Kinase Inhibitors from Marine Sponges


Abstract
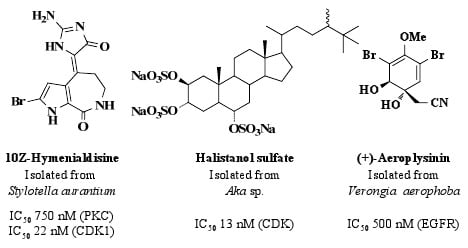
:
1. Introduction
2. Reviews
3. Protein Kinase C (PKC, EC 2.7.11.13)
4. Cyclin Dependent Kinases (CDK, EC 2.7.11.22)
4.1. Cyclin Dependent Kinase-1
4.2. Cyclin Dependent Kinase-4
5. Tyrosine Protein Kinase (TPK, EC 2.7.10.1)
Tyrosine Kinase pp60V-SRC
6. Epidermal Growth Factor Receptor (EC 2.7.10.1)
7. Mitogen-Activated Protein Kinase (EC 2.7.11.24)
Raf (EC 2.7.11.1)/MAP Kinase Kinase (EC 2.7.12.2)/MAPK (EC 2.7.11.24)
8. Glycogen Synthase Kinase-3 (GSK-3, EC 2.7.11.26)
9. Other Kinases
10. Conclusions
Acknowledgments
References
- Newman, DJ; Cragg, GM. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod 2007, 70, 461–477. [Google Scholar]
- Faulkner, DJ. Marine pharmacology. Antonie Leeuwenhoek 2000, 77, 135–145. [Google Scholar]
- Haefner, B. Drugs from the deep: Marine natural products as drug candidates. Drug Discov. Today 2003, 8, 536–544. [Google Scholar]
- Sharma, P; Sharma, R; Tyagi, R. Inhibitors of cyclin dependent kinases: Useful targets for cancer treatment. Curr. Cancer Drug Targets 2008, 8, 53–75. [Google Scholar]
- Novak, K. Conference Report—Protein Kinase Inhibitors in Cancer Treatment: Mixing and Matching? Proceedings of the Keystone Symposium on Protein Kinases and Cancer, Lake Tahoe, CA, USA, 24–29 February 2004, Medscape General Medicine: Lake Tahoe, CA, USA, 2004. [Google Scholar]
- Goldstein, D; Gray, N; Zarrinkar, P. High-throughput kinase profiling as a platform for drug discovery. Nat. Rev. Drug Discov 2008, 7, 391–397. [Google Scholar]
- Norman, P. Overview: Kinase Therapeutics Pipelines: An Assessment of Targets and Agents in Development; Cambridge Healthtech Institute: Needham, MA, USA, 2007. [Google Scholar]
- Marston, A. Natural products as a source of protein kinase activators and inhibitors. Curr. Top. Med. Chem 2011, 11, 1333–1339. [Google Scholar]
- Nakao, Y; Fusetani, N. Enzyme inhibitors from marine invertebrates. J. Nat. Prod 2007, 70, 689–710. [Google Scholar]
- Deslandes, S; Chassaing, S; Delfourne, E. Marine pyrrolocarbazoles and analogues: Synthesis and kinase inhibition. Mar. Drugs 2009, 7, 754–786. [Google Scholar]
- Nguyen, TNT; Tepe, JJ. Preparation of hymenialdisine, analogues and their evaluation as kinase inhibitors. Curr. Med. Chem 2009, 16, 3122–3143. [Google Scholar]
- Carter, CA; Kane, CJM. Therapeutic potential of natural compounds that regulate the activity of protein kinase C. Curr. Med. Chem 2004, 11, 2883–2902. [Google Scholar]
- Newton, AC. Protein kinase C: Structure, function and regulation. J. Biol. Chem 1995, 270, 28495–28498. [Google Scholar]
- Kortmansky, J; Schwartz, GK. Bryostatin-1: A Novel PKC inhibitor in clinical development. Cancer Investig 2003, 21, 924–936. [Google Scholar]
- Fusetani, N; Nakao, Y. Enzyme inhibitors from marine invertebrates. J. Nat. Prod 2007, 70, 689–710. [Google Scholar]
- Tamaoki, T; Nomoto, H; Takahashi, I; Kato, Y; Morimoto, M; Tomita, F. Staurosporine, a potent inhibitor of phospholipid/Ca++ dependent protein kinase. Biochem. Biophys. Res. Commun 1986, 135, 397–402. [Google Scholar]
- Kinnel, RB; Scheuer, PJ. 11-Hydroxystaurosporine: A highly cytotoxic, powerful protein kinase C inhibitor from a tunicate. J. Org. Chem 1992, 57, 6327–6329. [Google Scholar]
