1. Introduction
Many studies in the field of forensic science have shown the benefits of using single-nucleotide polymorphism (SNP) data in DNA investigation and intelligence [
1,
2,
3,
4]. In the last 30 years, standard forensic DNA-based methods have been built on short tandem repeats (STR). Such STR databases exist in most countries and a direct comparison between DNA samples and these databases is admissible in court for human identification purposes. However, STRs were originally selected to be forensically relevant for identification, but not necessarily include traits that can be used for phenotype and ancestry inference [
5]. The advances in DNA high-throughput sequencing (HTS) have improved the study of SNPs and their potential uses for forensics purposes [
6]. The ability to use millions of available SNPs, as opposed to a limited number of STR sites (13–26), opens the door for new forensics applications. Forensically relevant SNPs have been categorized into four groups of potential uses: (1) individual identification, (2) kinship search, (3) biogeographical ancestry, and (4) external visible characteristics [
7]. The main SNP genotyping technique for forensics has been customized SNP arrays, intended specifically for forensic use, such as SNaPshot [
8]. Such assays have been published for a wide range of markers, some of them combining STRs and SNPs in the same assay, using next generation sequencing [
9]. These assays, despite their lower costs, are limited to the SNPs they were designed for. Alternatively, as the cost of sequencing decreases, whole genome sequencing (WGS) of the sample can be conducted and then analyzed for all relevant markers, as suggested in [
10,
11,
12]. Currently this is not done, mostly due to considerations of ethics, time of analysis, and budget, but in the future it might be resolved.
The two primary challenges in forensic DNA are highly degraded samples and mixed samples (containing two or more individuals) [
13,
14]. When addressing degraded samples, SNPs have an advantage over STRs, as they require shorter amplicon lengths, and can overcome missingness as there are many sites that are spread across the whole genome. Regarding the mixture analysis, many studies have focused on the individualization problem, i.e., inferring the presence or absence of a known individual (POI—Person Of Interest) from a mixed DNA sample. STR-based methods such as STRmix [
15], LRmix Studio [
16] and EuroForMix [
17] achieve forensically sufficient discrimination when handling mixtures with 2–3 contributors, but lose power when this number increases [
18]. Gill et al. [
19] and Bleka et al. [
20] show how these STR packages, based on the widely used LR method, can be adapted to a SNP scenario. They are still limited to 2–3 person cases and lose accuracy when the contribution from the POI is low in uneven mix ratios. Other SNP based methods achieve good results even with complex mixtures of three or more contributors and various mixture ratios, outperforming STR-based methods. These algorithms rely on deep sequencing and on specific assumptions regarding the minor allele frequency (MAF) of the examined SNPs [
21] and the mix ratios [
18,
22]. It should be noted that since most SNPs are bi-allelic [
23], it is harder to recognize the presence of a mix in an “SNP only” profile. STRs, on the other hand, are multi-allelic, enabling easier recognition of the presence of more than one donor. To confront the shortcomings of bi-allelic SNPs, Kidd et al. [
24] offered the use of a new marker type, microhaplotype, a region with two or more SNPs that occur within the length of an HTS read, effectively creating a multi-allelic marker. This “long-read” based approach was applied in Voskoboinik et al. [
25].
