Mitochondrial-Directed Antioxidant Reduces Microglial-Induced Inflammation in Murine In Vitro Model of TC-83 Infection

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses and Cell Lines
2.2. Infection and Plaque Assay
2.3. Inhibitors and Cell Viability
2.4. Quantitative Real-Time Polymerase Chain Reaction
2.5. Caspase Quantitation
2.6. Cellular Reactive Oxygen Species
2.7. Mitochondrial Membrane Potential
2.8. Live Cell Imaging
2.9. Direct Flow Cytometry
2.10. Confocal Microscopy
2.11. Inflammatory Cytokine Array
2.12. Statistical Analysis
3. Results
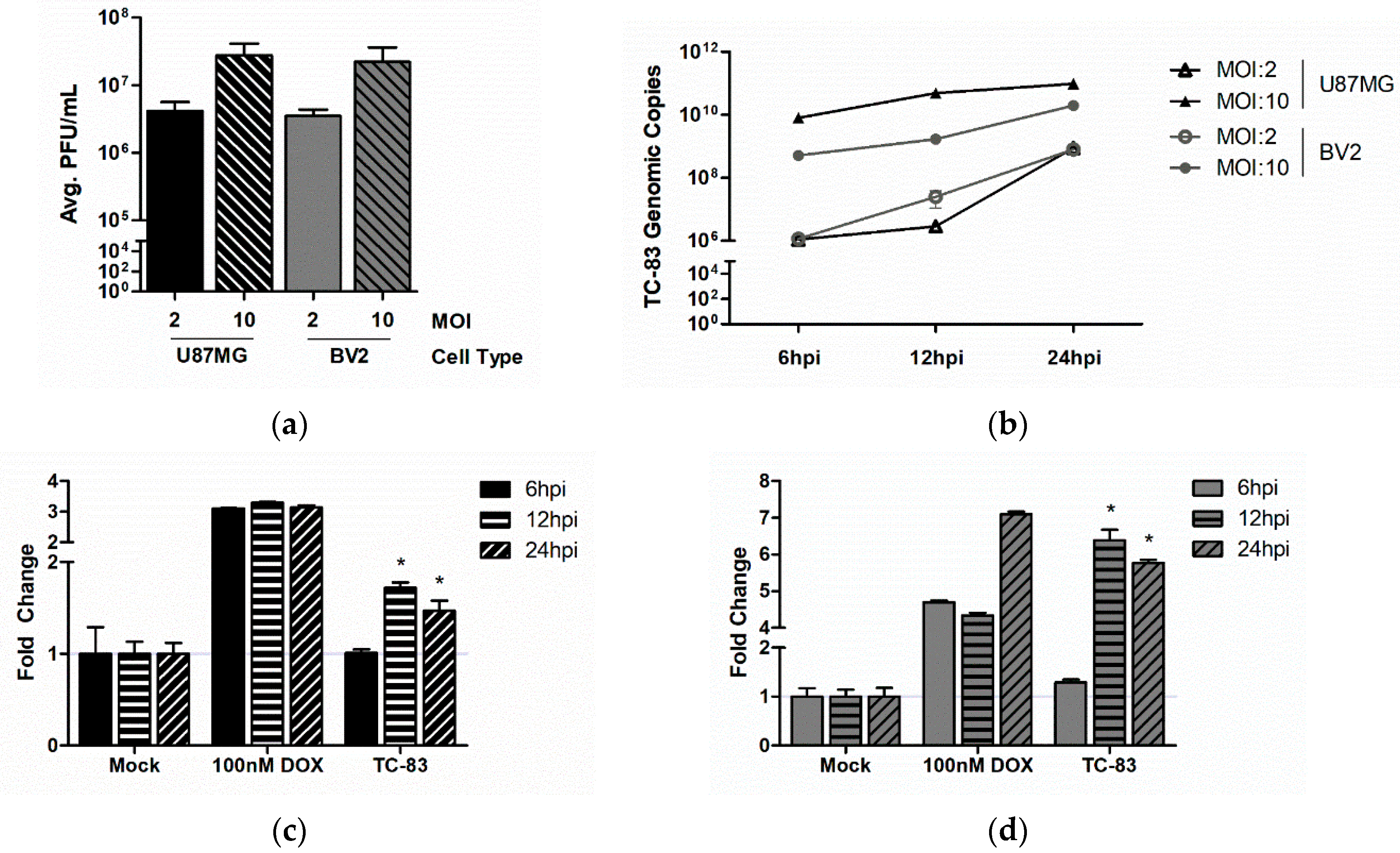
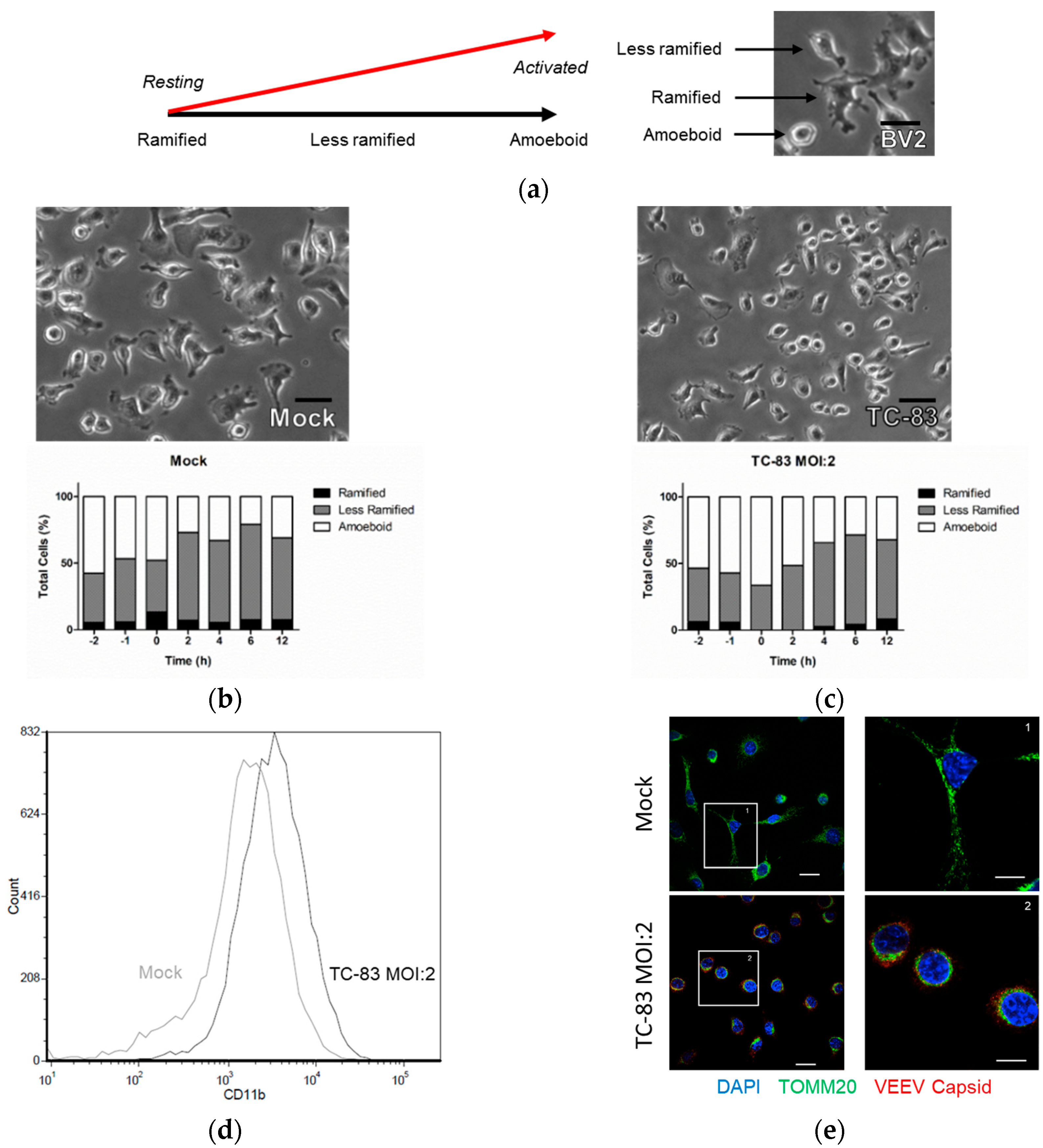
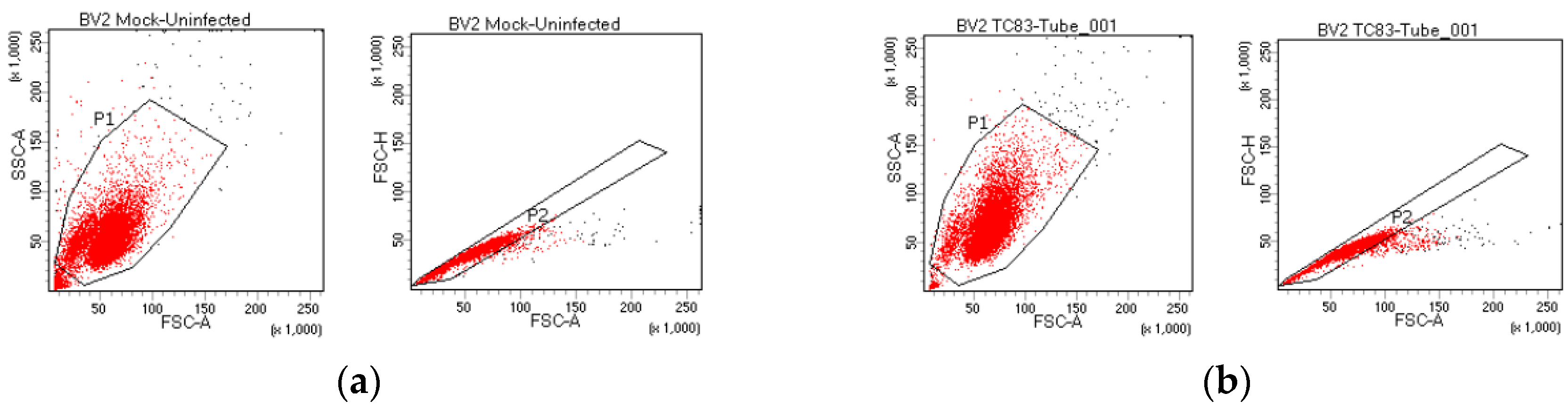
3.1. Murine Microglia are Susceptible to VEEV Infection
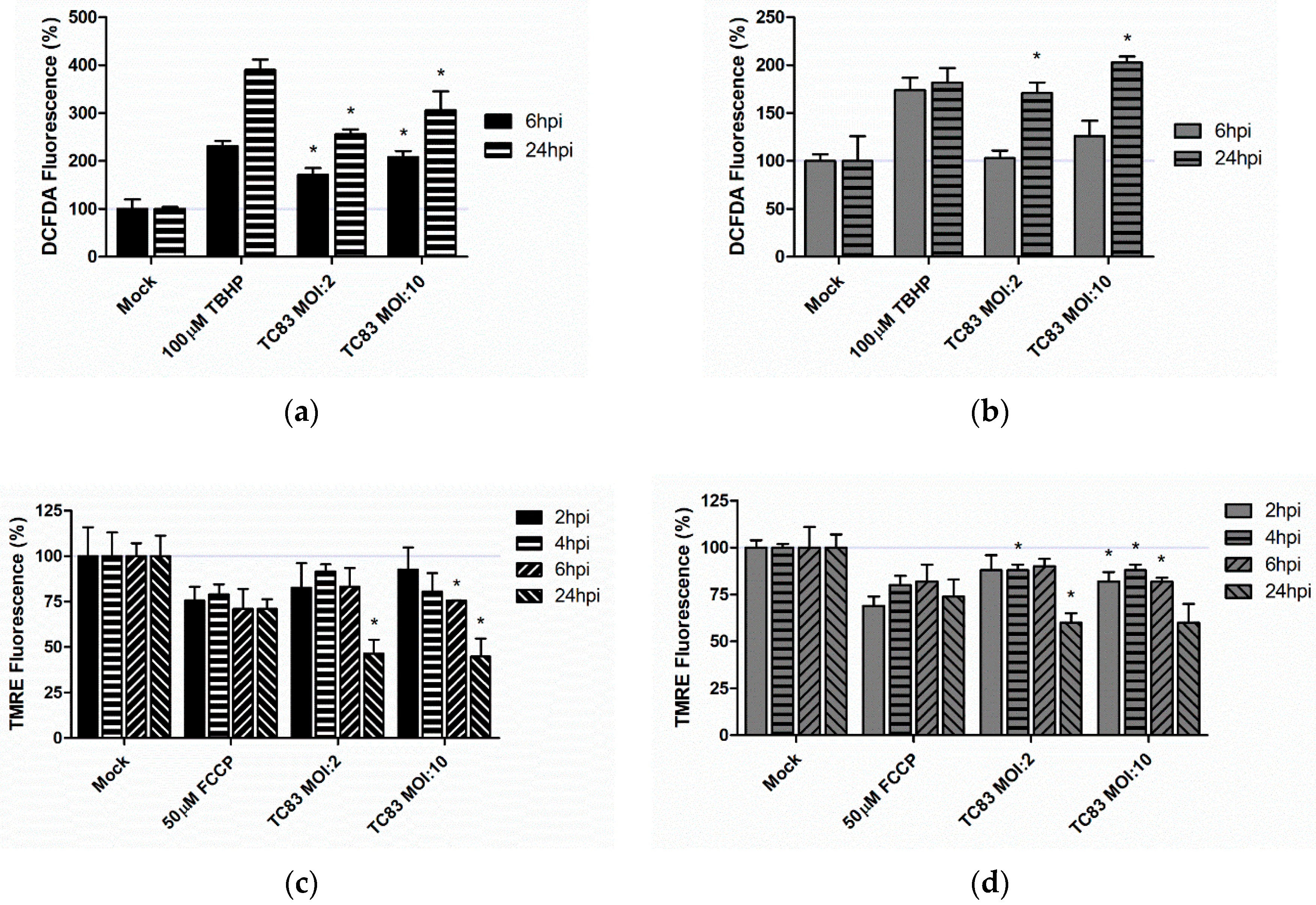
3.2. TC-83 Infection Induces Mitochondrial Dysfunction in Murine Microglia
3.3. Microglial Activation as the Result of TC-83 Infection
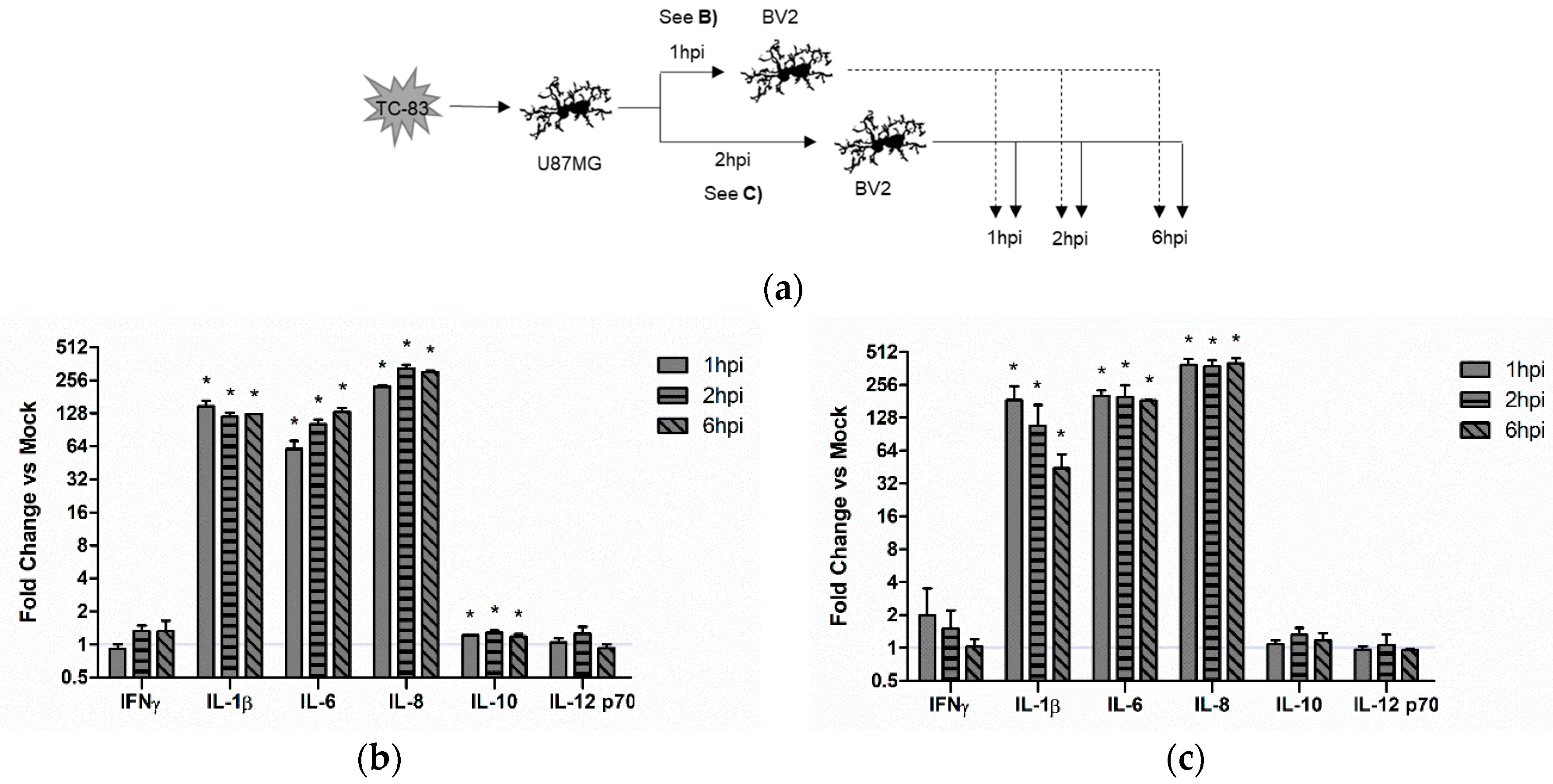
3.4. Microglial-Mediated Inflammatory Response to TC-83 Infection
3.4.1. Direct TC-83 Infection of Microglia Produces Altered Inflammatory Cytokine Profile
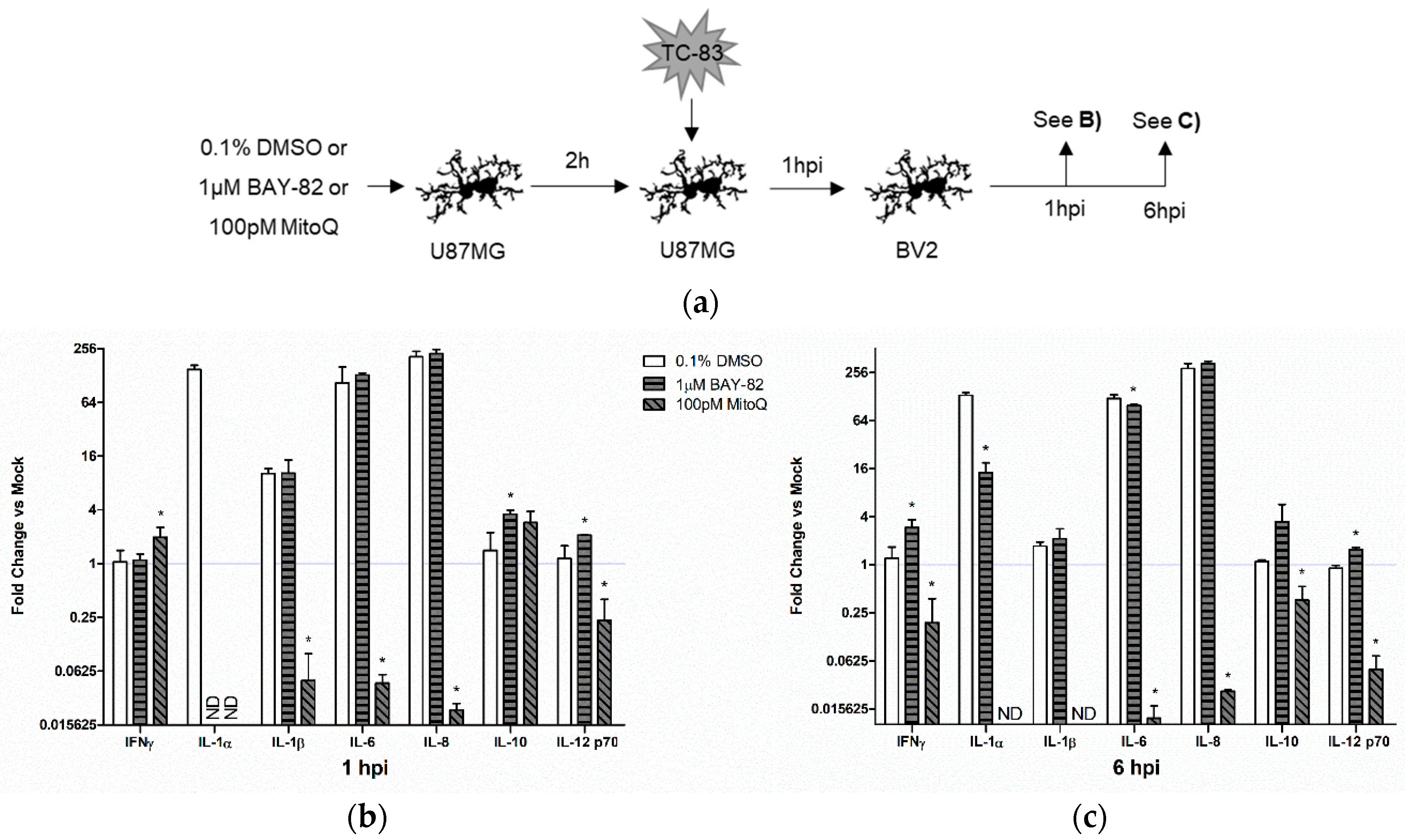
3.4.2. Indirect Inflammatory Response Significantly Contributes to Overall Inflammatory Burden
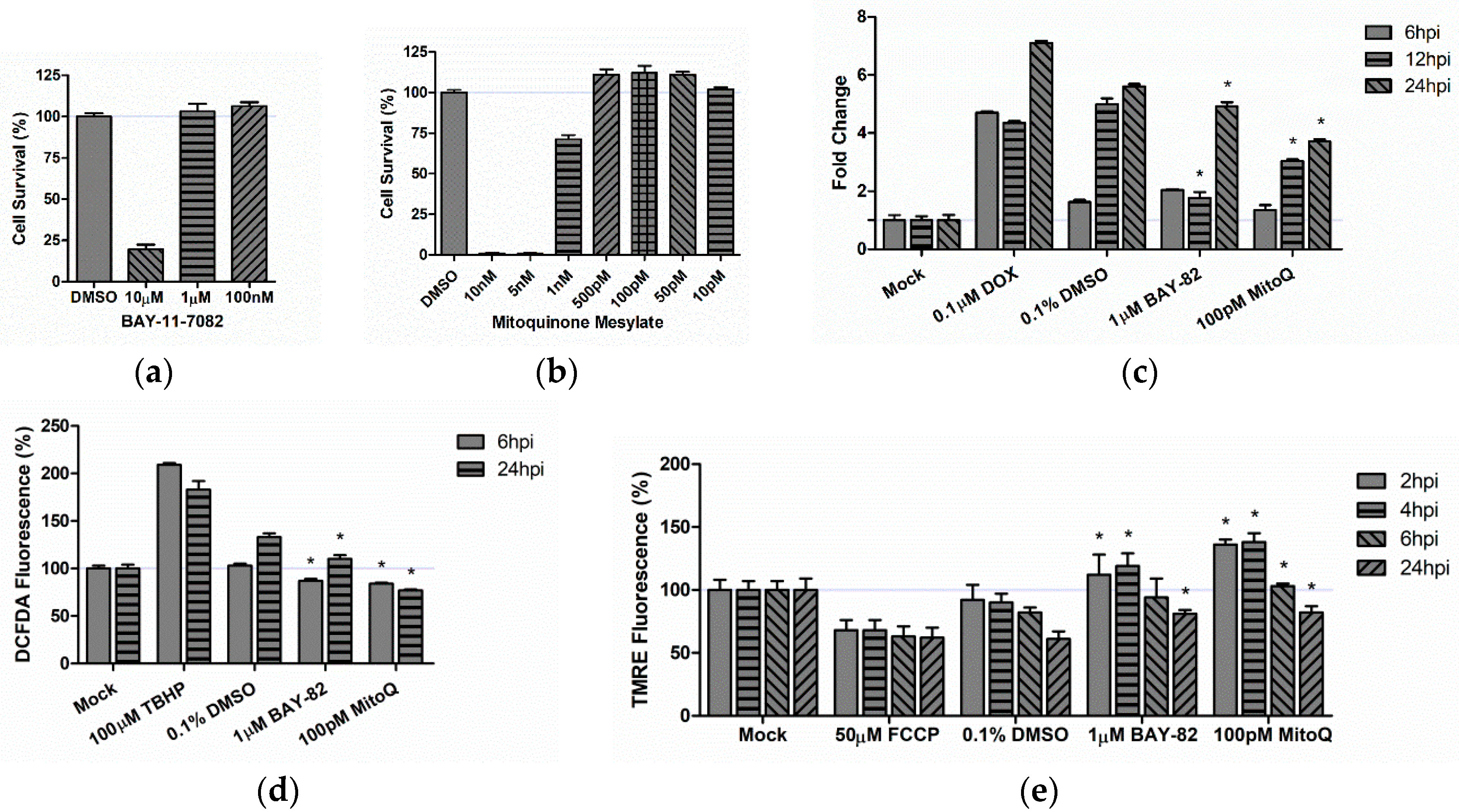
3.5. Therapeutic Intervention Rescues Mitochondrial Function in TC-83 Infected Microglia
3.6. Role of Mitochondrial Oxidative Stress in TC-83 Induced Inflammation
3.6.1. Mitochondrial Dysfunction Contributes to Direct Inflammatory Burden
3.6.2. Mitochondrial Dysfunction Contributes to Indirect Inflammatory Burden
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

References
- Aguilar, P.V.; Estrada-Franco, J.G.; Navarro-Lopez, R.; Ferro, C.; Haddow, A.D.; Weaver, S.C. Endemic Venezuelan equine encephalitis in the Americas: Hidden under the dengue umbrella. Future Virol. 2011, 6, 721–740. [Google Scholar] [CrossRef] [PubMed]