- Pettit, GR; Herald, CL; Doubek, DL; Herald, DL; Arnold, E; Clardy, J. Isolation and structure of bryostatin 1. J. Am. Chem. Soc 1982, 104, 6846–6848. [Google Scholar]
- Rodriguez, J; Peters, BM; Kurz, L; Schatzman, RC; McCarley, D; Lou, L; Crews, P. An alkaloid protein kinase C inhibitor, xestocyclamine A, from the marine sponge Xestospongia sp. J. Am. Chem. Soc 1993, 115, 10436–10437. [Google Scholar]
- Chappell, M. Total Synthesis of Xestocyclamine A; Grant No. 1F32GM019972-01; National Institute of General Medical Sciences (NIGMS): Bethesda, MD, USA, 1999. [Google Scholar]
- Yun, H; Gagnon, A; Danishefsky, SJ. Toward the synthesis of xestocyclamine A: Investigation of double Michael reaction and direct aza Diels-Alder reaction. Tetrahedron Lett 2006, 47, 5311–5315. [Google Scholar]
- Patil, AD; Freyer, AJ; Killmer, L; Hofmann, G; Johnson, RK. Z-Axinohydantoin and debromo-Z-axinohydantoin from the sponge Stylotella aurantium: Inhibitors of protein kinase C. Nat. Prod. Res 1997, 9, 201–207. [Google Scholar]
- Freyer, AJ; Patil, AD; Killmer, L; Offen, P; Carte, B; Jurewicz, AJ; Johnson, RK. Frondosins, five new sesquiterpene hydroquinone derivatives with novel skeletons from the sponge Dysidea frondosa: Inhibitors of interleukin-8 receptors. Tetrahedron 1997, 53, 5047–5060. [Google Scholar]
- Boyd, MR; Hallock, YF; Cardellina, JH. (−)-Frondosins A and D, HIV-inhibitory sesquiterpene hydroquinone derivatives from Euryspongia sp. Nat. Prod. Res 1998, 11, 153–160. [Google Scholar]
- Trost, BM; Hu, Y; Horne, DB. Total synthesis of (+)-frondosin A. Application of the Ru-catalyzed [5 + 2] cycloaddition. J. Am. Chem. Soc 2007, 129, 11781–11790. [Google Scholar]
- Inoue, M; Frontier, AJ; Danishefsky, SJ. The total synthesis of frondosin B. Angew. Chem. Int. Ed 2000, 39, 761–764. [Google Scholar]
- Li, X; Kynea, RE; Ovaska, TV. Total syntheses of (±)-frondosin C and (±)-8-epi-frondosin C via a tandem anionic 5-exo dig cyclization—Claisen rearrangement sequence. Tetrahedron 2007, 63, 1899–1906. [Google Scholar]
- Willis, RH; de Vries, DJ. BRS1, A C30 BIS-amino, BIS-hydroxy polyunsaturated lipid from an Australian calcareous sponge that inhibits protein kinase C. Toxicon 1997, 35, 1125–1129. [Google Scholar]
- Shigemori, H; Madono, T; Sasaki, T; Mikami, Y; Kobayashi, J. Nakijiquinones A and B, new antifungal sesquiterpenoid quinones with an amino acid residue from an Okinawan marine sponge. Tetrahedron 1994, 50, 8347–8354. [Google Scholar]
- Kobayashi, J; Madono, T; Shigemori, H. Nakijiquinones C and D, new sesquiterpenoid quinones with a hydroxy amino acid residue from a marine sponge inhibiting c-erbB-2 kinase. Tetrahedron 1995, 51, 10867–10874. [Google Scholar]
- Takahashi, Y; Kubota, T; Ito, J; Mikami, Y; Fromont, J; Kobayashi, J. Nakijiquinones G–I, new sesquiterpenoid quinones from marine sponge. Bioorg. Med. Chem 2008, 16, 7561–7564. [Google Scholar]
- Kissau, L; Stahl, P; Mazitschek, R; Giannis, A; Waldmann, H. Development of natural product-derived receptor tyrosine kinase inhibitors based on conservation of protein domain fold. J. Med. Chem 2003, 46, 2917–2931. [Google Scholar]
