
Hypervalence: A Useful Concept or One That Should Be Gracefully Retired?

Abstract
:1. Introduction
“Remarkably, there is not a single chemist among the authors, thus reinforcing the misconception that chemistry lacks any philosophical substance or profound intellectual content. The steady growth in interest in the philosophy of chemistry since the early 1990s demands that this situation be rectified by considering what chemical ideas may also be in need of ‘killing off’.”
2. Some Definitions and Examples
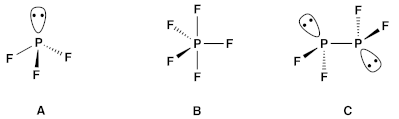
3. Historical Origins and Early Bonding Models

“The extension of the valence space to d-orbitals, which was earlier suggested to explain the stability of so-called hypervalent molecules, was discarded on the basis of quantum chemical calculations. Atomic orbitals with higher angular momentum such as d and f functions only serve as polarization functions for the sp space, but they are not genuine valence orbitals in main-group compounds.”
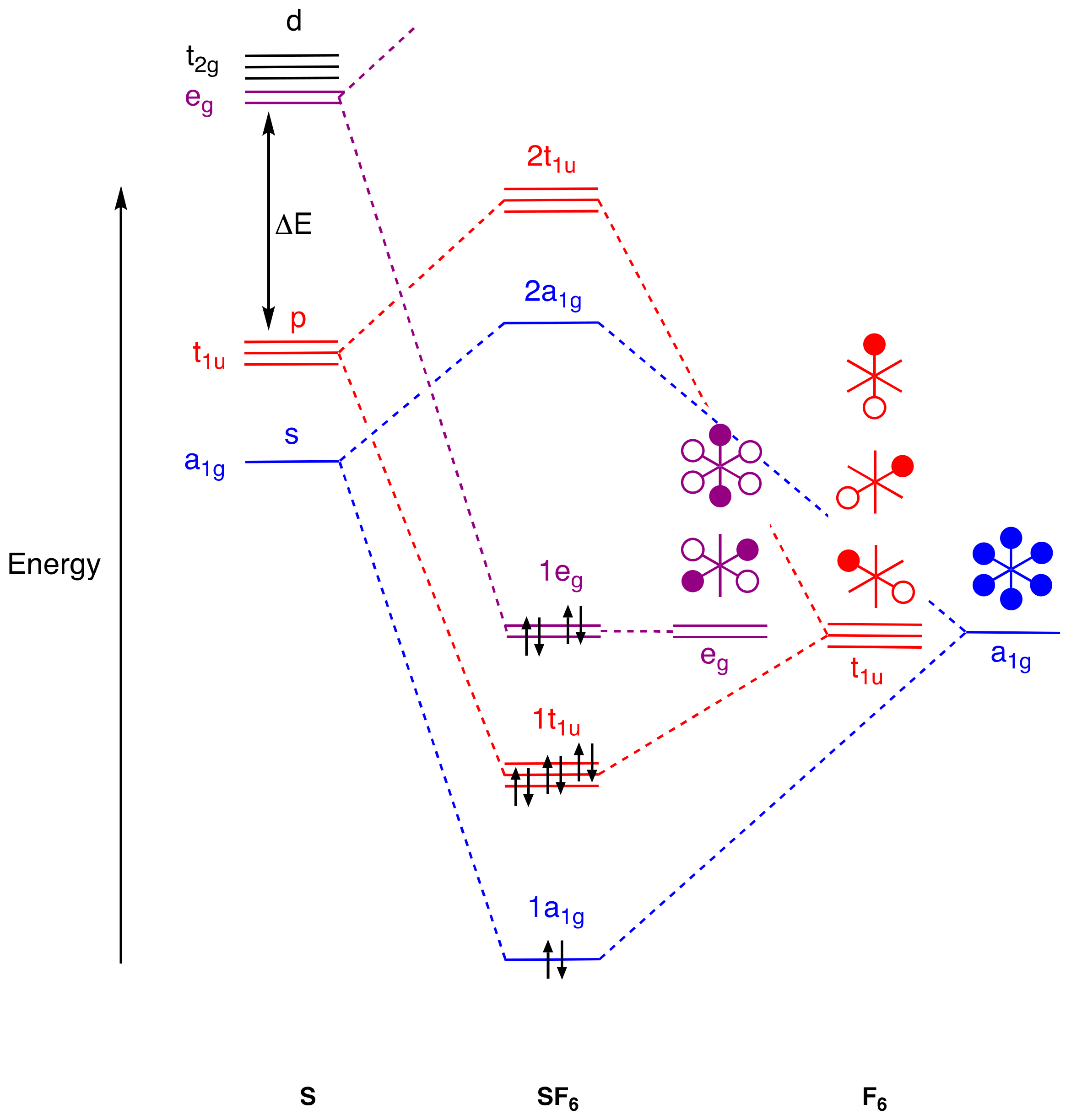
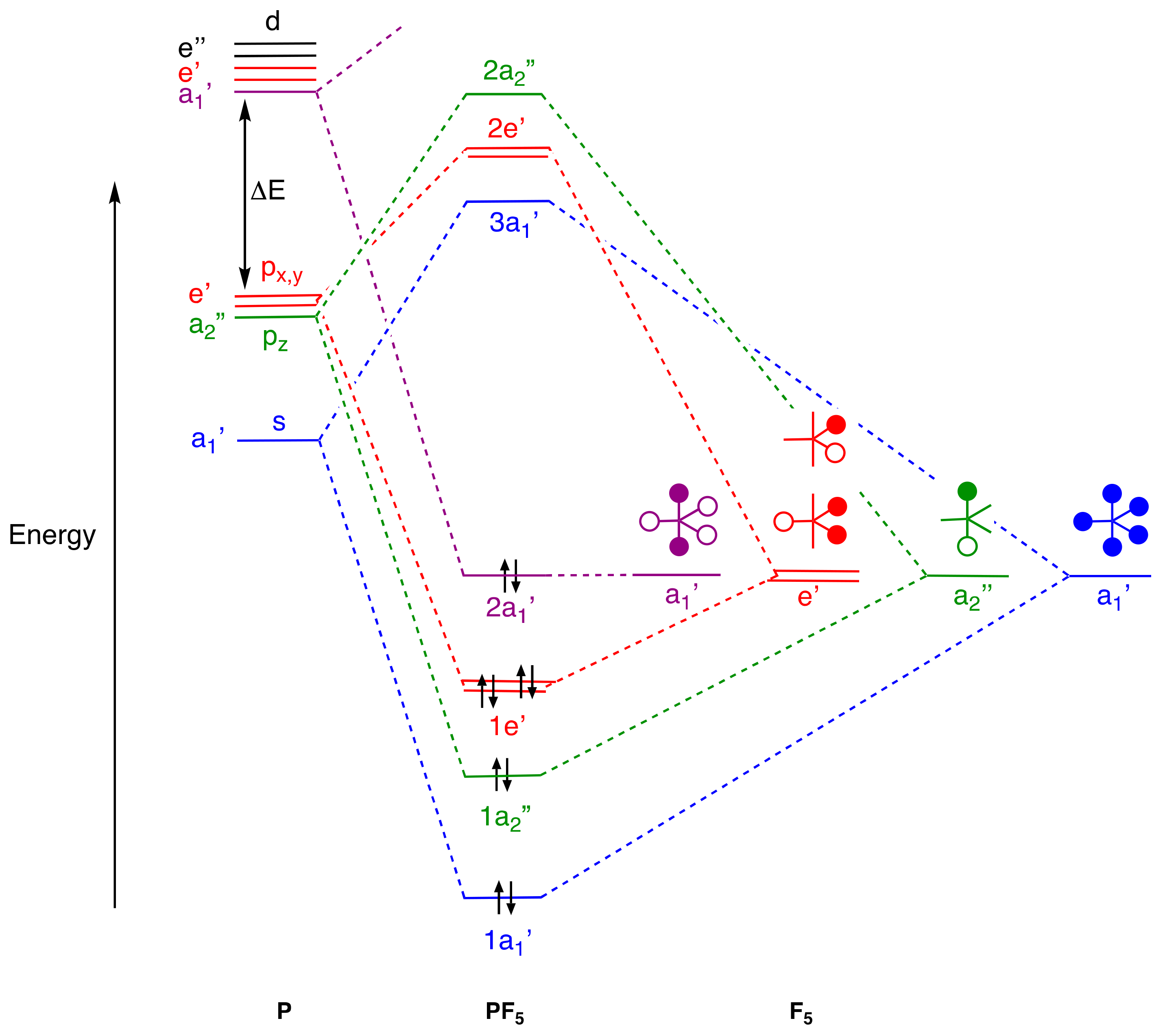
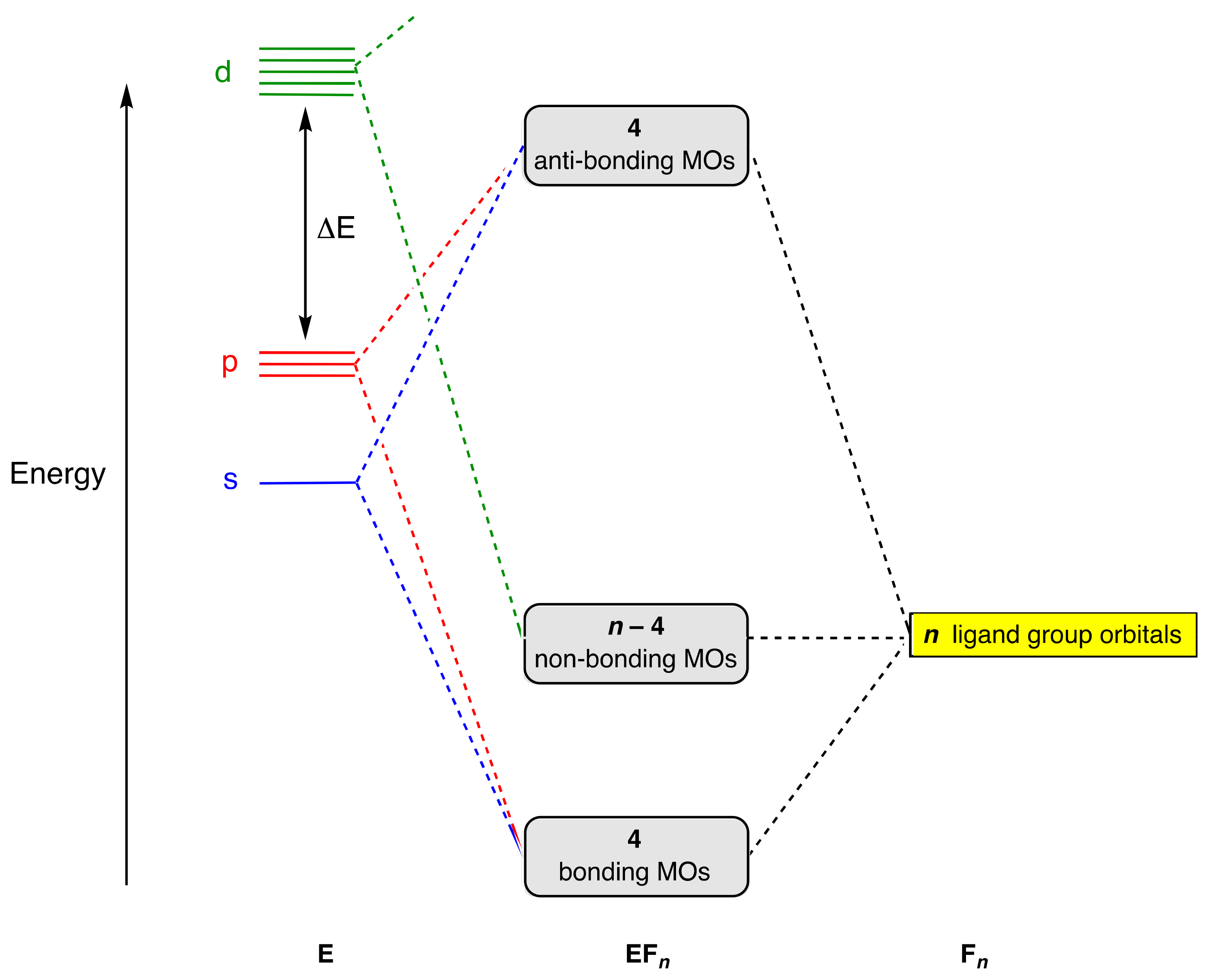
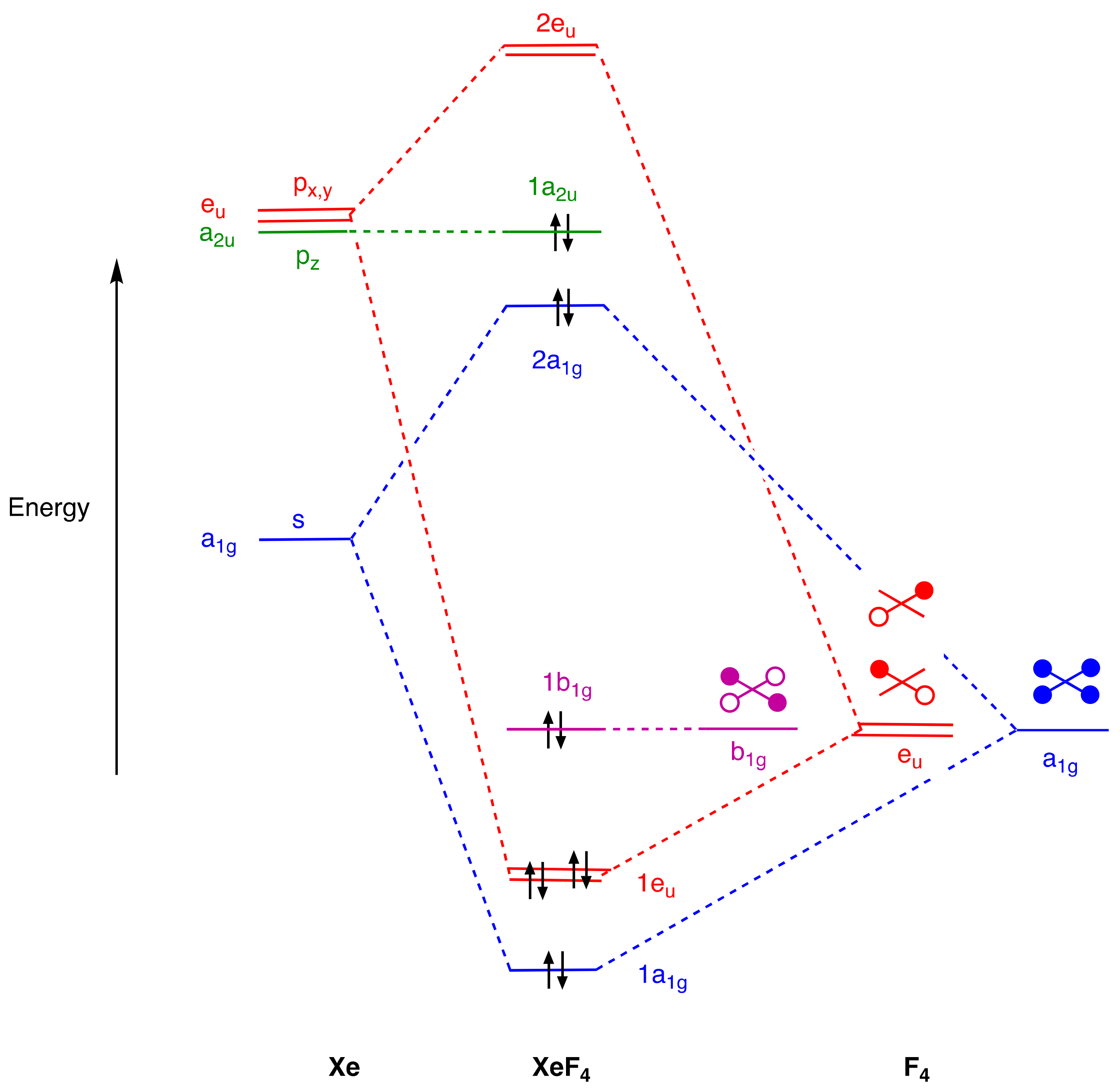
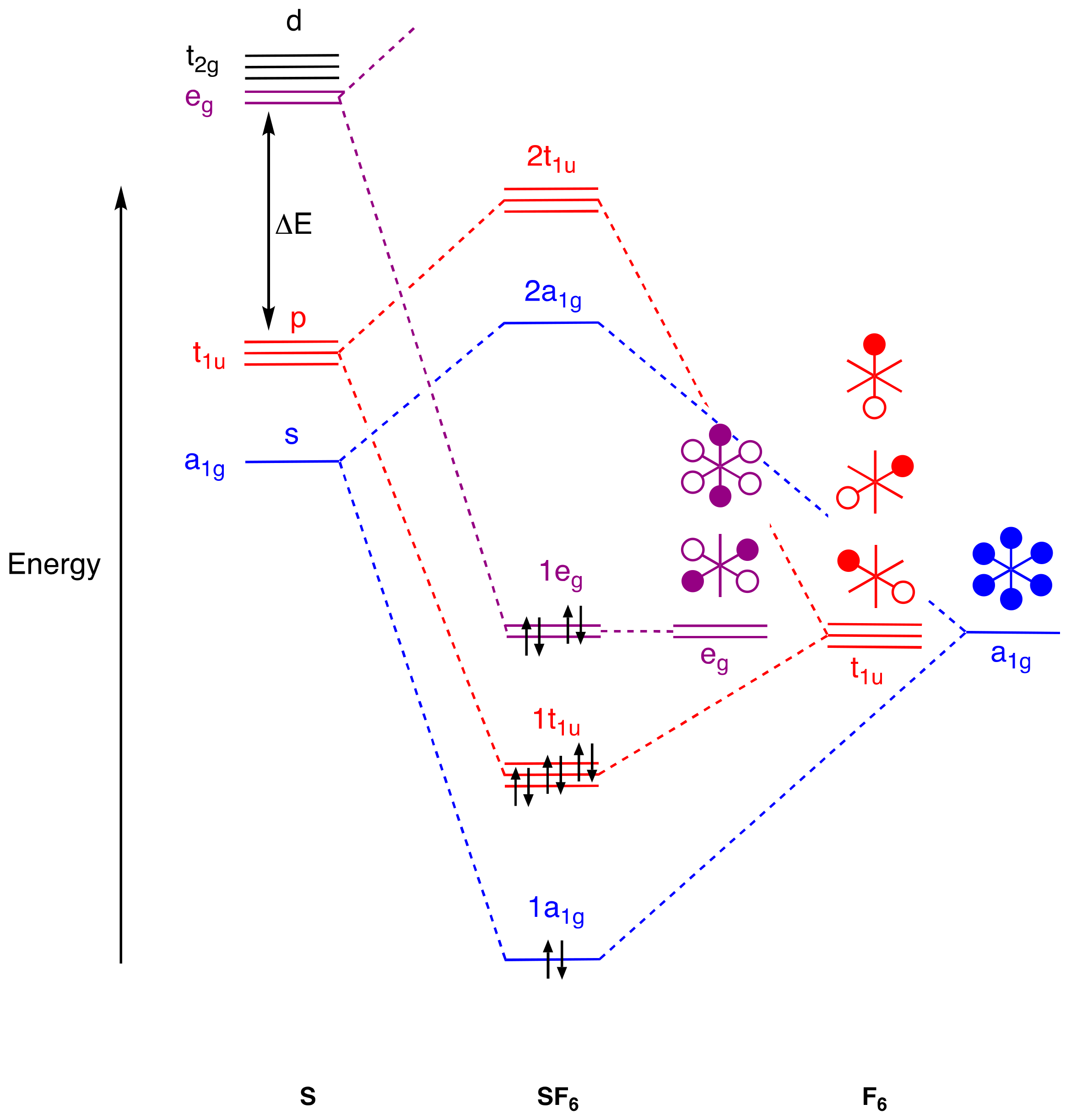
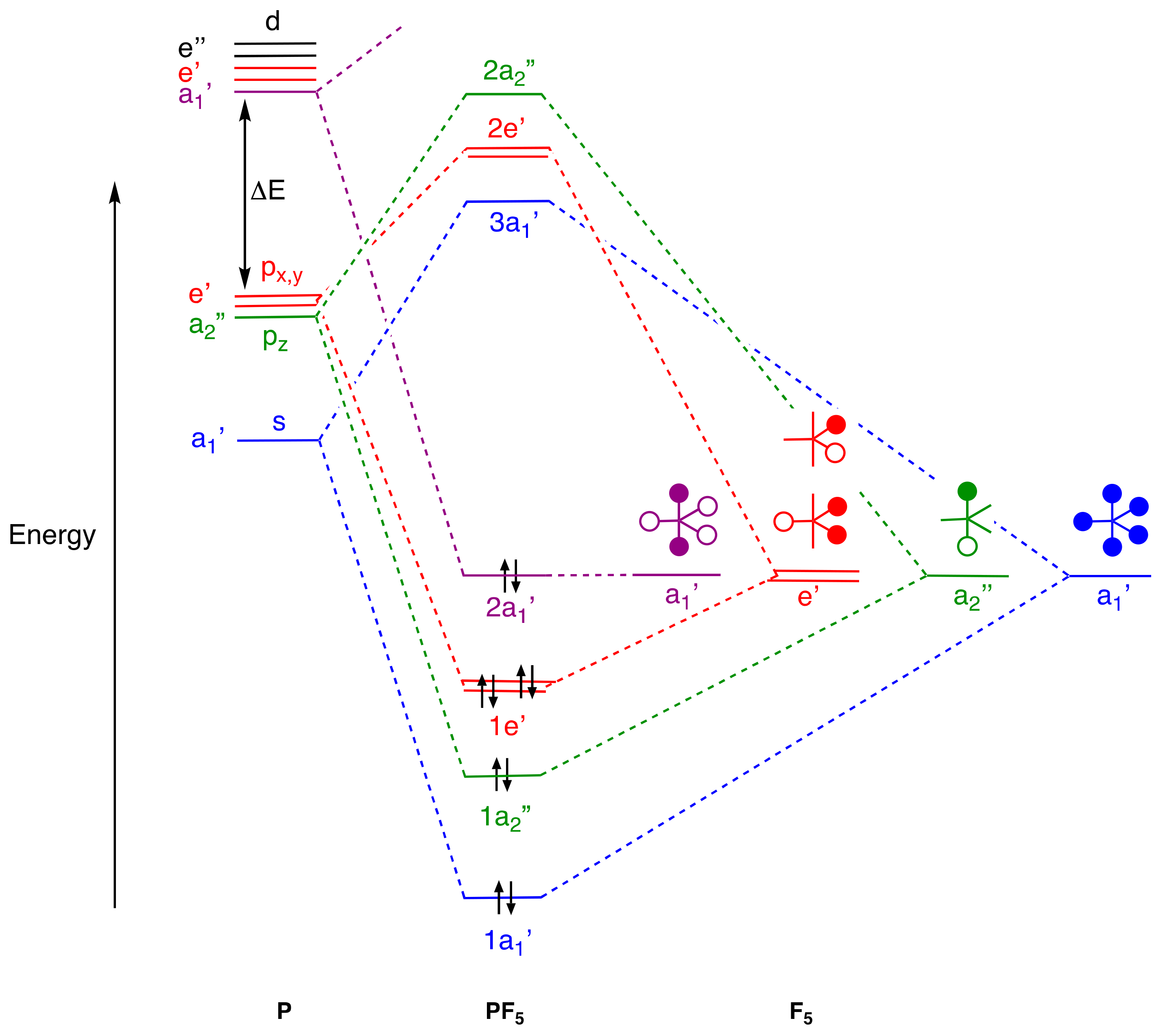
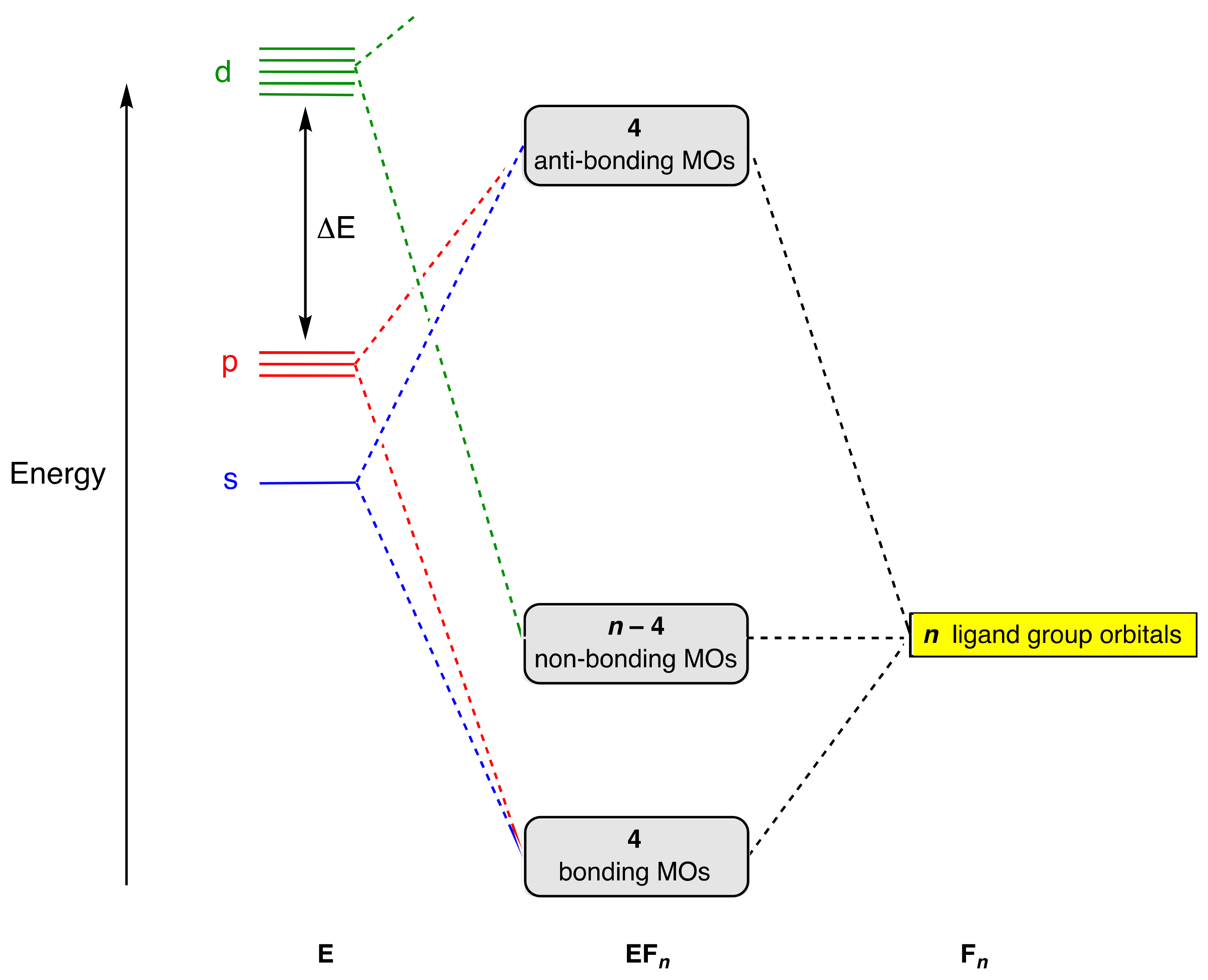
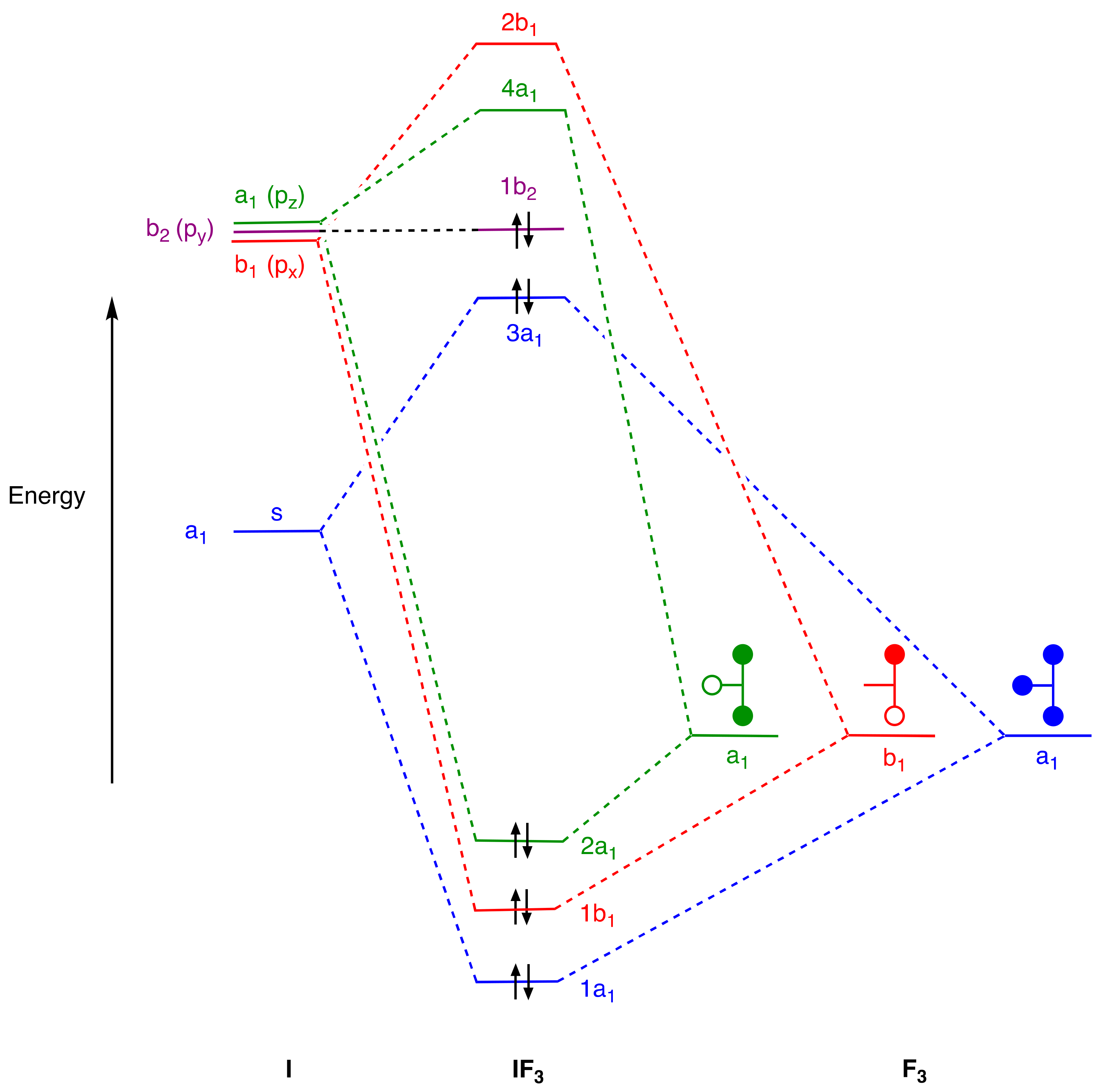
4. Molecular Orbital Analyses
5. Historical Criticisms

“In spite of its continuing overwhelming appeal we suggest that the familiar octet rule should be demoted. We retain only an eight-electron rule (cf. the 18-electron rule of transition metal chemistry), which indicates that a formal electron count of eight around a central atom is favorable. We assert