- Hawley, R.J.; Eitzen, E.M., Jr. Biological Weapons—A Primer for Microbiologists. Annu. Rev. Microbiol. 2001, 55, 235–253. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Ferro, C.; Barrera, R.; Boshell, J.; Navarro, J.C. Venezuelan Equine Encephalitis. Annu. Rev. Entomol. 2004, 49, 141–174. [Google Scholar] [CrossRef] [PubMed]
- Paessler, S.; Weaver, S.C. Vaccines for Venezuelan equine encephalitis. Vaccine 2009, 27, D80–D85. [Google Scholar] [CrossRef] [PubMed]
- Strauss, J.H.; Strauss, E.G. The alphaviruses: Gene expression, replication, and evolution. Microbiol. Rev. 1994, 58, 491–562. [Google Scholar] [PubMed]
- Ryzhikov, A.B.; Ryabchikova, E.I.; Sergeev, A.N.; Tkacheva, N.V. Spread of Venezuelan equine encephalitis virus in mice olfactory tract. Arch. Virol. 1995, 140, 2243–2254. [Google Scholar] [CrossRef] [PubMed]
- Charles, P.C.; Trgovcich, J.; Davis, N.L.; Johnston, R.E. Immunopathogenesis and Immune Modulation of Venezuelan Equine Encephalitis Virus-Induced Disease in the Mouse. Virology 2001, 284, 190–202. [Google Scholar] [CrossRef] [PubMed]
- Durrant, D.M.; Ghosh, S.; Klein, R.S. The Olfactory Bulb: An Immunosensory Effector Organ during Neurotropic Viral Infections. ACS Chem. Neurosci. 2016, 7, 464–469. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.C.; SenGupta, S.K.; Smith, J.F. Pathogenesis of Venezuelan Equine Encephalitis Virus Infection in Mice and Hamsters. Vet. Pathol. 1991, 28, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.C.; Rossiter, J.P. Apoptotic cell death is an important cause of neuronal injury in experimental Venezuelan equine encephalitis virus infection of mice. Acta Neuropathol. 1997, 93, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Schoneboom, B.A.; Catlin, K.M.; Marty, A.M.; Grieder, F.B. Inflammation is a component of neurodegeneration in response to Venezuelan equine encephalitis virus infection in mice. J. Neuroimmunol. 2000, 109, 132–146. [Google Scholar] [CrossRef]