- Stahl, P; Waldmann, H. Asymmetric synthesis of the nakijiquinones—Selective inhibitors of the Her-2/Neu protooncogene. Angew. Chem. Int. Ed 1999, 38, 3710–3713. [Google Scholar]
- Stahl, P; Kissau, L; Mazitschek, R; Huwe, A; Furet, P; Giannis, A; Waldmann, H. Total synthesis and biological evaluation of the nakijiquinones. J. Am. Chem. Soc 2001, 123, 11586–11593. [Google Scholar]
- Slamon, DJ; Clark, GM; Wong, SG; Levin, WJ; Ullrich, A; McGuire, WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/Neu oncogene. Science 1987, 235, 177–182. [Google Scholar]
- He, H; Kulanthaivel, P; Baker, BJ. New cytotoxic sesterterpenes from the marine sponge Spongia sp. Tetrahedron Lett 1994, 35, 7189–7192. [Google Scholar]
- Kulciţki, V. A biomimetic approach to some specifically functionalized cyclic terpenoids. Acta Biochim. Pol 2007, 54, 679–693. [Google Scholar]
- Longley, RE; Harmody, D. A rapid colorimetric microassay to detect agonist/antagonists of protein-kinase-C based on adherence of EL-4.IL-2 cells. J. Antibiot 1991, 44, 93–102. [Google Scholar]
- Horton, PA; Koehn, FE; Longley, RE; McConnell, OJ. Lasonolide A, a new cytotoxic macrolide from the marine sponge Forcepia sp. J. Am. Chem. Soc 1994, 116, 6015–6016. [Google Scholar]
- Isbrucker, RA; Guzman, EA; Pitts, TP; Wright, AE. Early effects of lasonolide A on pancreatic cancer cells. J. Pharmacol. Exp. Ther 2009, 331, 733–739. [Google Scholar]
- Alvi, KA; Jaspars, M; Crews, P; Strulovici, B; Oto, E. Penazetidine-A, an alkaloid inhibitor of protein kinase C. Bioorg. Med. Chem. Lett 1994, 4, 2447–2450. [Google Scholar]
- Chan, JA; Freyer, AJ; Carte, BK; Hemling, ME; Hofmann, GA; Mattern, MR; Mentzer, MA; Westley, JW. Protein kinase C inhibitors: Novel spirosesquiterpene aldehydes from a marine sponge Aka (=Siphonodictyon) coralliphagum. J. Nat. Prod 1994, 57, 1543–1548. [Google Scholar]
- Morgan, D. The Cell-Cycle Control System. In The Cell Cycle: Principles of Control; Oxford University Press: Oxford, UK, 2007; pp. 30–31. [Google Scholar]
- Morris, D; Bramwell, V; Turcotte, R; Figueredo, A; Blackstein, M; Verma, S; Matthews, S; Eisenhauer, E. A phase II study of flavopiridol in patients with previously untreated advanced soft tissue sarcoma. Sarcoma 2006, 1, 1–7. [Google Scholar]
- Muhtasib, H. Cyclin-dependent kinase inhibitors from natural sources: Recent advances and future prospects for cancer treatment. Adv. Phytomed 2006, 2, 155–167. [Google Scholar]
- Castedo, M; Perfettini, J-L; Roumier, T; Kroemer, G. Cyclin-dependent kinase-1: Linking apoptosis to cell cycle and mitotic castastrophe. Cell Death Differ 2002, 9, 1287–1293. [Google Scholar]
- Cimino, G; de Rosa, S; de Stefano, S; Mazzarella, L; Puliti, R; Sodano, G. Isolation and X-ray crystal structure of a novel bromo-compound from two marine sponges. Tetrahedron Lett 1982, 23, 767–768. [Google Scholar]
- Meijer, L; Thunnissen, AM; White, AW; Garnier, M; Nikolic, M; Tsai, LH; Walter, J; Cleverley, KE; Salinas, PC; Wu, YZ; et al. Inhibition of cyclin-dependent kinases, GSK-3[beta] and CK1 by hymenialdisine, a marine sponge constituent. Chem. Biol 2000, 7, 51–63. [Google Scholar]
- Goga, A; Yang, D; Tward, A; Morgan, D; Bishop, J. Inhibition of CDK1 as a potential therapy for tumours over-expressing MYC. Nat. Med 2007, 13, 820–827. [Google Scholar]