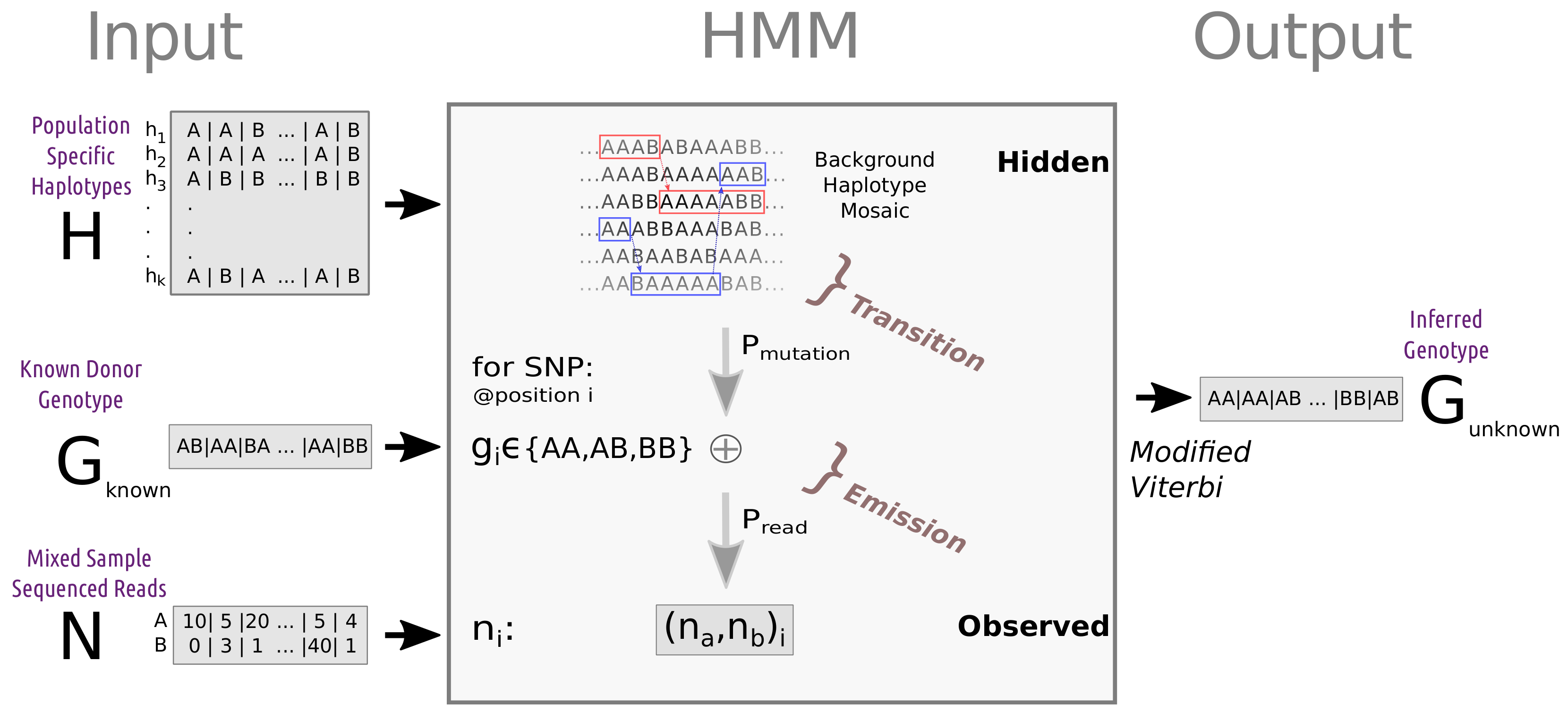
An important challenge with many potential forensic use cases is de-novo reconstruction of an unknown SNP profile from a DNA mixture. DEploid [
26], a framework designed for deconvolution of malaria haplotype strains in a mixture, has been adapted to separate mtDNA from a two person mixed sample [
27], potentially for future forensic use. To address a case in which WGS data is needed, we here introduce AH-HA, a novel approach to infer an unknown SNP profile from a DNA mixture of two individuals. Our method receives as an input WGS data of a two-person mixture, in which one genotype is known (“victim”) and the other is unknown (“suspect”). AH-HA is compared with other computational approaches over varying sequencing depths. It is designed to cope with low coverage (~5×) and missing reads by adapting the Li and Stephens model [
28], as described further in the Methods Section. This model is widely used for ancestral inference [
29], haplotype phasing [
30], and imputation [
31]. By using a population-specific reference panel with a hidden Markov model (HMM), AH-HA can infer the genotype for the unknown individual in each SNP.