here the new democracy principle, which, stated very simply, suggests that “it is the democratic right of every valence electron to take part in chemical bonding if it wants!””
“In order to calculate the overall electron count … we may now define a parameter called the valence electron equivalent, γ, as ‘the formal shared electron count at a given atom, obtained by any combination of valid ionic and covalent resonance forms that reproduces the observed charge distribution’.”
“It follows that for any given atom X, if γ(X) = 8, the atom obeys the original Lewis octet rule. If γ(X) < 8, the atom obeys the ‘modified octet rule’, but if γ(X) > 8, neither form of the octet rule is obeyed and the atom is hypervalent.”
6. A Brief Mention of VSEPR and the Treatment of Hypervalence in Modern Texts
7. A Summary of the Arguments So Far
8. The Question of π-Bonding

9. Hypervalence and the 2p Elements
10. Notation for Hypervalent Species
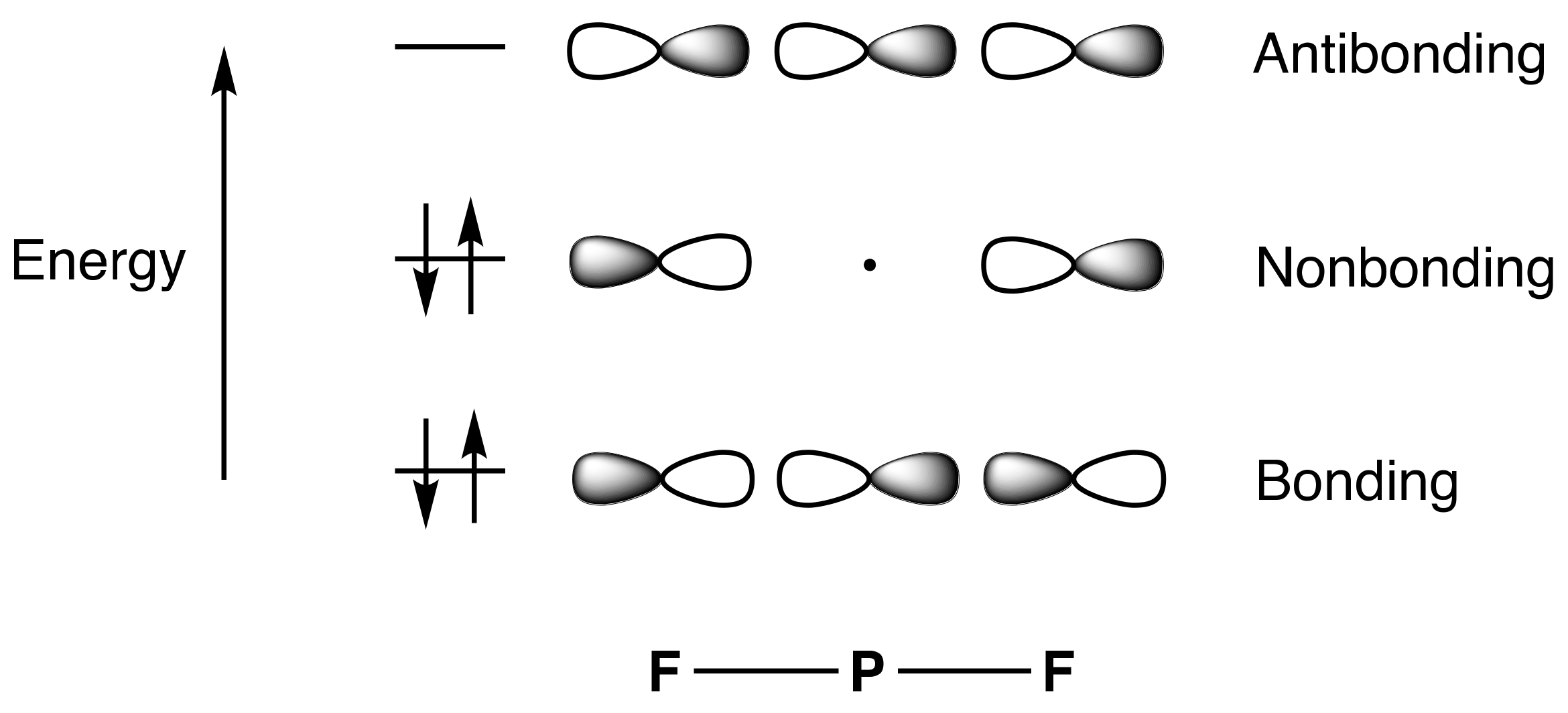
11. Depictions of 3c,4e Bonds

12. Conclusions and Recommendations
- (i)
- Introduce the definition of the term valence and stress that it is a classical concept, albeit a very useful one, which is most easily determined for a given element from a simple resonance or Lewis structure of the compound in which that element resides. It should be stressed that although prefixes which denote a number, such as in trivalent and pentavalent, are useful, those of a more subjective nature, such as hyper-, hypo- and sub-, afford no tangible benefit to any classification or description and should therefore not be used. The case against hyper- is further reinforced in some of the recommendations which follow.
