48 hours at 37°C, 5% CO2 after which they were analyzed by flow cytometry. Infection rates were normalized so that the number of GFP expressing cells in the HIV infected control equaled 100.
4.7 Flow Cytometry
Flow cytometry was performed using a FACScan instrument (Becton Dickinson, Franklin Lakes, NJ). Cells were pelleted by centrifugation at 1,700 x g, washed with fluorescence-activated cell sorter (FACS) buffer (2% fetal bovine serum and 0.02% sodium azide in PBS) and resuspended in FACS buffer for analysis. The flow cytometer was set to analyze a total of 10,000 cells, gating for GFP producing cells. Analysis was performed by Cell Quest Alias software (BD Biosciences, San Jose, CA). Percent of total cells expressing GFP indicates percent cells infected. Background counts were averaged and subtracted from the infected and treated cell values.
4.8 Time dependent inhibition of infection
Infection of CEM-SS cells with pseudotype virus expressing IIINL4env (X4 tropic envelope) or VSV envelope was performed as described above with the exception of 0.5 mL of virus added with 1 mL of fresh medium. This allows for consistency between treatments because medium does not have to be changed after spinoculation. Initial time-dependency experiments were performed with IIINL4env pseudotype virus. Papuamide A (178 nM), AZT (3.7 μM) or C34 (233 nM) was added at 0, 1 and 2 hours after virus addition. To determine effect of cell pretreatment, cells were treated with papuamide A overnight, pelleted by centrifugation at 1,700 × g and papuamide A containing medium aspirated. A second experiment with delayed addition of papuamide A to VSV and IIINL4env pseudotype virus was also performed.
4.10 Effect of VSV pseudotype virus pretreatment
VSV pseudotype virus was incubated with or without 178 nM papuamide A overnight in a 37°C, 5% CO2 incubator. Virus was centrifuged at 25,000 × g for 2 hours at 4°C to pellet the virus. Virus was resuspended in fresh RPMI, 10% FBS medium. Non-pretreated virus, non-pretreated virus plus 178 nM papuamide A and virus pretreated with 178 nM papuamide A were then used to infect CEM-SS cells by spinoculation and infectivity determined by flow cytometry after 24 hours.
4.11 Analysis
Image J (NIH) was used to quantify fluorescence from microscope images. Prism software (GraphPad Software) was used to generate and analyze dose response graph. Statistical analysis was performed using the paired Student t-test with a significance level at p < 0.05.
Acknowledgments
The authors would like to thank Drs. David Williams and Raymond Andersen for generously providing papuamide A. We thank Dr. David Myzska, University of Utah, Protein Interactions facility, for his assistance with the surface plasmon resonance studies. We also thank Drs. Jason DeHart and Orly Ardon for their technical assistance.
This work was supported by funding provided by NIH through the ICBG 5UO1TW006671 to Louis R. Barrows, NIAID grant AI49057 to Vicente Planelles and pre-doctoral fellowships awarded to Cynthia D. Andjelic by the PhRMA Foundation and the American Foundation for Pharmaceutical Education.
References
- Gochfeld, DJ; El Sayed, KA; Yousaf, M; Hu, JF; Bartyzel, P; Dunbar, DC; Wilkins, SP; Zjawiony, JK; Schinazi, RF; Schlueter-Wirtz, S; Tharnish, PM; Hamann, MT. Marine natural products as lead anti-HIV agents. Mini Rev. Med. Chem 2003, 3(5), 401–424. [Google Scholar]
- Tziveleka, LA; Vagias, C; Roussis, V. Natural products with anti-HIV activity from marine organisms. Curr. Top. Med. Chem 2003, 3(13), 1512–1535. [Google Scholar]
- Oku, N; Gustafson, KR; Cartner, LK; Wilson, JA; Shigematsu, N; Hess, S; Pannell, LK; Boyd, MR; McMahon, JB; Neamphamide, A. A new HIV-inhibitory depsipeptide from the Papua New Guinea marine sponge Neamphius huxleyi. J. Nat. Prod 2004, 67(8), 1407–1411. [Google Scholar]
- NIAID, Division of AIDS, HIV/OI/TB Therapeutics Database, Vol. September, 2007.