- Schoneboom, B.A.; Fultz, M.J.; Miller, T.H.; McKinney, L.C.; Grieder, F.B. Astrocytes as targets for Venezuelan equine encephalitis virus infection. J. Neurovirol. 1999, 5, 342–354. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Maheshwari, R.K. Oligonucleotide array analysis of Toll-like receptors and associated signalling genes in Venezuelan equine encephalitis virus-infected mouse brain. J. Gen. Virol. 2009, 90, 1836–1847. [Google Scholar] [CrossRef] [PubMed]
- Kehn-Hall, K.; Narayanan, A.; Lundberg, L.; Sampey, G.; Pinkham, C.; Guendel, I.; Duyne, R.V.; Senina, S.; Schultz, K.L.; Stavale, E.; et al. Modulation of GSK-3β Activity in Venezuelan Equine Encephalitis Virus Infection. PLoS ONE 2012, 7, e34761. [Google Scholar] [CrossRef] [PubMed]
- Schoneboom, B.A.; Lee, J.S.; Grieder, F.B. Early expression of IFN-alpha/beta and iNOS in the brains of Venezuelan equine encephalitis virus-infected mice. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2000, 20, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Peng, B.-H.; Borisevich, V.; Popov, V.L.; Zacks, M.A.; Estes, D.M.; Campbell, G.A.; Paessler, S. Production of IL-8, IL-17, IFN-gamma and IP-10 in human astrocytes correlates with alphavirus attenuation. Vet. Microbiol. 2013, 163, 223–234. [Google Scholar] [CrossRef] [PubMed]
- Andreasson, K.I.; Bachstetter, A.D.; Colonna, M.; Ginhoux, F.; Holmes, C.; Lamb, B.; Landreth, G.; Lee, D.C.; Low, D.; Lynch, M.A.; et al. Targeting innate immunity for neurodegenerative disorders of the central nervous system. J. Neurochem. 2016, 138, 653–693. [Google Scholar] [CrossRef] [PubMed]
