CRISPR–Cas9 Genetic Analysis of Virus–Host Interactions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
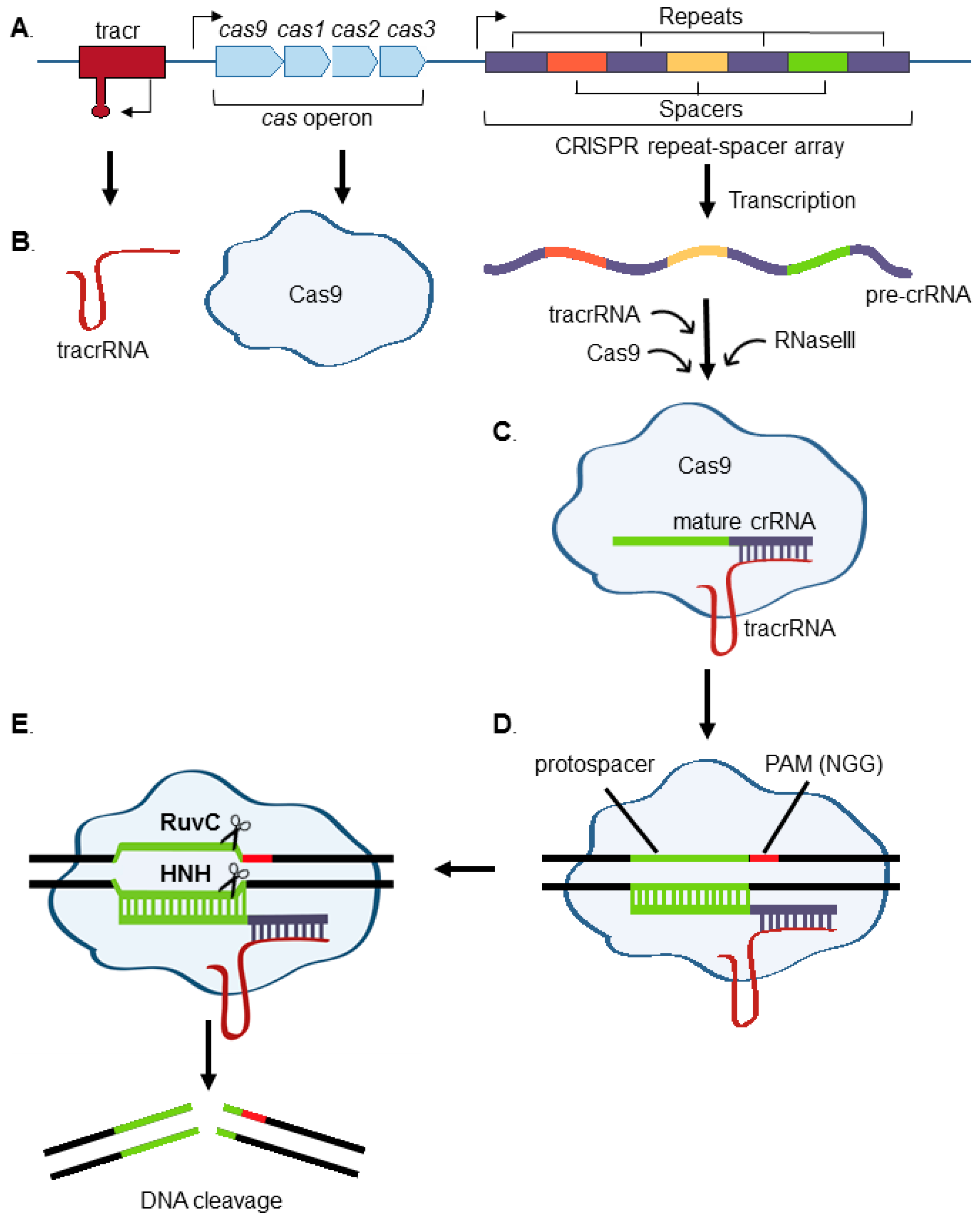