- Wan, Y; Hur, W; Cho, C; Liu, Y; Adrian, F; Lozach, O; Bach, S; Mayer, T; Fabbro, D; Meijer, L; et al. Synthesis and target identification of hymenialdisine analogs. Chem. Biol 2004, 11, 247–259. [Google Scholar]
- Tepe, J. Preparation of Hymenialdisine Derivatives and Use Thereof. US Patent 7,193,079, 20 March 2007. [Google Scholar]
- Killday, B; Yarwood, D; Sills, M; Murphy, P; Hooper, J; Wright, A. Microxine, a new cdc2 kinase inhibitor from the Australian marine sponge Microxima species. J. Nat. Prod 2001, 64, 525–526. [Google Scholar]
- Walker, SR; Carter, EJ; Huff, BC; Morris, JC. Variolins and related alkaloids. Chem. Rev 2009, 109, 3080–3098. [Google Scholar]
- Trimurtulu, G; Faulkner, DJ; Perry, NB; Ettouati, L; Litaudon, M; Blunt, JW; Munro, MHG; Jameson, GB. Alkaloids from the antarctic sponge Kirkpatrickia varialosa. Part 2: Variolin A and N(3′)-methyl tetrahydrovariolin B. Tetrahedron 1994, 50, 3993–4000. [Google Scholar]
- Anderson, RJ; Hill, JB; Morris, JC. Concise total syntheses of variolin B and deoxyvariolin B. J. Org. Chem 2005, 70, 6204–6212. [Google Scholar]
- Baeza, A; Mendiola, J; Burgos, C; Alvarez-Builla, J; Vaquero, JJ. Palladium-mediated C-N, C-C, and C-O functionalization of azolopyrimidines: A new total synthesis of variolin B. Tetrahedron Lett 2008, 49, 4073–4077. [Google Scholar]
- Ahaidar, A; Fernandez, D; Danelon, G; Cuevas, C; Manzanares, I; Albericio, F; Joule, JA; Alvarez, M. Total syntheses of variolin B and deoxyvariolin B. J. Org. Chem 2003, 68, 10020–10029. [Google Scholar]
- Molina, P; Fresneda, PM; Delgado, S; Bleda, JA. Synthesis of the potent antitumoral marine alkaloid variolin B. Tetrahedron Lett 2002, 43, 1005–1007. [Google Scholar]
- Sherr, CJ; Roberts, JM. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev 1999, 13, 1501–1512. [Google Scholar]
- Lin, J; Yan, X-J; Chen, H-M. Fascaplysin, a selective CDK4 inhibitor, exhibit anti-angiogenic activity in vitro and in vivo. Cancer Chemother. Pharmacol 2007, 59, 439–445. [Google Scholar]
- Soni, R; Muller, L; Furet, P; Schoepfer, J; Stephan, C; Zumstein-Mecker, S; Fretz, H; Chaudhuri, B. Inhibition of cyclin-dependent kinase 4 (Cdk4) by fascaplysin, a marine natural product. Biochem. Biophys. Res. Commun 2000, 275, 877–884. [Google Scholar]
- Kobayashi, J; Suzuki, M; Tsuda, M. Konbu’acidin A, a new bromopyrrole alkaloid with cdk4 inhibitory activity from hymeniacidon sponge. Tetrahedron 1997, 53, 15681–15684. [Google Scholar]
- Mukku, V; Edrada, RA; Schmitz, FJ; Shanks, MK; Chaudhuri, B; Fabbro, D. New sesquiterpene quinols from a micronesian sponge, Aka sp. J. Nat. Prod 2003, 66, 686–689. [Google Scholar]
- Levitzki, A; Mishani, E. Tyrphostins and other tyrosine kinase inhibitors. Annu. Rev. Biochem 2006, 75, 93–109. [Google Scholar]
- Carapancea, M. Strategies to Increase Effectiveness of Growth Factor Receptors-Targeted Therapy in Glioblastoma. Licentiate Thesis, Karolinska Institutet, Stockholm, Sweden, December 2007. [Google Scholar]
- Bifulco, G; Bruno, I; Minale, L; Riccio, R; Debitus, C; Bourdy, G; Vassas, A; Lavayre, J. Bioactive prenylhydroquinone sulfates and a novel C31 furanoterpene alcohol sulfate from the marine sponge, Ircinia sp. J. Nat. Prod 1995, 58, 1444–1449. [Google Scholar]