- Lannes, N.; Eppler, E.; Etemad, S.; Yotovski, P.; Filgueira, L. Microglia at center stage: A comprehensive review about the versatile and unique residential macrophages of the central nervous system. Oncotarget 2017, 8, 114393–114413. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M.; Khoury, J.E. Microglia in Health and Disease. Cold Spring Harb. Perspect. Biol. 2016, 8. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guajardo, V.; Barnum, C.J.; Tansey, M.G.; Romero-Ramos, M. Neuroimmunological processes in Parkinson’s disease and their relation to α-synuclein: Microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, K.; Schneider, S.; Bertsch, T.; Schlueter, D.; Fatar, M.; Ragoschke, A.; Kühl, S.; Kischka, U.; Hennerici, M. Temporal profile of release of interleukin-1β in neurotrauma. Neurosci. Lett. 2000, 284, 135–138. [Google Scholar] [CrossRef]
- Hanisch, U.-K. Microglia as a source and target of cytokines. Glia 2002, 40, 140–155. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-1, Interleukin-1 Receptors and Interleukin-1 Receptor Antagonist. Int. Rev. Immunol. 1998, 16, 457–499. [Google Scholar] [CrossRef] [PubMed]
- Solovjov, D.A.; Pluskota, E.; Plow, E.F. Distinct Roles for the α and β Subunits in the Functions of Integrin αMβ2. J. Biol. Chem. 2005, 280, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Rahmani, Z.; Huh, K.W.; Lasher, R.; Siddiqui, A. Hepatitis B Virus X Protein Colocalizes to Mitochondria with a Human Voltage-Dependent Anion Channel, HVDAC3, and Alters Its Transmembrane Potential. J. Virol. 2000, 74, 2840–2846. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Huh, K.-W.; Siddiqui, A. Mitochondrially Associated Hepatitis B Virus X Protein Constitutively Activates Transcription Factors STAT-3 and NF-κB via Oxidative Stress. Mol. Cell. Biol. 2001, 21, 7721–7730. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Khan, M.; Quan, J.; Till, A.; Subramani, S.; Siddiqui, A. Hepatitis B Virus Disrupts Mitochondrial Dynamics: Induces Fission and Mitophagy to Attenuate Apoptosis. PLOS Pathog. 