:1. Introduction to CRISPR
2. Use of Nonhomologous End-Joining (NHEJ) to Generate Functional Knockouts
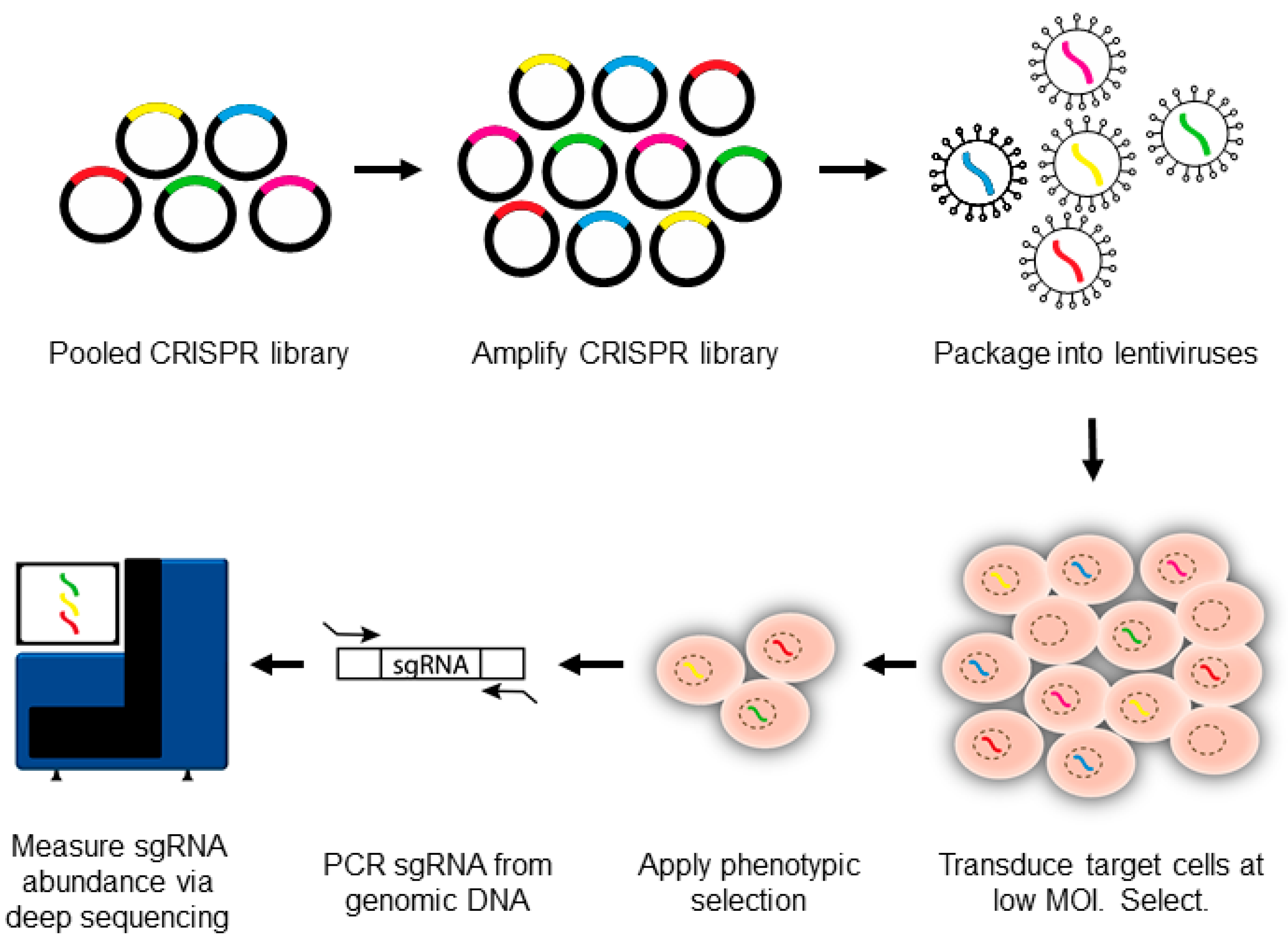
3. Introduction to CRISPR-Pooled Screens for Host–Virus Studies
4. Pooled Screens for RNA Virus