- Zampella, A; D'Auria, MV; Paloma, LG; Casapullo, A; Minale, L; Debitus, C; Henin, Y. Callipeltin A, an Anti-HIV Cyclic Depsipeptide from the New Caledonian Lithistida Sponge Callipelta sp. J. Am. Chem. Soc 1996, 118, 6202–6209. [Google Scholar]
- Plaza, A; Gustchina, E; Baker, HL; Kelly, M; Bewley, CA; Mirabamides, A-D. Depsipeptides from the sponge Siliquariaspongia mirabilis that inhibit HIV-1 fusion. J. Nat. Prod 2007, 70(11), 1753–1760. [Google Scholar]
- Ford, PW; Gustafson, KR; McKee, TC; Shigematsu, N; Maurizi, LK; Pannell, LK; Williams, DE; de Silva, ED; Lassota, P; Allen, TM; Van Soest, R; Andersen, RJ; Boyd, MR. Papuamides A-D, HIV-Inhibitory and Cytotoxic Depsipeptides from the Sponges Theonella mirabilis and Theonella swinhoei Collected in Papua New Guinea. J. Am. Chem. Soc 1999, 121, 5899–5909. [Google Scholar]
- Richman, DD; Morton, SC; Wrin, T; Hellmann, N; Berry, S; Shapiro, MF; Bozzette, SA. The prevalence of antiretroviral drug resistance in the United States. AIDS 2004, 18(10), 1393–1401. [Google Scholar]
- Dalgleish, AG; Beverley, PC; Clapham, PR; Crawford, DH; Greaves, MF; Weiss, RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature 1984, 312(5996), 763–767. [Google Scholar]
- McDougal, JS; Kennedy, MS; Sligh, JM; Cort, SP; Mawle, A; Nicholson, JK. Binding of HTLV-III/LAV to T4+ T cells by a complex of the 110K viral protein and the T4 molecule. Science 1986, 231(4736), 382–385. [Google Scholar]
- Rizzuto, CD; Wyatt, R; Hernandez-Ramos, N; Sun, Y; Kwong, PD; Hendrickson, WA; Sodroski, J. A conserved HIV gp120 glycoprotein structure involved in chemokine receptor binding. Science 1998, 280(5371), 1949–1953. [Google Scholar]
- Feng, Y; Broder, CC; Kennedy, PE; Berger, EA. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272(5263), 872–877. [Google Scholar]
- Deng, H; Liu, R; Ellmeier, W; Choe, S; Unutmaz, D; Burkhart, M; Di Marzio, P; Marmon, S; Sutton, RE; Hill, CM; Davis, CB; Peiper, SC; Schall, TJ; Littman, DR; Landau, NR. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381(6584), 661–666. [Google Scholar]
- Choe, H; Farzan, M; Sun, Y; Sullivan, N; Rollins, B; Ponath, PD; Wu, L; Mackay, CR; LaRosa, G; Newman, W; Gerard, N; Gerard, C; Sodroski, J. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 1996, 85(7), 1135–1148. [Google Scholar]
- Dragic, T; Litwin, V; Allaway, GP; Martin, SR; Huang, Y; Nagashima, KA; Cayanan, C; Maddon, PJ; Koup, RA; Moore, JP; Paxton, WA. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR-5. Nature 1996, 381(6584), 667–673. [Google Scholar]
- Wu, L; Gerard, NP; Wyatt, R; Choe, H; Parolin, C; Ruffing, N; Borsetti, A; Cardoso, AA; Desjardin, E; Newman, W; Gerard, C; Sodroski, J. CD4-induced interaction of primary HIV-1 gp120 glycoproteins with the chemokine receptor CCR-5. Nature 1996, 384(6605), 179–183. [Google Scholar]
- Doms, RW; Moore, JP. HIV-1 membrane fusion: targets of opportunity. J. Cell Biol 2000, 151(2), F9–14. [Google Scholar]
- Chan, DC; Fass, D; Berger, JM; Kim, PS. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89(2), 263–273. [Google Scholar]
- Tan, K; Liu, J; Wang, J; Shen, S; Lu, M. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc. Natl. Acad. Sci. USA 1997, 94(23), 12303–12308. [Google Scholar]