- (ii)
- Recognise the value of the octet rule as an important organising principle. As with all electron-counting rules, it is a simple heuristic but one of considerable scope and merit. Moreover, because the valence orbitals for p-block elements are the ns and three np orbitals, we should expect an 8-electron rule for much the same reason that we expect (and observe) an 18-electron rule for many d-block element compounds. There will always be exceptions, but it is a very good place to start. The fact that an octet-based description does not always survive an analysis involving more detailed MO methods is not a reason to ignore its merits at a simpler level.
- (iii)
- Stress that vacant d orbitals play no significant role in p-block element chemistry because their energies, according to calculations, are too high, and bonding models based on d orbital occupancy or any use of dnsp3-type hybrids for compounds such as PF5 and SF6 should be abandoned [61] (Appendix A Note 27).
- (iv)
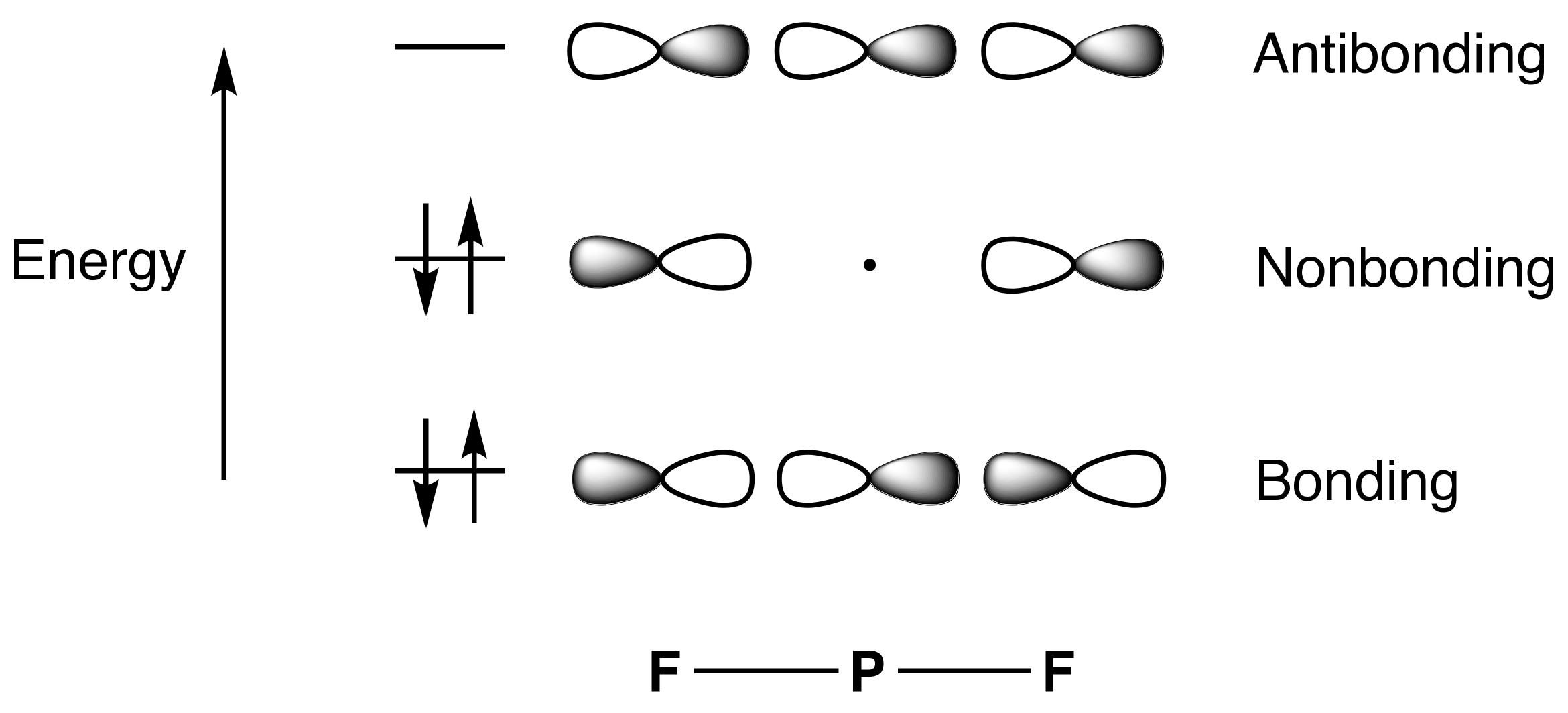
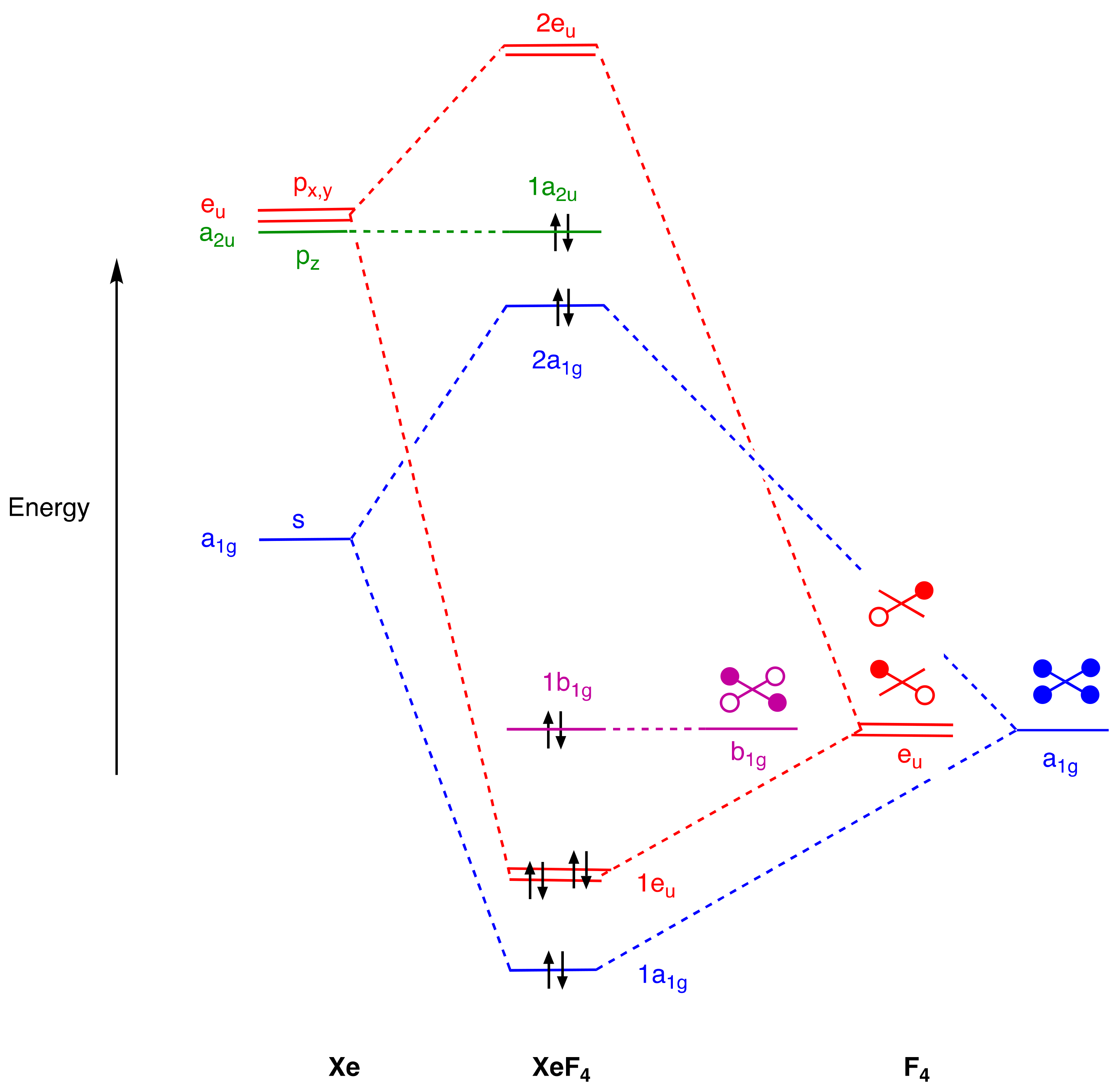
- For compounds to which simple valence electron counting methods assign more than eight electrons and which contain linear X–E–X units such as PF5, SF4, XeF2, and XeF4, the 3c,4e bonding model for this unit should be introduced, emphasising that because the non-bonding orbital is not localised on the central atom, the actual electron counts in these species do not exceed eight.
- (v)
- Recognise that for compounds where the coordination number exceeds four, some degree of multi-centre bonding will always be a feature, but any reference to the term hypercoordinate (as a possible alternative to hypervalent) is unhelpful because multi-centre bonding is also present in species with coordination numbers of four, or even less such as in SF4, XeF4, XeF2 IF3, or [I3]−, all of which contain linear X–E–X units.