4. Discussion
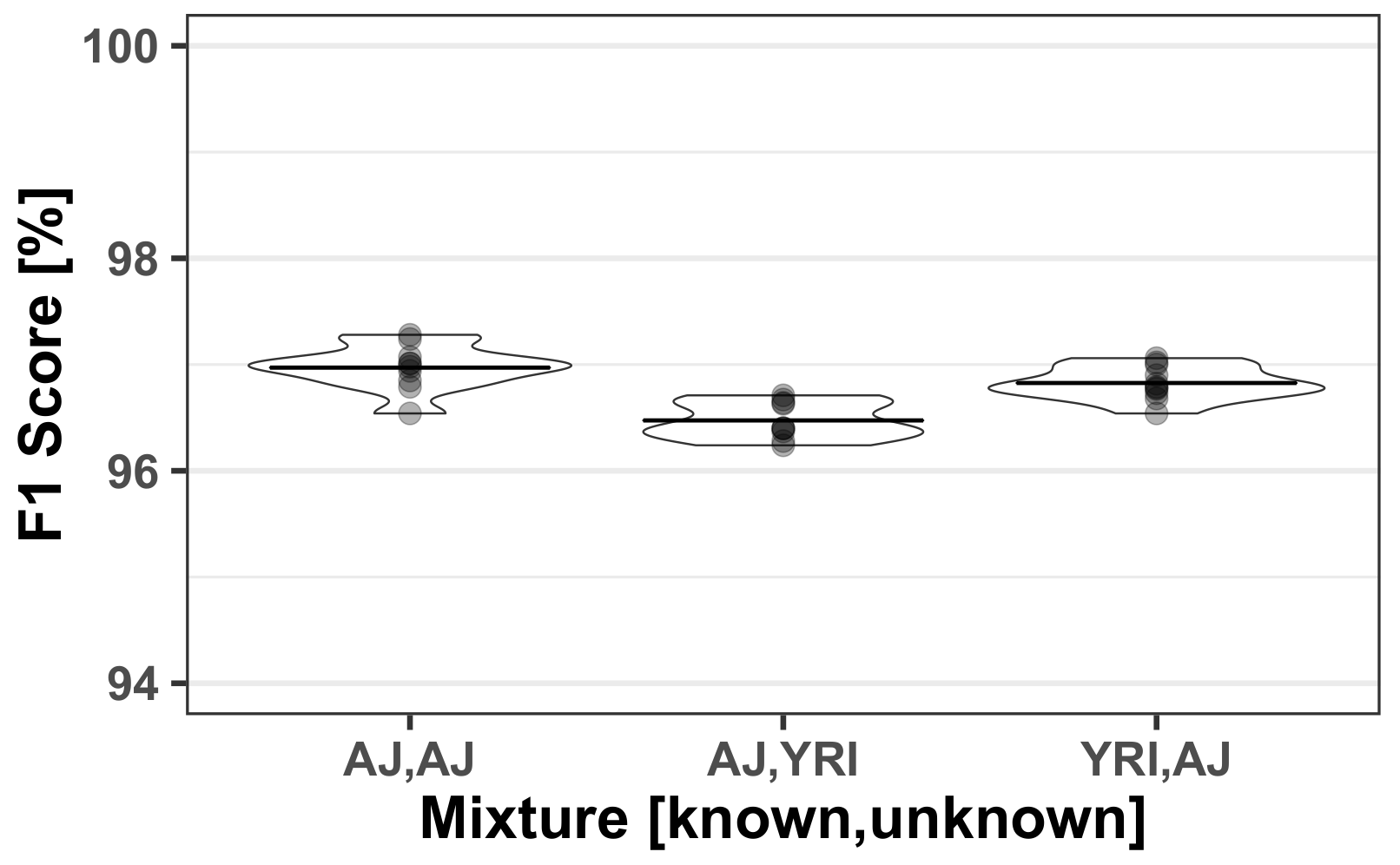
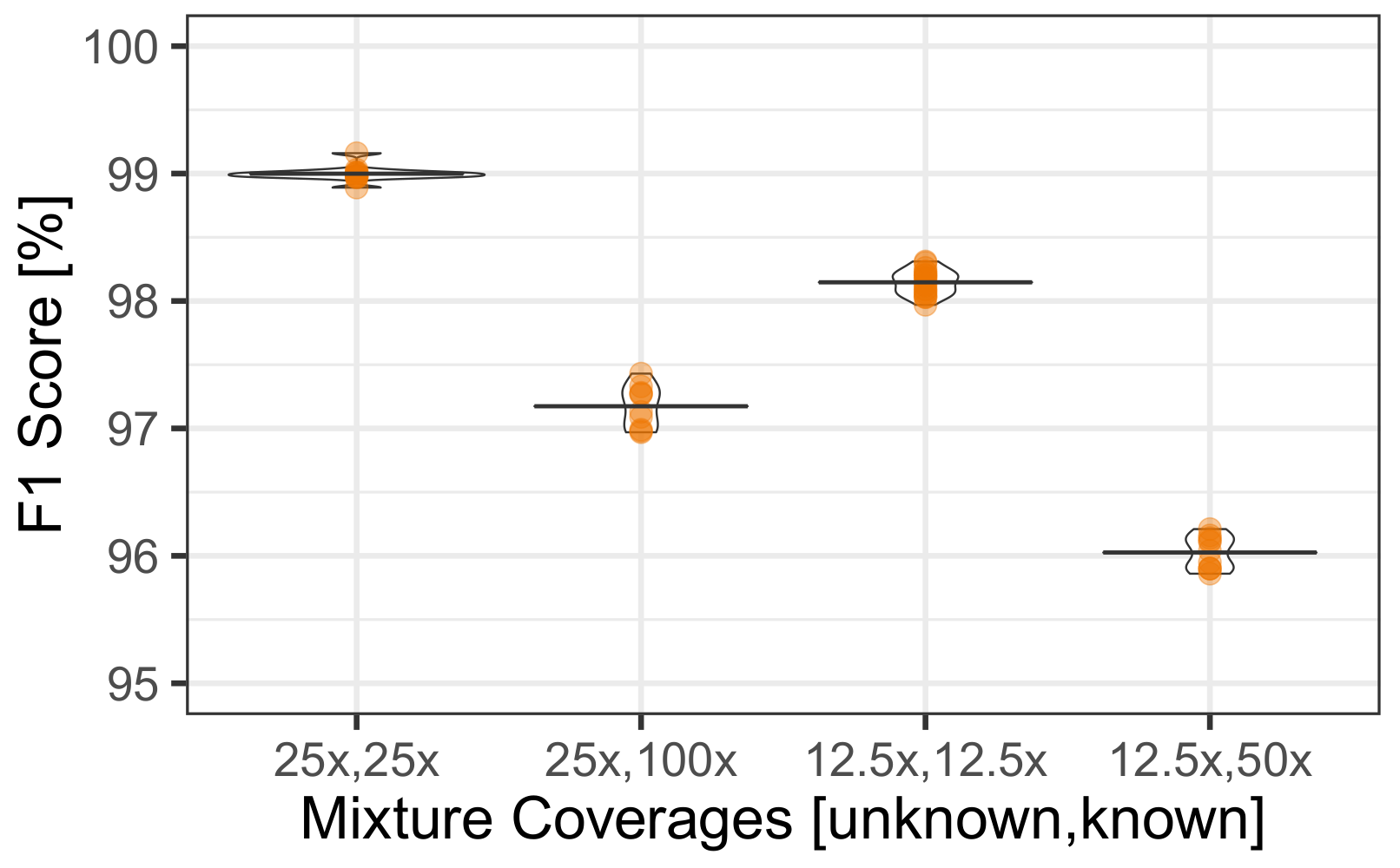
In this paper we introduced AH-HA, an approach to infer the SNP profile of an unknown individual from an HTS of a mixed sample. It outperforms other methods’ F1 scores over varying coverage rates, as demonstrated in
Figure 2B. In particular, the performance for low coverage is superior compared with more naïve algorithms, as shown in
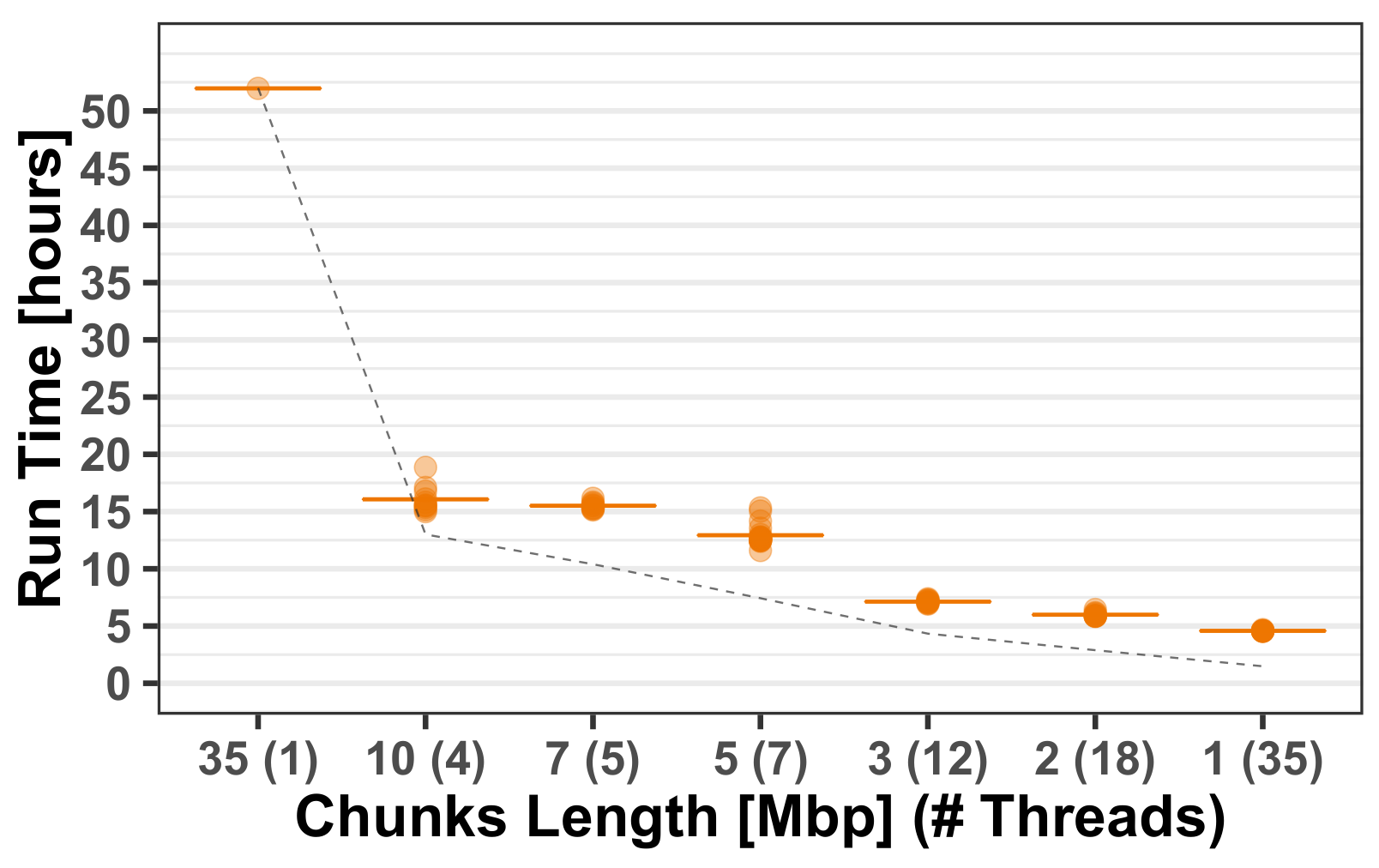