- Gray, G; Macara, I. The pp60V-SRC tyrosine kinase desensitizes epidermal growth factor binding to 3T3 fibroblasts by two distinct protein kinase C-independent mechanisms. J. Biol. Chem 1988, 263, 10714–10719. [Google Scholar]
- Alvi, KA; Diaz, MC; Crews, P; Slate, DL; Lee, RH; Moretti, R. Evaluation of new sesquiterpene quinones from two Dysidea sponge species as inhibitors of protein tyrosine kinase. J. Org. Chem 1992, 57, 6604–6607. [Google Scholar]
- Lee, RH; Slate, DL; Moretti, R; Alvi, KA; Crews, P. Marine sponge polyketide inhibitors of protein tyrosine kinase. Biochem. Biophys. Res. Commun 1992, 184, 765–772. [Google Scholar]
- Laurent, D; Jullian, V; Parenty, A; Knibiehler, M; Dorin, D; Schmitt, S; Lozach, O; Lebouvier, N; Frostin, M; Alby, F; et al. Antimalarial potential of xestoquinone, a protein kinase inhibitor isolated from a Vanuatu marine sponge Xestospongia sp. Bioorg. Med. Chem 2006, 14, 4477–4482. [Google Scholar]
- Toyooka, N; Nagaoka, M; Sasaki, E; Qin, H; Kakuda, H; Nemoto, H. Model studies toward the total synthesis of halenaquinol and halenaquinone. Tetrahedron 2002, 58, 6097–6101. [Google Scholar]
- Cao, S; Foster, C; Brisson, M; Lazo, JS; Kingston, DGI. Halenaquinone and xestoquinone derivatives, inhibitors of Cdc25B phosphatase from a Xestospongia sp. Bioorg. Med. Chem 2005, 13, 999–1003. [Google Scholar]
- Alvi, KA; Rodriguez, J; Diaz, MC; Moretti, R; Wilhelm, RS; Lee, RH; Slate, DL; Crews, P. Protein tyrosine kinase inhibitory properties of planar polycyclics obtained from the marine sponge Xestospongia cf. carbonaria and from total synthesis. J. Org. Chem 1993, 58, 4871–4880. [Google Scholar]
- Carpenter, G; Cohen, S. Epidermal growth factor. Annu. Rev. Biochem 1979, 48, 193–216. [Google Scholar]
- Kobayashi, J; Inaba, K; Tsuda, M. Tauroacidins A and B, new bromopyrrole alkaloids possessing a taurine residue from hymeniacidon sponge. Tetrahedron 1997, 53, 16679–16682. [Google Scholar]
- Kobayashi, J; Hirano, K; Kubota, T; Tsuda, M; Watanabe, K; Fromont, J. Ma’edamines A and B, cytotoxic bromotyrosine alkaloids with a unique 2(1H)pyrazinone ring from sponge Suberea sp. Tetrahedron 2000, 56, 8107–8110. [Google Scholar]
- Inaba, K; Sato, H; Tsuda, M; Kobayashi, J. Spongiacidins A–D, new bromopyrrole alkaloids from hymeniacidon sponge. J. Nat. Prod 1998, 61, 693–695. [Google Scholar]
- Gossauer, A. Monopyrollic Natural Compounds Including Tetramic Acid Derivatives; Springer: Berlin, Germany, 2003. [Google Scholar]
- Kreuter, M-H; Leake, RE; Rinaldi, F; Müller-Klieser, W; Maidhof, A; Müller, WEG; Schröder, HC. Inhibition of intrinsic protein tyrosine kinase activity of EGF-receptor kinase complex from human breast cancer cells by the marine sponge metabolite (+)-aeroplysinin-1. Comp. Biochem. Physiol. B Biochem. Mol. Biol 1990, 97, 151–158. [Google Scholar]
- Rodriguez-Nieto, S; Gonzalez-Iriarte, M; Carmona, R; Munoz-Chapuli, R; Medina, MA; Quesada, AR. Antiangiogenic activity of aeroplysinin-1, a brominated compound isolated from a marine sponge. FASEB J 2002, 16, 261–263. [Google Scholar]