2013, 9, e1003722. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.-J.; Syed, G.H.; Khan, M.; Chiu, W.-W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, M.J.; Wang, L.H.; Schneider, R.J. Calcium Signaling by HBx Protein in Hepatitis B Virus DNA Replication. Science 2001, 294, 2376–2378. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, A.; Amaya, M.; Voss, K.; Chung, M.; Benedict, A.; Sampey, G.; Kehn-Hall, K.; Luchini, A.; Liotta, L.; Bailey, C.; et al. Reactive oxygen species activate NFκB (p65) and p53 and induce apoptosis in RVFV infected liver cells. Virology 2014, 449, 270–286. [Google Scholar] [CrossRef] [PubMed]
- Terasaki, K.; Won, S.; Makino, S. The C-Terminal Region of Rift Valley Fever Virus NSm Protein Targets the Protein to the Mitochondrial Outer Membrane and Exerts Antiapoptotic Function. J. Virol. 2013, 87, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Keck, F.; Brooks-Faulconer, T.; Lark, T.; Ravishankar, P.; Bailey, C.; Salvador-Morales, C.; Narayanan, A. Altered mitochondrial dynamics as a consequence of Venezuelan Equine encephalitis virus infection. Virulence 2017, 8, 1849–1866. [Google Scholar] [CrossRef] [PubMed]
- Keck, F.; Kortchak, S.; Bakovic, A.; Roberts, B.; Agrawal, N.; Narayanan, A. Direct and Indirect Pro-Inflammatory Cytokine Response Resulting from TC-83 Infection of Glial Cells. Virulence 2018. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Bhomia, M.; Honnold, S.P.; Maheshwari, R.K. Role of adhesion molecules and inflammation in Venezuelan equine encephalitis virus infected mouse brain. Virol. J. 2011, 8, 197. [Google Scholar] [CrossRef] [PubMed]