- Weissenhorn, W; Dessen, A; Harrison, SC; Skehel, JJ; Wiley, DC. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387(6631), 426–430. [Google Scholar]
- Cavrois, M; De Noronha, C; Greene, WC. A sensitive and specific enzyme-based assay detecting HIV-1 virion fusion in primary T lymphocytes. Nat. Biotechnol 2002, 20(11), 1151–1154. [Google Scholar]
- Kiser, R; Makovsky, S; Terpening, SJ; Laing, N; Clanton, DJ. Assessment of a cytoprotection assay for the discovery and evaluation of anti-human immunodeficiency virus compounds utilizing a genetically-impaired virus. J. Virol. Methods 1996, 58(1–2), 99–109. [Google Scholar]
- Hahnefeld, C; Drewianka, S; Herberg, FW. Determination of kinetic data using surface plasmon resonance biosensors. Methods Mol. Med 2004, 94, 299–320. [Google Scholar]
- Jonsson, U; Fagerstam, L; Ivarsson, B; Johnsson, B; Karlsson, R; Lundh, K; Lofas, S; Persson, B; Roos, H; Ronnberg, I; et al. Real-time biospecific interaction analysis using surface plasmon resonance and a sensor chip technology. Biotechniques 1991, 11(5), 620–627. [Google Scholar]
- Jonsson, U; Fagerstam, L; Lofas, S; Stenberg, E; Karlsson, R; Frostell, A; Markey, F; Schindler, F. Introducing a biosensor based technology for real-time biospecific interaction analysis. Ann. Biol. Clin. (Paris) 1993, 51(1), 19–26. [Google Scholar]
- Malmqvist, M. Biospecific interaction analysis using biosensor technology. Nature 1993, 361(6408), 186–187. [Google Scholar]
- Andersen, JL; DeHart, JL; Zimmerman, ES; Ardon, O; Kim, B; Jacquot, G; Benichou, S; Planelles, V. HIV-1 Vpr-induced apoptosis is cell cycle dependent and requires Bax but not ANT. PLoS Pathog 2006, 2(12), e127. [Google Scholar]
- Aiken, C. Pseudotyping human immunodeficiency virus type 1 (HIV-1) by the glycoprotein of vesicular stomatitis virus targets HIV-1 entry to an endocytic pathway and suppresses both the requirement for Nef and the sensitivity to cyclosporin A. J. Virol 1997, 71(8), 5871–5877. [Google Scholar]
- Schlegel, R; Dickson, RB; Willingham, MC; Pastan, IH. Amantadine and dansylcadaverine inhibit vesicular stomatitis virus uptake and receptor-mediated endocytosis of alpha 2-macroglobulin. Proc. Natl. Acad. Sci. USA 1982, 79(7), 2291–2295. [Google Scholar]
- Superti, F; Seganti, L; Ruggeri, FM; Tinari, A; Donelli, G; Orsi, N. Entry pathway of vesicular stomatitis virus into different host cells. J. Gen. Virol 1987, 68, 387–399. [Google Scholar]
- Beer, C; Andersen, DS; Rojek, A; Pedersen, L. Caveola-dependent endocytic entry of amphotropic murine leukemia virus. J. Virol 2005, 79(16), 10776–11787. [Google Scholar]
- von Laer, D; Thomsen, S; Vogt, B; Donath, M; Kruppa, J; Rein, A; Ostertag, W; Stocking, C. Entry of amphotropic and 10A1 pseudotyped murine retroviruses is restricted in hematopoietic stem cell lines. J. Virol 1998, 72(2), 1424–1430. [Google Scholar]
- Este, JA; Telenti, A. HIV entry inhibitors. Lancet 2007, 370(9581), 81–88. [Google Scholar]
- Akkina, RK; Walton, RM; Chen, ML; Li, QX; Planelles, V; Chen, IS. High-efficiency gene transfer into CD34+ cells with a human immunodeficiency virus type 1-based retroviral vector pseudotyped with vesicular stomatitis virus envelope glycoprotein G. J. Virol 1996, 70(4), 2581–2585. [Google Scholar]