Host Dependency Factors
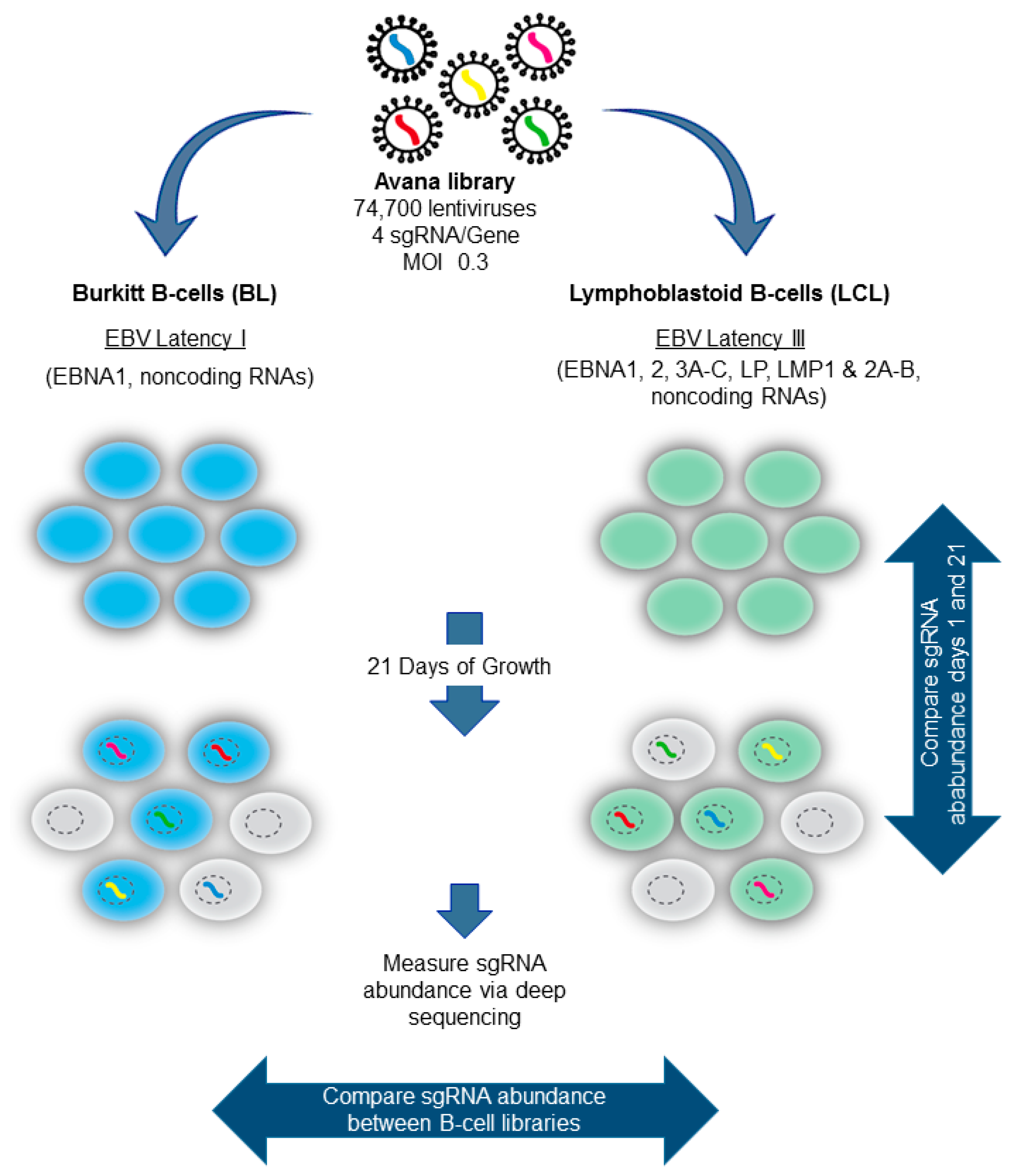
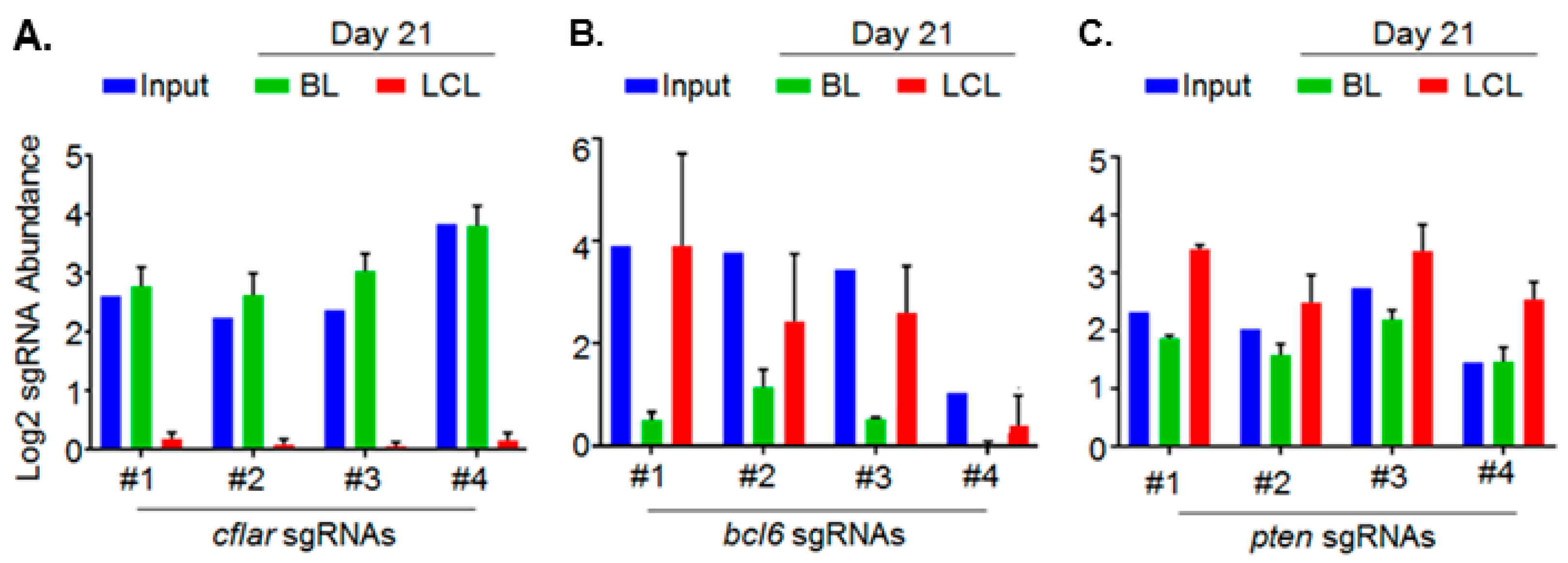
5. Genomewide Screen for