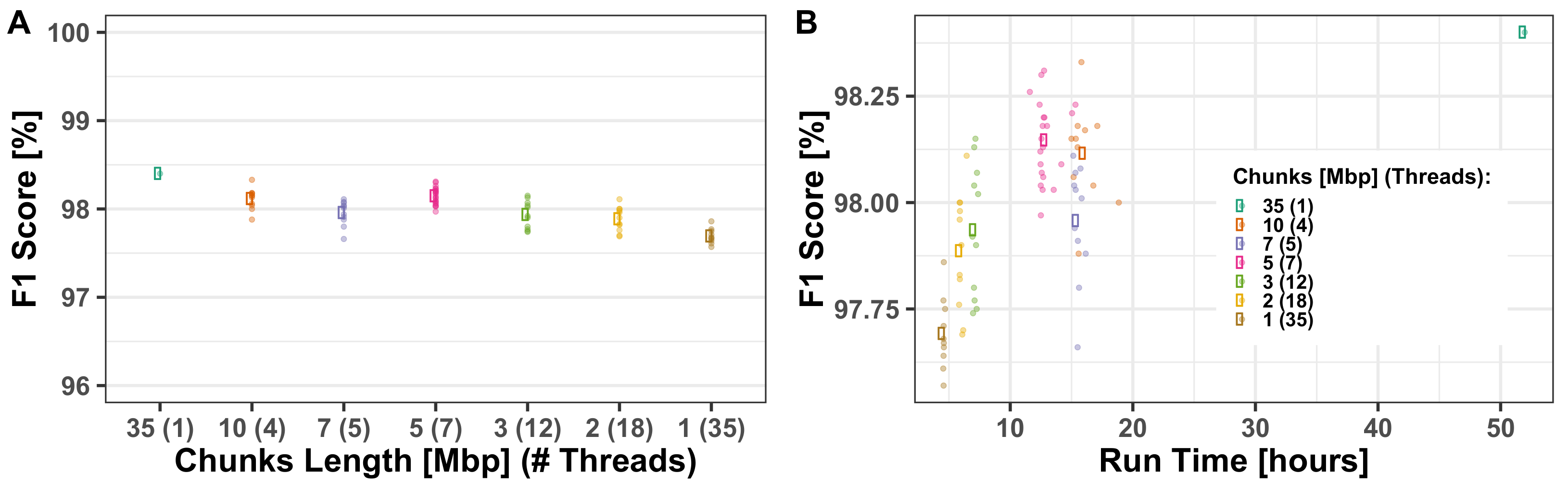
Section 3.1.1. The robustness of AH-HA was shown over all chromosomes, mixed population cases and different hyper-parameters. AH-HA’s run-time and memory was improved by utilizing multi-thread parallelization, splitting the chromosome into chunks and processing them simultaneously, as demonstrated in
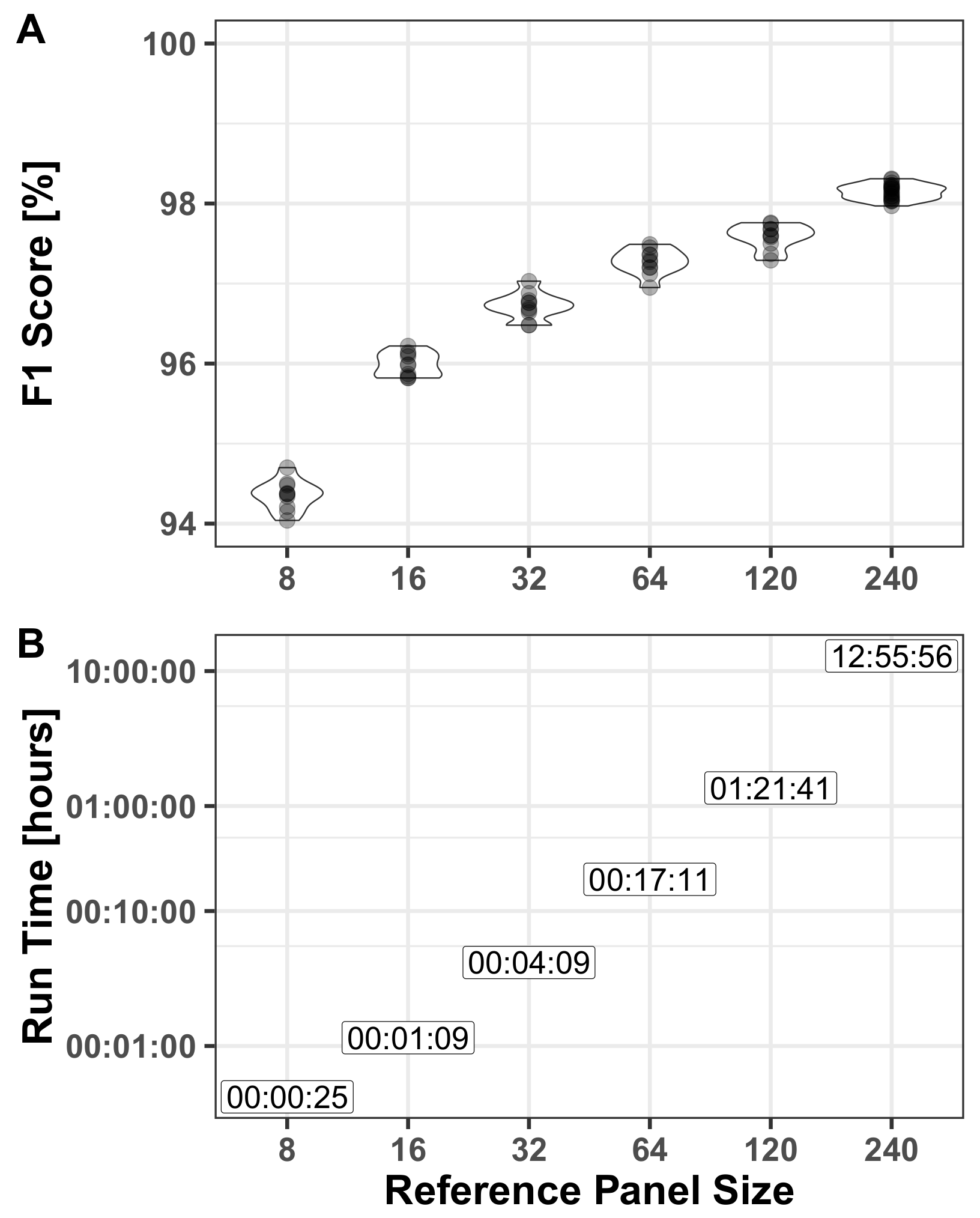
Section 3.1.2. The size of the reference panel strongly affects the run time but has a minor impact on the model’s performance, as demonstrated in
Section 3.2.2. Consequently, AH-HA performs better than naïve methods, while maintaining comparable run times. Our approach uses two sources of information: one based only on read counts (without any LD consideration) and the other leaning heavily on LD. This means that when the read count for the observed allele is low, there is a higher weight to the LD model, essentially imputing the genotype.
Other algorithms dealing with SNPs in forensic DNA mixtures focus only on individual identification in a complex mixtures [
14], mainly for inclusion or exclusion of a specific known individual from a mixture. For example, Voskoboinik et al. [
21] showed that using 1000 SNPs with relatively low minor allele frequencies (~0.05–0.1), the presence of a known person in a ten-person mixture can be identified with high confidence. Ricke et al. [
18] introduced a method that utilizes 2655 SNPs for identifying sub profiles from a mixture with uneven mixture ratios. These other algorithms focus on a different problem (identifying the presence of a known person). They are using a customized SNP panel of several thousand markers and require high coverage. AH-HA, on the contrary, works on hundreds of thousands of SNPs inferred from NGS data with moderate to low coverage (5×). Also, these algorithms require certain minor allele frequencies (MAF) in their data and uneven mixture ratios, while in AH-HA all of the called SNPs were used.