- Miller, AD. Human gene therapy comes of age. Nature 1992, 357(6378), 455–460. [Google Scholar]
- Parsons, AB; Lopez, A; Givoni, IE; Williams, DE; Gray, CA; Porter, J; Chua, G; Sopko, R; Brost, RL; Ho, CH; Wang, J; Ketela, T; Brenner, C; Brill, JA; Fernandez, GE; Lorenz, TC; Payne, GS; Ishihara, S; Ohya, Y; Andrews, B; Hughes, TR; Frey, BJ; Graham, TR; Andersen, RJ; Boone, C. Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 2006, 126(3), 611–625. [Google Scholar]
- Callahan, MK; Popernack, PM; Tsutsui, S; Truong, L; Schlegel, RA; Henderson, AJ. Phosphatidylserine on HIV envelope is a cofactor for infection of monocytic cells. J. Immunol 2003, 170(9), 4840–4845. [Google Scholar]
- Volpon, L; Besson, F; Lancelin, JM. NMR structure of active and inactive forms of the sterol-dependent antifungal antibiotic bacillomycin L. Eur. J. Biochem 1999, 264(1), 200–210. [Google Scholar]
- Campbell, SM; Crowe, SM; Mak, J. Lipid rafts and HIV-1: from viral entry to assembly of progeny virions. J. Clin. Virol 2001, 22(3), 217–227. [Google Scholar]
- Raulin, J. Human immunodeficiency virus and host cell lipids. Interesting pathways in research for a new HIV therapy. Prog. Lipid Res 2002, 41(1), 27–65. [Google Scholar]
- Simons, K; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 387(6633), 569–572. [Google Scholar]
- Maddon, PJ; Dalgleish, AG; McDougal, JS; Clapham, PR; Weiss, RA; Axel, R. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell 1986, 47(3), 333–348. [Google Scholar]
- Wei, X; Decker, JM; Liu, H; Zhang, Z; Arani, RB; Kilby, JM; Saag, MS; Wu, X; Shaw, GM; Kappes, JC. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother 2002, 46(6), 1896–1905. [Google Scholar]
- Derdeyn, CA; Decker, JM; Sfakianos, JN; Wu, X; O'Brien, WA; Ratner, L; Kappes, JC; Shaw, GM; Hunter, E. Sensitivity of human immunodeficiency virus type 1 to the fusion inhibitor T-20 is modulated by coreceptor specificity defined by the V3 loop of gp120. J. Virol 2000, 74(18), 8358–8367. [Google Scholar]
- Platt, EJ; Wehrly, K; Kuhmann, SE; Chesebro, B; Kabat, D. Effects of CCR5 and CD4 cell surface concentrations on infections by macrophagetropic isolates of human immunodeficiency virus type 1. J. Virol 1998, 72(4), 2855–2864. [Google Scholar]
- Nara, PL; Fischinger, PJ. Quantitative infectivity assay for HIV-1 and-2. Nature 1988, 332(6163), 469–470. [Google Scholar]
- Nara, PL; Hatch, WC; Dunlop, NM; Robey, WG; Arthur, LO; Gonda, MA; Fischinger, PJ. Simple, rapid, quantitative, syncytium-forming microassay for the detection of human immunodeficiency virus neutralizing antibody. AIDS Res. Hum. Retroviruses 1987, 3(3), 283–302. [Google Scholar]
- Lyerly, HK; Reed, DL; Matthews, TJ; Langlois, AJ; Ahearne, PA; Petteway, SR, Jr; Weinhold, KJ. Anti-GP 120 antibodies from HIV seropositive individuals mediate broadly reactive anti-HIV ADCC. AIDS Res. Hum. Retroviruses 1987, 3(4), 409–422. [Google Scholar]
- Trkola, A; Matthews, J; Gordon, C; Ketas, T; Moore, JP. A cell line-based neutralization assay for primary human immunodeficiency virus type 1 isolates that use either the CCR5 or the CXCR4 coreceptor. J. Virol 1999, 73(11), 8966–8974. [Google Scholar]