- (vi)
- Where more detailed insights into molecular electronic structure are required, this will be by means of simple, qualitative, MO energy-level diagrams derived from ligand group orbitals or, ultimately, by recourse to more quantitative electronic structure calculations, albeit sometimes at the expense of simple octet-based descriptions. This offers no reprieve for hypervalence, however, because too strict an adherence to classifying compounds as hypervalent if their electron count exceeds eight leads to a greatly increased catalogue of compounds, many of which would not normally be considered as such; see also point (ix) below.
- (vii)
- With examples such as [SiF5]− and [SiF6]2−, it can be demonstrated that use of the term hypervalent is logically inconsistent because, according to the definition of the term valence, the silicon centre in these anions is tetravalent, as is the silicon in SiF4 itself. To describe these anions as hypervalent is therefore a misnomer.
- (viii)
- For compounds with formal multiple bonds such as the P–O bond in POF3, the fact that the charge-separated +P–O− single bond representation allocates 8 electrons to the phosphorus centre whereas the P=O and −P≡O+ multiple bond depictions assign 10 and 12 electrons, respectively, highlights another inconsistency within the concept of hypervalence since in one portrayal of the bonding, the molecule would not be considered hypervalent, whereas in the other two, it would be described as such. The +P–O− representation is therefore preferred in this context (and with bonds to a terminal oxygen more generally) and brings a further advantage in that it reflects the polarity of the P–O bond. It should, nevertheless, be recognised that using the P=O form is useful in other contexts.
- (ix)
- An additional advantage of the +P–O− representation (more generally, +E–O−) is that it provides a straightforward introduction to π-type negative hyperconjugation interactions which admits of the possibility of additional electron density at the central element, albeit not restricted to compounds that are traditionally classified as hypervalent, which further undermines the value of the term [62]. (Appendix A Note 28, 29)
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Notes
- Sidgwick notes that whilst the definition he advocates is appropriate in the great majority of cases, it is not universally applicable, but the exceptions are not germane to this discussion.
- An alternative definition of valence taken from the IUPAC “Gold Book” is as follows: ‘The maximum number of univalent atoms (originally hydrogen or chlorine atoms) that may combine with an atom of the element under consideration, or with a fragment, or for which an atom of this element can be substituted’. This is best seen as referring to the maximum valence an element can adopt [9].
- Low- and high- are also used as prefixes for valent which we accept have some value, at least in a relative sense.
- See also [11].
- Musher used the older periodic table group numbering of V-VIII rather than the modern 15-18 used here.
- In terms of the trigonal bipyramidal structure of PF5, this would involve the two axial P–F bonds being considered hypervalent (HV) with those to the three equatorial fluorine atoms treated as ‘normal’ covalent (CV) bonds.
- In fact, Musher classified hypervalent molecules as either HVI or HVII according to whether the valence s orbital was involved in bonding or not. Thus, SF2 would be classified as CV, SF4 as HVI, since the S 3s orbital was not considered to be involved in bonding to the fluorine atoms, and SF6 as HVII since the S 3s orbital does contribute to the bonding. PF5 is therefore HVII according to this definition.
- The Rundle/Pimentel 3c,4e bonding model has also been described as ‘hyperbonding’ by Weinhold and Landis [19], following Musher, and additionally as ω-bonding, an ω bond denoting the entirety of the 3c,4e interaction.
- As many authors have pointed out, notably Reed and Schleyer [32], accurate calculations do employ d orbitals as polarisation functions to better determine the precise electronic and molecular structure, but this is not the same as d orbital occupancy.
- [36] offers an instructive comparison of MO and VB approaches to the bonding in SF6 without recourse to d2sp3 hybridisation.
- Compounds such as [SbCl6]3– and [TeCl6]2– are often described as having a stereochemically inactive lone pair and therefore cited as exceptions to the rules of VSEPR which are largely predicated on lone pairs being stereochemically active. Compounds with stereochemically inactive lone pairs are not especially common but where they are encountered, drawing a satisfactory Lewis structure becomes problematic.
- An alternative view of the bonding in IF3 and XeF4 can be formulated using 3c,4e bonding. Thus, for IF3, the axial IF2 unit could be viewed in a similar way to the axial PF2 unit in PF5 and this would lead to an 8-electron count for the iodine in IF3. However, this would require a non-bonding pair of electrons to be localised on the two axial F atoms which is not compatible with the MO diagram in Figure 5. For XeF4, the two orthogonal XeF2 units could also be viewed in a similar way to the axial PF2 unit in PF5 and this would also lead to an 8-electron count for the Xe in XeF4. However, this would require two non-bonding pairs of electrons to be localised on the four F atoms which is likewise not compatible with the MO diagram in Figure 6. The incompatibility of the 3c,4e model with the MO diagrams in many EFn species (including SF6) is clear from the diagrams collected in the Supplementary Material.
- [44] covers much of the early history of the octet rule developed by Lewis and Langmuir.
- An alternative hybridisation scheme for PF5 could involve a pair of sp hybrids which bond to the axial fluorine atoms whilst the equatorial fluorines would be bonded in a multicentre arrangement using the remaining two p orbitals. Axial P–F bond orders would therefore be 1 whereas the equatorial P–F bonds would have a bond order of 2/3. This is no better in terms of rationalising the P–F bond lengths in PF5 but we should not expect simple models to necessarily accommodate small differences in bond lengths for which more quantitative electronic structure calculations are required.
- The question of the P–F(ax) and P–F(eq) relative bond orders has been much debated because, while the simple 3c,4e model predicts the P–F(ax) bond order to be 0.5 and the P–F(eq) bond order to be 1.0 (but see Appendix A Note 18), the P–F bond lengths are, as noted in the text, rather similar (difference of <0.05 Å). However, using the simple MO treatment for PF5 given in Figure 2, which reveals 4 pairs of electrons in bonding orbitals, the 1a1’ orbital is shared equally amongst the P–F bonds contributing 0.2 to the bond order in each. Additionally, the 1a2” orbital is shared between the two axial P–F bonds contributing 0.5 to the bond order of the two P–F(ax) whilst the 1e’ orbital is shared between the three equatorial P–F bonds contributing 0.66 to the bond order of each. Assuming that all of these P–F orbital interactions are similar results in an overall bond order for P–F(ax) of 0.7 and for P-F(eq) of 0.86. The conclusion is that a simple MO analysis predicts the P–F(ax) bond order to be 0.8 of that for the P–F(eq) bonds which is rather more consistent with the small difference in the P–F bond lengths.
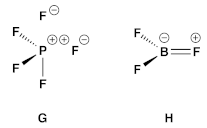
- Although BF3 is nominally assigned 6 electrons, π-donation from filled F orbitals to the vacant p orbital on boron raises the electron count as depicted in H.
- Although simple VSEPR methods assume that an oxygen contributes two electrons to the central atom resulting in a formal double bond, one can also start from a structure of type J in which an O− contributes only one electron to a central element which now carries a formal positive charge. This just serves to emphasise that at its core, VSEPR rationalisations of structure are based simply on the numbers of bonded atoms (or groups) plus the number of lone pairs.
- Hyperconjugation is the term used to describe σ→π* electron donation whereas negative hyperconjugation describes π→σ* donation. We will ignore the slight irony inherent in the term hyperconjugation and not argue with the use of the prefix hyper- in this context.
- Higher coordination numbers are encountered for first row atoms albeit in interstitial environments in clusters and as observed for carbon in [C(AuL)6]2+ (L = PPh3).
- A solid line between two atoms to represent bond was first used by Couper in 1858 and a few years later, and rather more extensively, by Crum-Brown in 1861. Following Lewis’ introduction of the electron pair bond, the solid line became widely associated with a 2c,2e bond but in many delocalised species, particularly clusters, it merely denotes a connectivity.
- There are various other descriptions of 3c,4e interactions, some also incorporating Z groups, which contribute zero electrons, such as X–X–L, L–Z–L and L–L–Z which are not relevant to the discussion in this paper but serve to usefully differentiate between different types of 3c,4e interactions.
- It is interesting to note that recent work on s-block element carbonyl complexes does make a strong case for (n–1)d orbital involvement alongside the ns orbital interactions, particularly for complexes of barium; see [61] and refs. therein.
- With further regard to species with formal multiple bonds, it can also be recognised that, for example in sulfate, SO42–, a simple Lewis dot structure shown in P, indeed a representation favoured by Lewis himself, avoids any concerns about violation of the octet rule for either oxygen or sulfur, and, as Weinhold and Landis have stated, such a structure is supported by the calculations they describe in [19].

- 29.