- Rateb, ME; Houssen, WE; Legrave, NM; Clements, C; Jaspars, M; Ebel, R. Dibenzofurans from the marine sponge-derived ascomycete Super1F1-09. Bot. Mar 2010, 53, 499–506. [Google Scholar]
- Rateb, ME; Houssen, WE; Schumacher, M; Harrison, WTA; Diederich, M; Ebel, R; Jaspars, M. Bioactive diterpene derivatives from the marine sponge Spongionella sp. J. Nat. Prod 2009, 72, 1471–1476. [Google Scholar]
- Kannan-Thulasiraman, P; Katsoulidis, E; Tallman, MS; Arthur, JSC; Platanias, LC. Activation of the mitogen- and stress-activated kinase 1 by arsenic trioxide. J. Biol. Chem 2006, 281, 22446–22452. [Google Scholar]
- Buchanan, MS; Edser, A; King, G; Whitmore, J; Quinn, RJ. Cheilanthane sesterterpenes, protein kinase inhibitors, from a marine sponge of the genus Ircinia. J. Nat. Prod 2001, 64, 300–303. [Google Scholar]
- Brown, J; Kesler, C; Neary, J; Fishman, L. Effects of marine sponge extracts on mitogen-activated protein kinase (MAPK/ERK1,2) activity in SW-13 huma adrenal carcinoma cells. Toxicon 2001, 39, 1835–1839. [Google Scholar]
- Kolch, W. Meaningful relationships: The regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem. J 2000, 351, 289–305. [Google Scholar]
- Tasdemir, D; Mallon, R; Greenstein, M; Feldberg, L; Kim, S; Collins, K; Wojciechowicz, D; Mangalindan, G; Concepcion, G; Harper, MK; Ireland, CM. Aldisine alkaloids from the Philippine sponge Stylissa massa are potent Inhibitors of mitogen-activated protein kinase kinase-1 (MEK-1). J. Med. Chem 2002, 45, 529–532. [Google Scholar]
- Segraves, NL; Crews, P. A Madagascar sponge Batzella sp. as a source of alkylated iminosugars. J. Nat. Prod 2005, 68, 118–121. [Google Scholar]
- Freitas, J; Malpezzi, E; Costa, L; Berlinck, R; Almeida, A; Ogawa, C; Sanchez, M; Hajdu, E. Cytotoxic and neurotoxic effects induced by halitoxin isolated from Amphimedon viridis (Porifera). Toxicon 1996, 34, 335. [Google Scholar]
- Lee, K-H; Nishimura, S; Matsunaga, S; Fusetani, N; Horinouchi, S; Yoshida, M. Inhibition of protein synthesis and activation of stress-activated protein kinases by onnamide A and theopederin B, antitumor marine natural products. Cancer Sci 2005, 96, 357–364. [Google Scholar]
- Fedorov, S; Bode, A; Stonik, V; Gorshkova, I; Schmid, P; Radchenko, O; Berdyshev, E; Dong, Z. Marine alkaloid polycarpine and its synthetic derivative dimethylpolycarpine induce apoptosis in JB6 cells through p53- and caspase 3-dependent pathways. Pharm. Res 2004, 21, 2307–2319. [Google Scholar]
- Fusetani, N; Sugawara, T; Matsunaga, S. Bioactive marine metabolites. 41. Theopederins A–E, potent antitumor metabolites from a marine sponge, Theonella sp. J. Org. Chem 1992, 57, 3828–3832. [Google Scholar]
- Sakemi, S; Ichiba, T; Kohmoto, S; Saucy, G; Higa, T. Isolation and structure elucidation of onnamide A, a new bioactive metabolite of a marine sponge, Theonella sp. J. Am. Chem. Soc 1988, 110, 4851–4853. [Google Scholar]
- Andreasen, P. PAI-1—A Potential therapeutic target in cancer. Curr. Drug Targets 2007, 8, 1030–1041. [Google Scholar]
- Williams, DE; Telliez, JB; Liu, J; Tahir, A; van Soest, R; Andersen, RJ. Meroterpenoid MAPKAP (MK2) inhibitors isolated from the Indonesian marine sponge Acanthodendrilla sp. J. Nat. Prod 2004, 67, 2127–2129. [Google Scholar]