- Murakami, T; Freed, EO. The long cytoplasmic tail of gp41 is required in a cell type-dependent manner for HIV-1 envelope glycoprotein incorporation into virions. Proc. Natl. Acad. Sci. USA 2000, 97(1), 343–348. [Google Scholar]
- Freed, EO; Englund, G; Martin, MA. Role of the basic domain of human immunodeficiency virus type 1 matrix in macrophage infection. J. Virol 1995, 69(6), 3949–3954. [Google Scholar]
- Han, JY; Zhao, Y; Anderson, WF; Cannon, PM. Role of variable regions A and B in receptor binding domain of amphotropic murine leukemia virus envelope protein. J. Virol 1998, 72(11), 9101–9108. [Google Scholar]
- Challita-Eid, PM; Klimatcheva, E; Day, BT; Evans, T; Dreyer, K; Rimel, BJ; Rosenblatt, JD; Planelles, V. Inhibition of HIV type 1 infection with a RANTES-IgG3 fusion protein. AIDS Res. Hum. Retroviruses 1998, 14(18), 1617–1624. [Google Scholar]
- Langlade-Demoyen, P; Michel, F; Hoffenbach, A; Vilmer, E; Dadaglio, G; Garicia-Pons, F; Mayaud, C; Autran, B; Wain-Hobson, S; Plata, F. Immune recognition of AIDS virus antigens by human and murine cytotoxic T lymphocytes. J. Immunol 1988, 141(6), 1949–1957. [Google Scholar]
- Gallo, SA; Sackett, K; Rawat, SS; Shai, Y; Blumenthal, R. The stability of the intact envelope glycoproteins is a major determinant of sensitivity of HIV/SIV to peptidic fusion inhibitors. J. Mol. Biol 2004, 340(1), 9–14. [Google Scholar]
- Tobiume, M; Lineberger, JE; Lundquist, CA; Miller, MD; Aiken, C. Nef does not affect he efficiency of human immunodeficiency virus type 1 fusion with target cells. J. Virol 2003, 77(19), 10645–10650. [Google Scholar]
- Garlickn, RL; Kirschner, RJ; Eckenrode, FM; Tarpley, WG; Tomich, CS. Escherichia coli expression, purification, and biological activity of a truncated soluble CD4. AIDS Res. Hum. Retroviruses 1990, 6(4), 465–479. [Google Scholar]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte
| |||
| sCD4 | gp120 | Peptide | |
| Molecular Weight | 26,000 | 120,000 | 1898 |
| Expected Response at Saturation (RU) | 9,743 | 44,968 | 712 |
| Observed Response (RU)at Maximum Analyte Concentration | 10 | 35 | 30 |
| Fold Difference of Responses | 974 | 1284 | 24 |
| KD (μM) | 0.08 | 2 | 176 |
| Maximum Analyte Concentration Tested (μM) | 0.05 | 0.08 | 5 |
| Fold Difference between KD and Analyte Concentration | 1.6 | 24 | 35 |
| Expected Response at Maximum Concentration Tested (RU) | 6089 | 1874 | 20 |
Binding interactions between papuamide A and the above listed analytes were measured using surface plasmon resonance (SPR). Above defined values were used for data analysis calculations. Formulas provided in text.
Share and Cite
Andjelic, C.D.; Planelles, V.; Barrows, L.R. Characterizing the Anti-HIV Activity of Papuamide A. Mar. Drugs 2008, 6, 528-549. https://doi.org/10.3390/md20080027
Andjelic CD, Planelles V, Barrows LR. Characterizing the Anti-HIV Activity of Papuamide A. Marine Drugs. 2008; 6(4):528-549. https://doi.org/10.3390/md20080027
Chicago/Turabian StyleAndjelic, Cynthia D., Vicente Planelles, and Louis R. Barrows. 2008. "Characterizing the Anti-HIV Activity of Papuamide A" Marine Drugs 6, no. 4: 528-549. https://doi.org/10.3390/md20080027
APA StyleAndjelic, C. D., Planelles, V., & Barrows, L. R. (2008). Characterizing the Anti-HIV Activity of Papuamide A. Marine Drugs, 6(4), 528-549. https://doi.org/10.3390/md20080027