It should be mentioned that AH-HA will not match the whole genome sequence of the unknown/suspect even if the deconvolution is accurate, as it does not infer sites that are not in the reference panel, which may include variants that are individual-specific. In our case, sites not included in TAGC128 were excluded. If a site does not appear in TAGC128, everyone has the exact same allele. Such sites are not informative and do not affect the HMM. These excluded sites can still be inferred from the mixture using the naïve BYS algorithm.
AH-HA requires prior knowledge about the ethnic origin of the suspect and an accurate genotype of the victim. In case of unknown ancestry of the suspect, available forensic and investigative methods can be used, for example by searching for unique markers in the mixture that do not belong to the victim and are indicative for deducing ancestry [
39,
40].
Currently, AH-HA handles only bi-allelic SNPs, which are the majority of SNPs [
23]. For uneven mixture ratios, assuming the ratio is deduced beforehand with another technique, the emission probabilities can be adjusted for the new ratios, as shown in
Section 3.4 for a 1:4 case. AH-HA can confront noisy reads by changing
. In case of an admixed unknown individual, we would need a multi-ancestry panel. Also, instead of the Viterbi solver that gives only the “best” path of the HMM, a softer solving method can be used. As discussed in Rabiner et al. [
38], combining probabilities from the forward and backward stages the probability for each state value can be calculated, enabling a soft decision over all state values (per SNP) and even a “confidence” measure.
In mixtures with more than two individuals, when only one of them is unknown, AH-HA will require all known genotypes, and the emission step should be adapted accordingly. Extending AH-HA to infer more than one unknown individual is a greater challenge for the currently used HMM. First of all, computation wise, the algorithm will need to process for every SNP all combinations of haplotype pairs for every unknown individual. This will increase computation cost by two polynomial orders for each additional unknown individual. E.g., if we have two unknown individuals to infer from the mixture instead of one, AH-HA will effectively be calculating a 4D hidden state matrix (2 × 2) instead of a 2D matrix. In the case of all haplotypes coming from the same reference panel with J haplotypes, this would mean calculating and saving hidden state probabilities for each SNP. The second challenge is assignment of haplotypes to individuals. The algorithm infers four haplotypes, but without additional information on the target individuals, it is difficult to assign these haplotypes into two genotypes correctly.
AH-HA can be extended by exploring new implementation methods. First, incorporating “read-based” inference to the algorithm. This approach has the ability to accurately “stitch” SNPs from the same haplotype by overlapping read sequences (containing two or more SNPs) [
41]. This will result in a better haplotype estimation for closely positioned SNPs, improving genotype inference. Second, inference can be made using the Markov chain Monte Carlo (MCMC) algorithm, similar to the method used by SHAPEIT [
33]. This has the potential to improve run time and memory, but maintain accuracy. Another approach for improving run times could be to scale up from looking at per SNP states into using “representative” haplotype chunks as state values, similar to the method used in BEAGLE [
42]. Also, adapting our model to fit the algorithm described by Lunter [
43] and solve based on a positional Burrows–Wheeler transform may significantly improve run time and will be a subject of future research.
AH-HA requires HTS as input and currently cannot use cheaper SNP typing alternatives. Adapting it to using such alternatives may be doable, but it should be more difficult to perform deconvolution of the mixture using such data. This is because the available information is in the form of intensities (i.e., "analog" information vs. the "digital" information in sequencing).
AH-HA is built on the principles of the Li and Stephens model [
28], which revolutionized phasing, imputation, and ancestry inference. However, in the context of DNA mixture analysis, this model’s potential has not been fully realized. AH-HA opens the door for future studies in DNA mixture analysis, which will develop as more and more HTS elements are being used for forensic work.


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}