References
- Brockman, J. (Ed.) This Idea Must Die: Scientific Theories That Are Blocking Progress; Harper Perennial: New York, NY, USA, 2015. [Google Scholar]
- Scerri, E.R. Five Ideas in Chemical Education That Must Die. Found. Chem. 2019, 21, 61–69. [Google Scholar] [CrossRef]
- Zhao, L.; Pan, S.; Holzmann, N.; Schwerdtfeger, P.; Frenking, G. Chemical Bonding and Bonding Models in Main-Group Chemistry. Chem. Rev. 2019, 119, 8781–8845. [Google Scholar] [CrossRef] [PubMed]
- Dewar, M.J.S. Chemical Implications of s Conjugation. J. Am. Chem. Soc. 1984, 106, 669–682. [Google Scholar] [CrossRef]
- Russell, C.A. The History of Valency; Leicester University Press: Leicester, UK, 1971. [Google Scholar]
- Brock, W.H. The Fontana History of Chemistry; Fontana Press: London, UK, 1992. [Google Scholar]
- Palmer, W.G. Valency: Classical and Modern, 2nd ed.; Cambridge University Press: Cambridge, UK, 1959. [Google Scholar]
- Sidgwick, N.V. The Electronic Theory of Valence; Oxford University Press: Oxford, UK, 1927. [Google Scholar]
- IUPAC. Compendium of Chemical Terminology, 2nd ed.; (the “Gold, Book”); McNaught, A.D., Wilkinson, A., Eds.; Blackwell Scientific Publications: Hoboken, NJ, USA, 1997; Available online: https://goldbook.iupac.org/terms/view/V06588 (accessed on 12 September 2022).
- Parkin, G. Valence, Oxidation Number, and Formal Charge: Three Related but Fundamentally Different Concepts. J. Chem. Educ. 2006, 83, 791–799. [Google Scholar] [CrossRef]
- Smith, D.W. Valence, Covalence, Hypervalence, Oxidation State and Coordination Number. J. Chem. Educ. 2005, 82, 1202–1204. [Google Scholar] [CrossRef]
- Shorter Oxford English Dictionary, 6th ed.; Oxford University Press: Oxford, UK, 2007.
- Norman, N.C.; Pringle, P.G. In Defence of Oxidation States. Dalton Trans. 2021, 51, 400–410. [Google Scholar] [CrossRef]
- Norman, N.C. Periodicity and the s- and p-Block Elements, 2nd ed.; Oxford University Press: Oxford, UK, 2021. [Google Scholar]
- Karen, P.; McArdle, P.; Takats, J. Toward a Comprehensive Definition of Oxidation State (IUPAC Technical Report). Pure Appl. Chem. 2014, 86, 1017–1081. [Google Scholar] [CrossRef] [Green Version]
- Karen, P.; McArdle, P.; Takats, J. Comprehensive Definition of Oxidation State (IUPAC Recommendations 2016). Pure Appl. Chem. 2016, 88, 831–839. [Google Scholar] [CrossRef] [Green Version]
- Jensen, W.B. The Origin of the Term Hypervalent. J. Chem. Educ. 2006, 83, 1751–1752. [Google Scholar] [CrossRef]
- Musher, J.L. The Chemistry of Hypervalent Molecules. Angew. Chem. Int. Ed. 1969, 8, 54–68. [Google Scholar] [CrossRef]
- Weinhold, F.; Landis, C.R. Valency and Bonding: A Natural Bond Orbital Donor-Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Lewis, G.N. The Atom and the Molecule. J. Am. Chem. Soc. 1916, 38, 762–785. [Google Scholar] [CrossRef] [Green Version]
- Langmuir, I. The Arrangement of Electrons in Atoms and Molecules. J. Am. Chem. Soc. 1919, 41, 868–934. [Google Scholar] [CrossRef] [Green Version]
- Lewis, G.N. Valence and the Structure of Atoms and Molecules; Chemical Catalogue Company: New York, NY, USA, 1923. [Google Scholar]
- Langmuir, I. Types of Valence. Science 1921, 54, 59–67. [Google Scholar] [CrossRef]
- Pauling, L. The Nature of the Chemical Bond and the Structure of Molecules and Crystals: An Introduction to Modern Structural Chemistry, 2nd ed.; Cornell University Press: Ithaca, NY, USA, 1940. [Google Scholar]
- Coulson, C.A. Valence, 2nd ed.; Oxford University Press: Oxford, UK, 1961. [Google Scholar]
- Sugden, S. The Parachor and Valency; Routledge: London, UK, 1930. [Google Scholar]
- Rundle, R.E. On the Probable Structure of XeF2 and XeF4. J. Am. Chem. Soc. 1963, 85, 112–113. [Google Scholar] [CrossRef]
- Pimentel, G.C. The Bonding of Trihalide and Bifluoride Ions by the Molecular Orbital Method. J. Chem. Phys. 1951, 19, 446–448. [Google Scholar] [CrossRef]
- Huheey, J.E.; Keiter, E.A.; Keiter, R.L. Inorganic Chemistry: Principles of Structure and Reactivity; Harper Collins: New York, NY, USA, 1993. [Google Scholar]
- Kutzelnigg, W. Chemical Bonding in Higher Main Group Elements. Angew. Chem. Int. Ed. Engl. 1984, 23, 272–295. [Google Scholar] [CrossRef]
- Magnusson, E. Hypercoordinate Molecules of Second-Row Elements: D Functions or d Orbitals? J. Am. Chem. Soc. 1990, 112, 7940–7951. [Google Scholar] [CrossRef]
- Reed, A.E.; Schleyer, P.v.R. Chemical Bonding in Hypervalent Molecules. The Dominance of Ionic Bonding and Negative Hyperconjugation over d-Orbital Participation. J. Am. Chem. Soc. 1990, 112, 1434–1445. [Google Scholar] [CrossRef]
- Gilheany, D.G. No d Orbitals but Walsh Diagrams and Maybe Banana Bonds: Chemical Bonding in Phosphines, Phosphine Oxides and Phosphonium Ylides. Chem. Rev. 1994, 94, 1339–1374. [Google Scholar] [CrossRef]
- Curnow, O.J. A Simple Qualitative Molecular-Orbital/Valence-Bond Description of the Bonding in Main Group “Hypervalent” Molecules. J. Chem. Educ. 1998, 75, 910–915. [Google Scholar] [CrossRef]
- Curnow, O.J. The Rise and Decline of d Orbitals: Bonding in Hypervalent Compounds. Chem. N. Z. 1996, 60, 10–14. [Google Scholar]
- Galbraith, J.M. On the Role of d Orbital Hybridisation in the Chemistry Curriculum. J. Chem. Educ. 2007, 84, 783–787. [Google Scholar] [CrossRef]
- Albright, T.A.; Burdett, J.K.; Whangbo, M.H. Orbital Interactions in Chemistry; Wiley: Hoboken, NJ, USA, 1985. [Google Scholar]
- Purcell, K.F.; Kotz, J.C. Inorganic Chemistry; W.B. Saunders Company: Philadelphia, PA, USA, 1977. [Google Scholar]
- Hoffmann, R.; Howell, J.M.; Muetterties, E.L. Molecular Orbital Theory of Pentacoordinate Phosphorus. J. Am. Chem. Soc. 1972, 94, 3047–3058. [Google Scholar] [CrossRef]
- Yang, D.-D.; Wang, F. Structures and Stabilities of Group 17 Fluorides EF3 (E = I, At, and Element 117) with Spin-Orbit Coupling. Phys. Chem. Chem. Phys. 2012, 14, 15816–15825. [Google Scholar] [CrossRef] [PubMed]
- Gillespie, R.J.; Robinson, E.A. Hypervalence and the Octet Rule. Inorg. Chem. 1995, 34, 978–979. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Silvi, B. The Octet Rule and Hypervalence: Two Misunderstood Concepts. Coord. Chem. Rev. 2002, 233, 53–62. [Google Scholar] [CrossRef]
- Noury, S.; Silvi, B.; Gillespie, R.J. Chemical Bonding in Hypervalent Molecules: Is the Octet Rule Relevant? Inorg. Chem. 2002, 41, 2164–2172. [Google Scholar] [CrossRef]