- Hamann, M; Alonso, D; Martin-Aparicio, E; Fuertes, A; Perez-Puerto, M; Castro, A; Morales, S; Navarro, M; Monte-Millan, M; Medina, M; et al. Glycogen synthase kinase-3 (GSK-3) inhibitory activity and structure activity relationship (SAR). Studies of the manzamine alkaloids. Potential for Alzheimer’s disease. J. Nat. Prod 2007, 70, 1397–1405. [Google Scholar]
- Ang, K; Holmes, M; Higa, T; Hamann, M; Kara, U. In vivo antimalarial activity of the beta-carboline alkaloid manzamine A. Antimicrob. Agents Chemother 2000, 44, 1645–1649. [Google Scholar]
- Kara, U; Higa, T; Holmes, M; Ang, K. Antimalarial activity of β-carboline alkaloids. US Patent 6,143,756, 7 November 2000. [Google Scholar]
- McCulloch, MWB; Bugni, TS; Concepcion, GP; Coombs, GS; Harper, MK; Kaur, S; Mangalindan, GC; Mutizwa, MM; Veltri, CA; Virshup, DM; et al. Carteriosulfonic acids A–C, GSK-3 beta inhibitors from a Carteriospongia sp. J. Nat. Prod 2009, 72, 1651–1656. [Google Scholar]
- Marion, F; Williams, DE; Patrick, BO; Hollander, I; Mallon, R; Kim, SC; Roll, DM; Feldberg, L; Van Soest, R; Andersen, RJ. Liphagal, a selective inhibitor of PI3 kinase alpha isolated from the sponge Aka coralliphaga: Structure elucidation and biomimetic synthesis. Org. Lett 2006, 8, 321–324. [Google Scholar]
- Alvarez-Manzaneda, E; Chahboun, R; Alvarez, E; Cano, MJ; Haidour, A; Alvarez-Manzaneda, R. Enantioselective total synthesis of the selective PI3 kinase inhibitor liphagal. Org. Lett 2010, 12, 4450–4453. [Google Scholar]
- Hertiani, T; Edrada-Ebel, RA; Kubbutat, MHG; van Soest, RWM; Proksch, P. Protein kinase inhibitors from Indonesian sponge Axynissa sp. Maj. Farm. Indones 2008, 19, 78–85. [Google Scholar]
- Zivanovic, A; Pastro, NJ; Fromont, J; Thomson, M; Skropeta, D. Kinase Inhibitory, haemolytic and cytotoxic activity of three deep-water sponges from North Western Australia and their fatty acid composition. Nat Prod Commun 2011, in press. [Google Scholar]
- Lebouvier, N; Jullian, V; Desvignes, I; Maurel, S; Parenty, A; Dorin-Semblat, D; Doerig, C; Sauvain, M; Laurent, D. Antiplasmodial activities of homogentisic acid derivative protein kinase inhibitors isolated from a vanuatu marine sponge Pseudoceratina sp. Mar. Drugs 2009, 7, 640–653. [Google Scholar]
- Sauleau, P; Retailleau, P; Nogues, S; Carletti, I; Marcourt, L; Raux, R; Al Mourabit, A; Debitus, C. Dihydrohymenialdisines, new pyrrole-2-aminoimidazole alkaloids from the marine sponge Cymbastela cantharella. Tetrahedron Lett 2011, 52, 2676–2678. [Google Scholar]
- Skropeta, D. Deep-sea natural products. Nat. Prod. Rep 2008, 25, 1131–1166. [Google Scholar]








{kind=link}
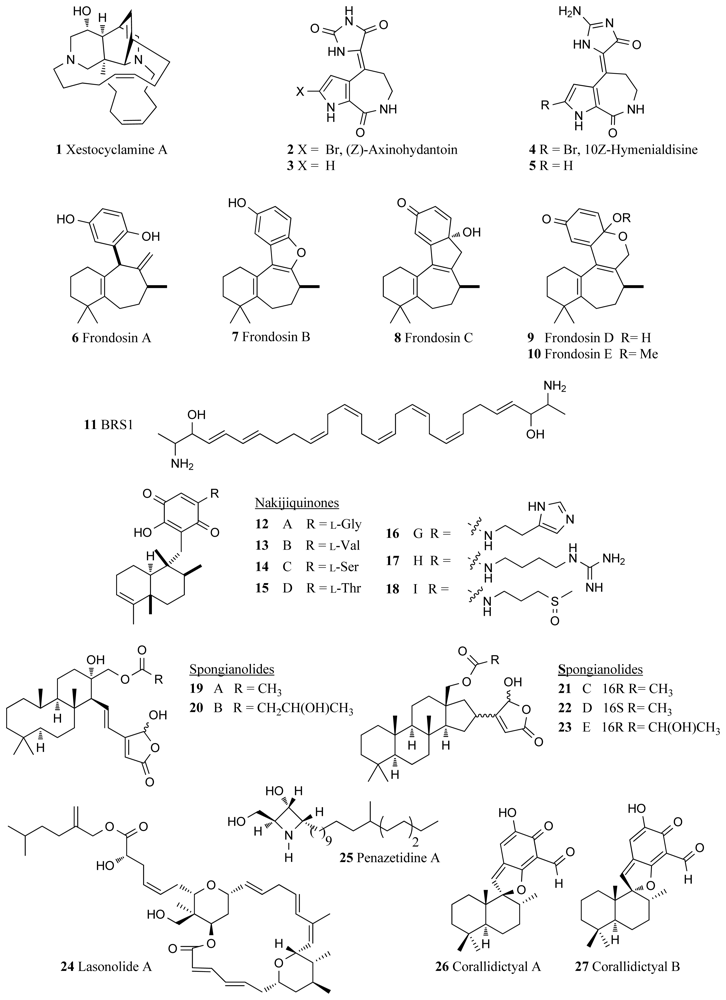{kind=link}
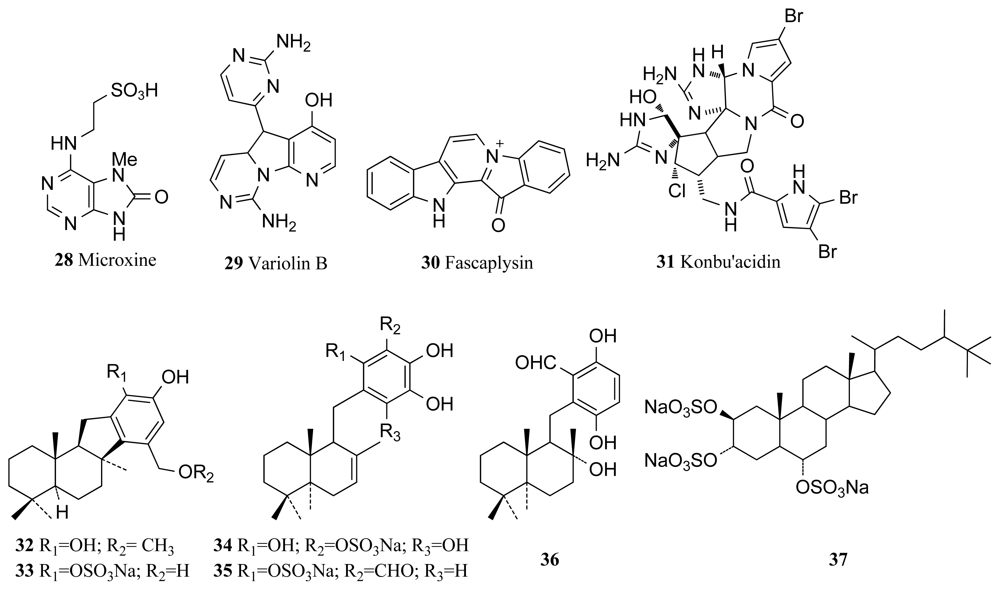{kind=link}
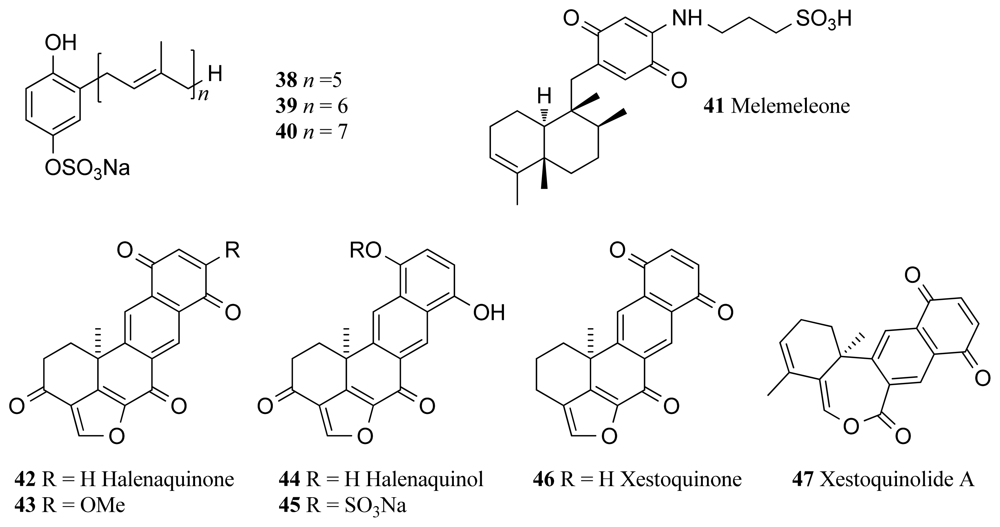{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Kinase | Sponge species | Natural product (or compound type) | IC50 (μM) ₤ (or % inhibition) | Ref. |
|---|---|---|---|---|
| PKC | Xestospongia sp. | Xestocyclamine A (1) | 10 | [19] |
| Stylotella aurantium | Axinohydantoins (2, 3) | 9–22 | [22] | |
| Stylotella aurantium | Hymenialdisines (4, 5) | 0.8–1.3 | [22] | |
| Dysidea frondosa | Frondosins A–E (6–10) | 2–31 | [23] | |
| Class Calcarea | BRS 1 (11) | 98 | [28] * | |