- Bhomia, M.; Balakathiresan, N.; Sharma, A.; Gupta, P.; Biswas, R.; Maheshwari, R. Analysis of microRNAs induced by Venezuelan equine encephalitis virus infection in mouse brain. Biochem. Biophys. Res. Commun. 2010, 395, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Sharma, A.; Han, J.; Yang, A.; Bhomia, M.; Knollmann-Ritschel, B.; Puri, R.K.; Maheshwari, R.K. Differential host gene responses from infection with neurovirulent and partially-neurovirulent strains of Venezuelan equine encephalitis virus. BMC Infect. Dis. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Goshima, F.; Daikoku, T.; Inagaki-Ohara, K.; Takakuwa, H.; Kato, K.; Nishiyama, Y. Mitochondrial distribution and function in herpes simplex virus-infected cells. J. Gen. Virol. 2000, 81, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Ambrosius, B.; Faissner, S.; Guse, K.; von Lehe, M.; Grunwald, T.; Gold, R.; Grewe, B.; Chan, A. Teriflunomide and monomethylfumarate target HIV-induced neuroinflammation and neurotoxicity. J. Neuroinflamm. 2017, 14. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, R.S.; Kothari, S.T.; Gohil, D.J.; D’Souza, M.; Chowdhary, A.S. Novel evidence of microglial immune response in impairment of Dengue infection of CNS. Immunobiology 2015, 220, 1170–1176. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Dutta, K.; Kumawat, K.L.; Ghoshal, A.; Adhya, D.; Basu, A. Abrogated Inflammatory Response Promotes Neurogenesis in a Murine Model of Japanese Encephalitis. PLoS ONE 2011, 6, e17225. [Google Scholar] [CrossRef] [PubMed]
- Jadhav, V.S.; Krause, K.-H.; Singh, S.K. HIV-1 Tat C modulates NOX2 and NOX4 expressions through miR-17 in a human microglial cell line. J. Neurochem. 2014, 131, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Ye, J.; Zhu, B.; Song, Y.; Chen, H.; Cao, S. Roles of TLR3 and RIG-I in mediating the inflammatory response in mouse microglia following Japanese encephalitis virus infection. J. Immunol. Res. 2014, 2014, 787023. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, L.; Carey, B.; Kehn-Hall, K. Venezuelan Equine Encephalitis Virus Capsid—The Clever Caper. Viruses 2017, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.; Voss, K.; Sampey, G.; Senina, S.; de la Fuente, C.; Mueller, C.; Calvert, V.; Kehn-Hall, K.; Carpenter, C.; Kashanchi, F.; et al. The role of IKKβ in Venezuelan equine encephalitis virus infection. PLoS ONE 2014, 9, e86745. [Google Scholar] [CrossRef] [PubMed]
- Koterski, J.; Twenhafel, N.; Porter, A.; Reed, D.S.; Martino-Catt, S.; Sobral, B.; Crasta, O.; Downey, T.; DaSilva, L. Gene expression profiling of nonhuman primates exposed to aerosolized Venezuelan equine encephalitis virus. FEMS Immunol. Med. Microbiol. 2007, 51, 462–472. [Google Scholar] [CrossRef] [PubMed]
- Jahrling, P.B.; Navarro, E.; Scherer, W.F. Interferon induction and sensitivity as correlates to virulence of Venezuelan encephalitis viruses for hamsters. Arch. Virol. 1976, 51, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Alevizatos, A.C.; McKinney, R.W.; Feigin, R.D. Live, Attenuated Venezuelan Equine Encephalomyelitis Virus Vaccine. Am. J. Trop. Med. Hyg. 1967, 16, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Vilhardt, F.; Haslund-Vinding, J.; Jaquet, V.; McBean, G. Microglia antioxidant systems and redox signalling. Br. J. Pharmacol. 2017, 174, 1719–1732. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Jana, A.; Yatish, K.; Freidt, M.B.; Fung, Y.K.; Martinson, J.A.; Pahan, K. Reactive oxygen species up-regulate CD11b in microglia via nitric oxide: Implications for neurodegenerative diseases. Free Radic. Biol. Med. 2008, 45, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Fung, Y.K.; Liu, X.; Pahan, K. Up-regulation of Microglial CD11b Expression by Nitric Oxide. J. Biol. Chem. 2006, 281, 14971–14980. [Google Scholar] [CrossRef] [PubMed]