- Gillespie, R.J.; Popelier, P.L.A. Chemical Bonding and Molecular Geometry: From Lewis to Electron Densities; Oxford University Press: Oxford, UK, 2001. [Google Scholar]
- Gillespie, R.J.; Robinson, E.A. Gilbert N. Lewis and the Chemical Bond: The Electron Pair and the Octet Rule from 1916 to the Present Day. J. Comput. Chem. 2007, 28, 87–97. [Google Scholar] [CrossRef]
- Robinson, E.A. The Duodecet Rule: Part 1. Valence Bond Structures of Oxo and Azo Species of Si(IV), P(V), S(VI) and Cl(VII). J. Mol. Struc. 1989, 186, 9–28. [Google Scholar] [CrossRef]
- Cioslowski, J.; Mixon, S.T. Rigorous Interpretation of Electronic Wave Functions. 2. Electronic Structures of Selected Phosphorus, Sulfur, and Chlorine Fluorides and Oxides. Inorg. Chem. 1993, 32, 3209–3216. [Google Scholar] [CrossRef]
- Cooper, D.L.; Cunningham, T.P.; Gerratt, J.; Karadakov, P.B.; Raimondi, M. Chemical Bonding to Hypercoordinate Second-Row Atoms: D Orbital Participation Versus Democracy. J. Am. Chem. Soc. 1994, 116, 4414–4426. [Google Scholar] [CrossRef]
- Durrant, M.C. A Quantitative Definition of Hypervalency. Chem. Sci. 2015, 6, 6614–6623. [Google Scholar] [CrossRef] [Green Version]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Oxford University Press: Oxford, UK, 2003. [Google Scholar]
- Gillespie, R.J.; Hargittai, I. The VSEPR Model of Molecular Geometry; Allyn and Bacon: Boston, MA, USA, 1991. [Google Scholar]
- Housecroft, C.E.; Sharpe, A.G. Inorganic Chemistry, 5th ed.; Pearson: London, UK, 2018. [Google Scholar]
- Weller, M.T.; Overton, T.L.; Rourke, J.P.; Armstrong, F.A. Inorganic Chemistry, 7th ed.; Oxford University Press: Oxford, UK, 2018. [Google Scholar]
- Keeler, J.; Wothers, P. Chemical Structure and Reactivity: An Integrated Approach, 2nd ed.; Oxford University Press: Oxford, UK, 2014. [Google Scholar]
- Dobado, J.A.; Martinez-Garcia, H.; Molina, J.; Sundberg, M.R. Chemical Bonding in Hypervalent Molecules Revised. Application of the Atoms in Molecules Theory to Y3XZ (Y = H or CH3; X = N, P or As; Z = O or S) Compounds. J. Am. Chem. Soc. 1998, 120, 8461–8471. [Google Scholar] [CrossRef]
- Perkins, C.W.; Martin, J.C.; Arduengo, A.J.; Lau, W.; Alegria, A.; Kochi, J.K. An Electrically Neutral s-Sulfuranyl Radical from the Homolysis of a Perester with Neighboring Sulfenyl Sulfur: 9-S-3 Species. J. Am. Chem. Soc. 1980, 102, 7753–7759. [Google Scholar] [CrossRef]
- Green, M.L.H.; Parkin, G. The Classification and Representation of Main Group Element Compounds that Feature Three-Centre Four-Electron Interactions. Dalton Trans. 2016, 45, 18784–18795. [Google Scholar] [CrossRef]
- Green, M.L.H. A New Approach to the Formal Classification of Covalent Compounds and Elements. J. Organomet. Chem. 1995, 500, 127–148. [Google Scholar] [CrossRef]
- Green, M.L.H.; Parkin, G. Application of the Covalent Bond Classification Method for the Teaching of Inorganic Chemistry. J. Chem. Educ. 2014, 91, 807–816. [Google Scholar] [CrossRef]
- Parkin, G. Classification of Organometallic Compounds. In Comprehensive Organometallic Chemistry III; Elsevier: Amsterdam, The Netherlands, 2007; Volume 1, pp. 1–57. [Google Scholar]
- Zhou, M.; Frenking, G. Transition-Metal Chemistry of the Heavier Alkaline Earth Atoms Ca, Sr, and Ba. Acc. Chem. Res. 2021, 54, 3071–3082. [Google Scholar] [CrossRef]
- Bernes, E.; Fronzoni, G.; Stener, M.; Guarnaccio, A.; Zhang, T.; Grazioli, C.; Johansson, F.O.L.; Coreno, M.; de Simone, M.; Puglia, C.; et al. S 2p and P 2p Core Level Spectroscopy of PPT Ambipolar Material and its Building Block Moieties. J. Phys. Chem. C 2020, 124, 14510–14520. [Google Scholar] [CrossRef]
- Wirth, T. (Ed.) Hypervalent Iodine Chemistry: Modern Development in Organic Synthesis. Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2003; Volume 224. [Google Scholar]
- Zhdankin, V.V. Hypervalent Iodine Chemistry: Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds; Wiley: Hoboken, NJ, USA, 2014. [Google Scholar]
- Yoshimura, A.; Zhdankin, V.V. Advances in Synthetic Applications of Hypervalent Iodine Compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef]
- Crabtree, R.H. Hypervalency, Secondary Bonding and Hydrogen Bonding: Siblings Under the Skin. Chem. Soc. Rev. 2017, 46, 1720–1729. [Google Scholar] [CrossRef]
- Jackson, B.A.; Harshman, J.; Miliordos, E. Addressing the Hypervalent Model: A Straightforward Explanation of Traditionally Hypervalent Molecules. J. Chem. Educ. 2020, 97, 3638–3646. [Google Scholar] [CrossRef]
- Munzarova, M.L.; Hoffmann, R. Electron-Rich Three-Centre Bonding: Role of s, p Interactions Across the p-Block. J. Am. Chem. Soc. 2002, 124, 4787–4795. [Google Scholar] [CrossRef] [PubMed]
- Braïda, B.; Hiberty, P.C. The Essential Role of Charge-Shift Bonding in Hypervalent Prototype XeF2. Nat. Chem. 2013, 5, 417–422. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
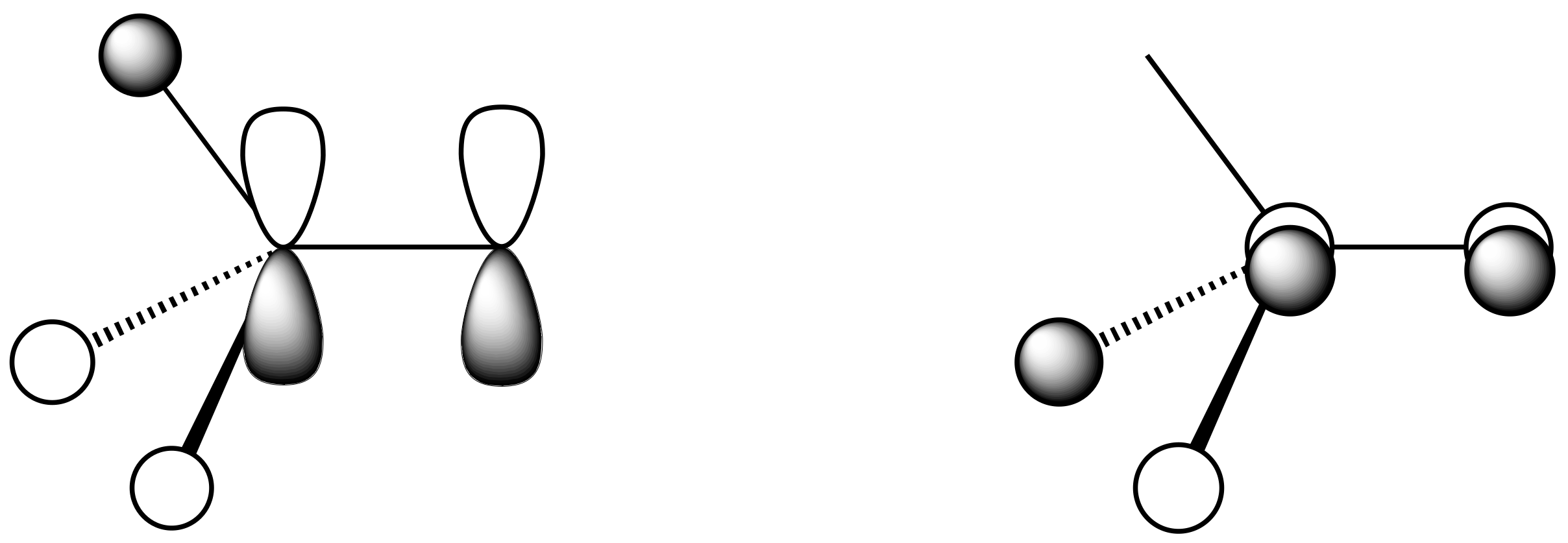{kind=link}
| Halides: | PF5 (10), AsCl5 (10), SF4 (10), SF6 (12), IF3 (10), IF5 (12), IF7 (14), I2Cl6 (12), |
| XeF2 (10), XeF4 (12), XeF6 (14) | |
| Halo-anions: | [SiF6]2− (12), [SnCl6]2− (12), [PF6]− (12), [ICl4]− (12), [IF8]− (16), [XeF5]− (14), [XeF8]2− (18) |
| Oxides: | P4O10 (10), SO2 (10), SO3 (12), ClO2 (11), Cl2O7 (14), I2O5 (12), XeO3 (14) |
| Oxyhalides: | POF3 (10), SOCl2 (10), SO2Cl2 (12) |
| Complexes: | [SiF4(pyridine)2] (12), [Bi2Cl6(PMe3)4] (14) |
| Alkyls and aryls: | BiMe5 (10), BiPh5 (10), TeMe4 (10), TeMe6 (12) |
| Miscellaneous: | PhICl2 (10), PhI(OAc)2 (10) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Norman, N.C.; Pringle, P.G. Hypervalence: A Useful Concept or One That Should Be Gracefully Retired? Chemistry 2022, 4, 1226-1249. https://doi.org/10.3390/chemistry4040082
Norman NC, Pringle PG. Hypervalence: A Useful Concept or One That Should Be Gracefully Retired? Chemistry. 2022; 4(4):1226-1249. https://doi.org/10.3390/chemistry4040082
Chicago/Turabian StyleNorman, Nicholas C., and Paul G. Pringle. 2022. "Hypervalence: A Useful Concept or One That Should Be Gracefully Retired?" Chemistry 4, no. 4: 1226-1249. https://doi.org/10.3390/chemistry4040082
APA StyleNorman, N. C., & Pringle, P. G. (2022). Hypervalence: A Useful Concept or One That Should Be Gracefully Retired? Chemistry, 4(4), 1226-1249. https://doi.org/10.3390/chemistry4040082