| Family Spongiidae | Nakijiquinones A–D, G–I (12–18) | 23–270 | [29,30] | |
| Spongia sp. | Spongianolides A–E (19–23) | 20–30 | [36] | |
| Forecpia sp. | Lasonolide A (24) | 0.03 | [38,40] * | |
| Penares sollesi | Penazetidine A (25) | 1 | [41] | |
| Aka coralliphaga | Corallidictyals A and B (26, 27) | 28 | [42] | |
| CDK | Axinella verrucosa | Hymenialdisine (4) | 0.02 | [48] * |
| Microxina sp. | Microxine (28) | 13 | [52] | |
| Kirkpatrickia varialosa | Variolin B (29) | 0.03 | [53] * | |
| Fascaplysinopsis sp. | Fascaplysin (30) | 0.4 | [61] * | |
| Hymeniacidon sp. | Konbu’acidin A (31) | 27 | [62] | |
| Aka sp. | Quinol derivative (34) | 0.019 | [63] | |
| Aka sp. | Halistanol sulfate (37) | 0.013 | [63] | |
| TPK | Ircinia sp. | Prenylhydroquinone 4-sulfates (38–40) | 7–15 | [66] |
| Dysidea sp. | Melemeleone B (41) | 28 | [68] | |
| Xestospongia sp. | Halenoquinone (42, 43) | 1.5–5 | [69,73] * | |
| Xestospongia sp. | Halenaquinols (44, 45) | 0.6–60 | [69] * | |
| Xestospongia sp. | Xestoquinone (46) | 28 | [69] * | |
| Xestospongia sp. | Xestoquinolide A (47) | 80 | [73] * | |
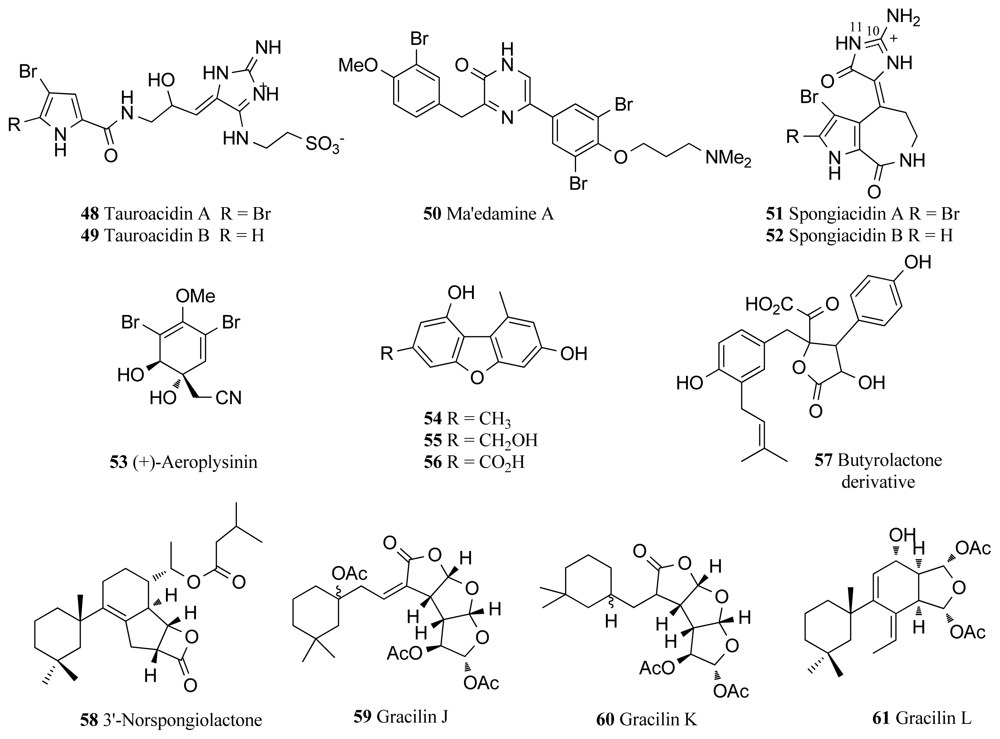
| EGFR | Hymeniacidon sp. | Tauroacidins A–B (48, 49) | 38–45 | [75] |
| Suberea sp. | Ma’edamine A (50) | 11 | [76] | |
| Hymeniacidon sp. | Spongiacidins A–B (51, 52) | 19–21 | [78] | |
| Verongia aerophoba | Aeroplysinin-1 (53) | 0.5 | [79] | |
| Acanthella cavernosa | Dibenzofurandiols (54–57) | 33–59 † | [81] | |
| Spongionella sp. | 3′-Norspongiolactone (58) | 25 † | [82] | |
| Spongionella sp. | Gracilins J–L (59–61) | 19–75 † | [82] | |
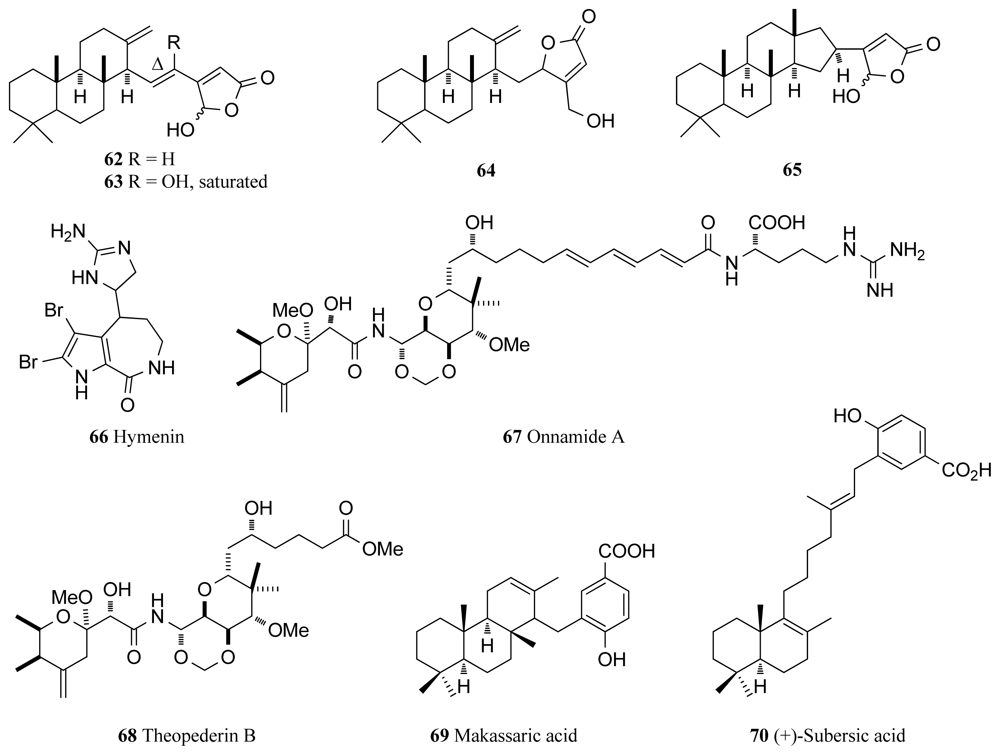
| MAPK | Ircinia sp. | Cheilanthene sesterpenoids (62–65) | 4–90 | [84] |
| Raf/MAP | Stylissa massa | Hymenialdisines (4, 5) | 0.003–0.006 | [87] * |
| Stylotella aurantium | Hymenin (66) | 129 | [87] * | |
| Theonella sp. | Theopederin B (68) | - ‡ | [90,92] * | |
| Theonella sp. | Onnamide A (67) | - ‡ | [90,93] * | |
| Acanthodendrilla sp. | (+)-Makassaric acid (69) | 20 | [95] | |
| Acanthodendrilla sp. | (+)-Subersic acid (70) | 9.6 | [95] | |
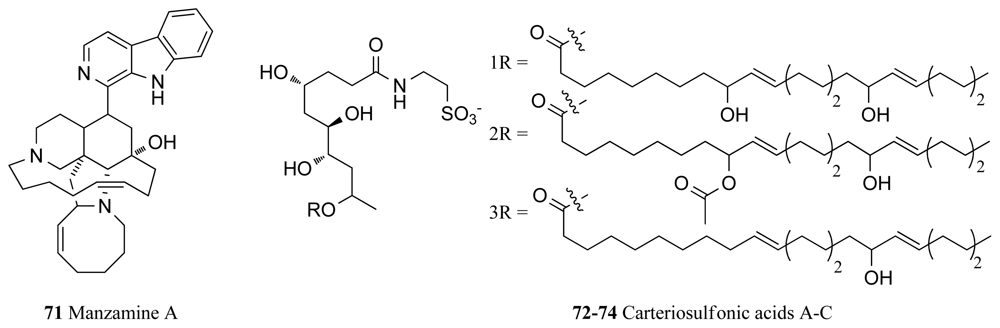
| GSK-3 | Haliclona sp. | Manzamine A (71) | 10 | [96] * |
| Unidentified sp. | Glycerol lipids (72–74) | 0.1–0.4 | [99] | |
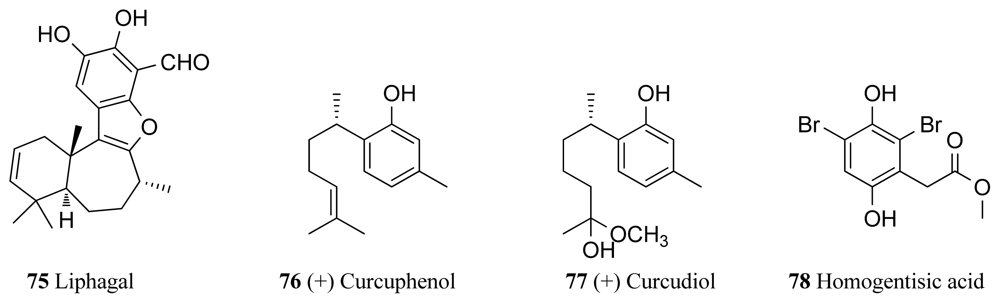
| Others | Aka coralliphaga | Liphagal (75) | 0.1 | [100,101] |
| Axynissa sp. | (+)-Curcuphenol (76) | 36 | [102] | |
| Axynissa sp. | (+)-Curcudiol (77) | 37 | [102] | |
| Pseudoceratina sp. | Homogentisic acid (78) | 1.8 | [104] |
© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Skropeta, D.; Pastro, N.; Zivanovic, A. Kinase Inhibitors from Marine Sponges. Mar. Drugs 2011, 9, 2131-2154. https://doi.org/10.3390/md9102131
Skropeta D, Pastro N, Zivanovic A. Kinase Inhibitors from Marine Sponges. Marine Drugs. 2011; 9(10):2131-2154. https://doi.org/10.3390/md9102131
Chicago/Turabian StyleSkropeta, Danielle, Natalie Pastro, and Ana Zivanovic. 2011. "Kinase Inhibitors from Marine Sponges" Marine Drugs 9, no. 10: 2131-2154. https://doi.org/10.3390/md9102131
APA StyleSkropeta, D., Pastro, N., & Zivanovic, A. (2011). Kinase Inhibitors from Marine Sponges. Marine Drugs, 9(10), 2131-2154. https://doi.org/10.3390/md9102131