- Rajalakshmy, A.R.; Malathi, J.; Madhavan, H.N. Hepatitis C Virus NS3 Mediated Microglial Inflammation via TLR2/TLR6 MyD88/NF-κB Pathway and Toll Like Receptor Ligand Treatment Furnished Immune Tolerance. PLoS ONE 2015, 10, e0125419. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Sheng, W.S.; Schachtele, S.J.; Lokensgard, J.R. Reactive oxygen species drive herpes simplex virus (HSV)-1-induced proinflammatory cytokine production by murine microglia. J. Neuroinflamm. 2011, 8, 123. [Google Scholar] [CrossRef] [PubMed]
- Steel, C.D.; Breving, K.; Tavakoli, S.; Kim, W.-K.; Sanford, L.D.; Ciavarra, R.P. Role of peripheral immune response in microglia activation and regulation of brain chemokine and proinflammatory cytokine responses induced during VSV encephalitis. J. Neuroimmunol. 2014, 267, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Das Sarma, J. Microglia-mediated neuroinflammation is an amplifier of virus-induced neuropathology. J. Neurovirol. 2014, 20, 122–136. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keck, F.; Khan, D.; Roberts, B.; Agrawal, N.; Bhalla, N.; Narayanan, A. Mitochondrial-Directed Antioxidant Reduces Microglial-Induced Inflammation in Murine In Vitro Model of TC-83 Infection. Viruses 2018, 10, 606. https://doi.org/10.3390/v10110606
Keck F, Khan D, Roberts B, Agrawal N, Bhalla N, Narayanan A. Mitochondrial-Directed Antioxidant Reduces Microglial-Induced Inflammation in Murine In Vitro Model of TC-83 Infection. Viruses. 2018; 10(11):606. https://doi.org/10.3390/v10110606
Chicago/Turabian StyleKeck, Forrest, Daud Khan, Brian Roberts, Nitin Agrawal, Nishank Bhalla, and Aarthi Narayanan. 2018. "Mitochondrial-Directed Antioxidant Reduces Microglial-Induced Inflammation in Murine In Vitro Model of TC-83 Infection" Viruses 10, no. 11: 606. https://doi.org/10.3390/v10110606
APA StyleKeck, F., Khan, D., Roberts, B., Agrawal, N., Bhalla, N., & Narayanan, A. (2018). Mitochondrial-Directed Antioxidant Reduces Microglial-Induced Inflammation in Murine In Vitro Model of TC-83 Infection. Viruses, 10(11), 606. https://doi.org/10.3390/v10110606