EBV-Transformed B Cell Host Dependency Factors
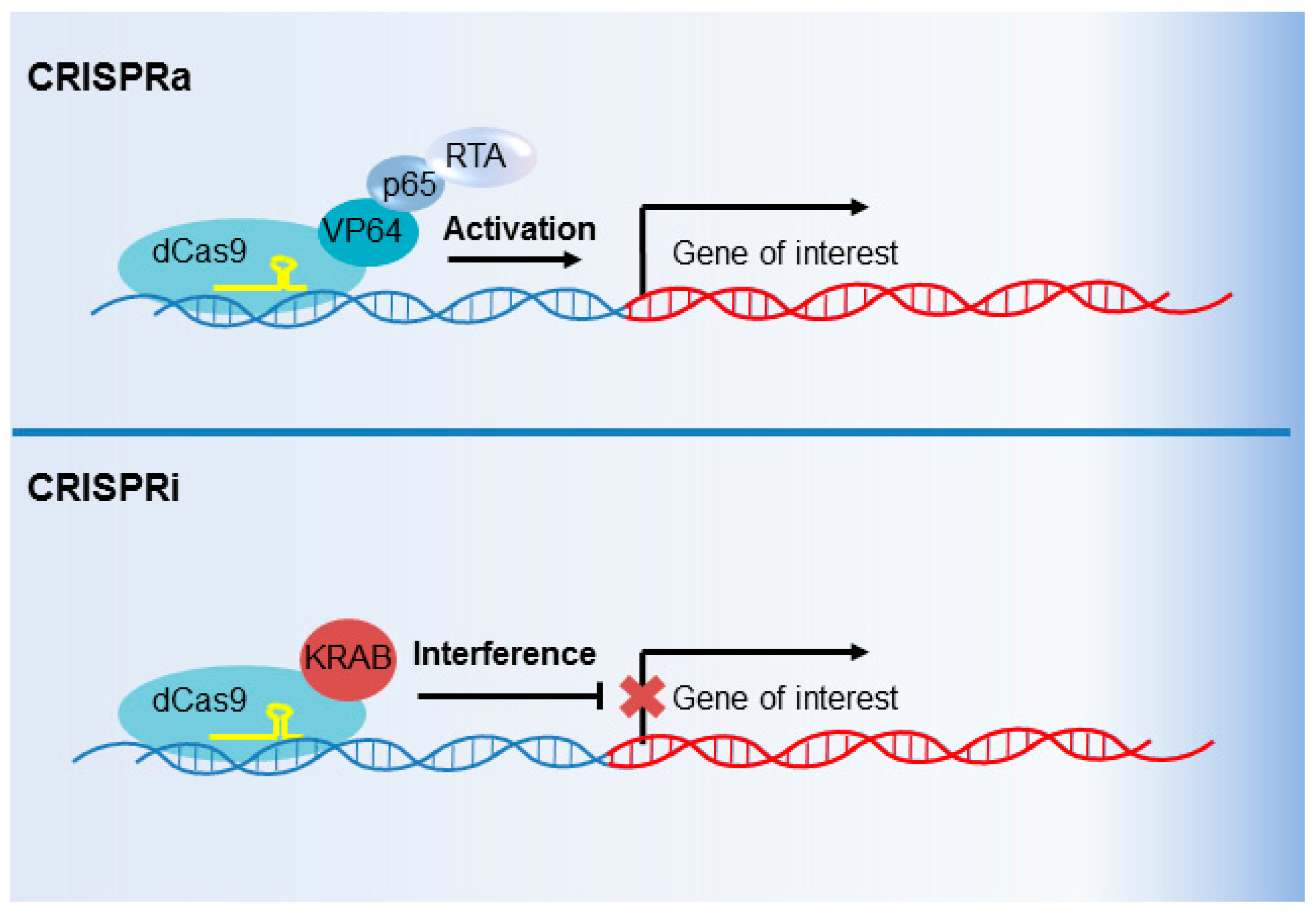
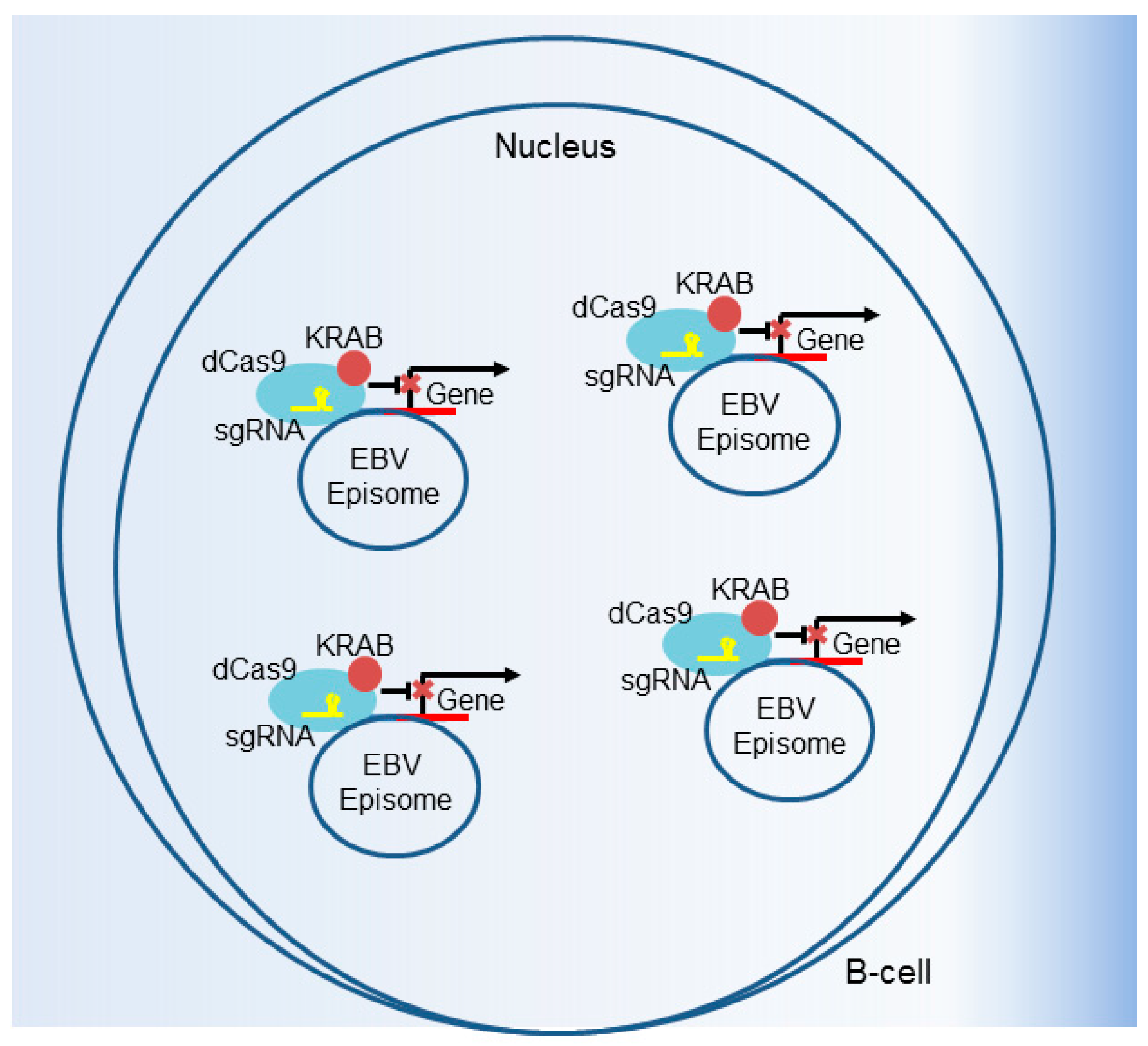
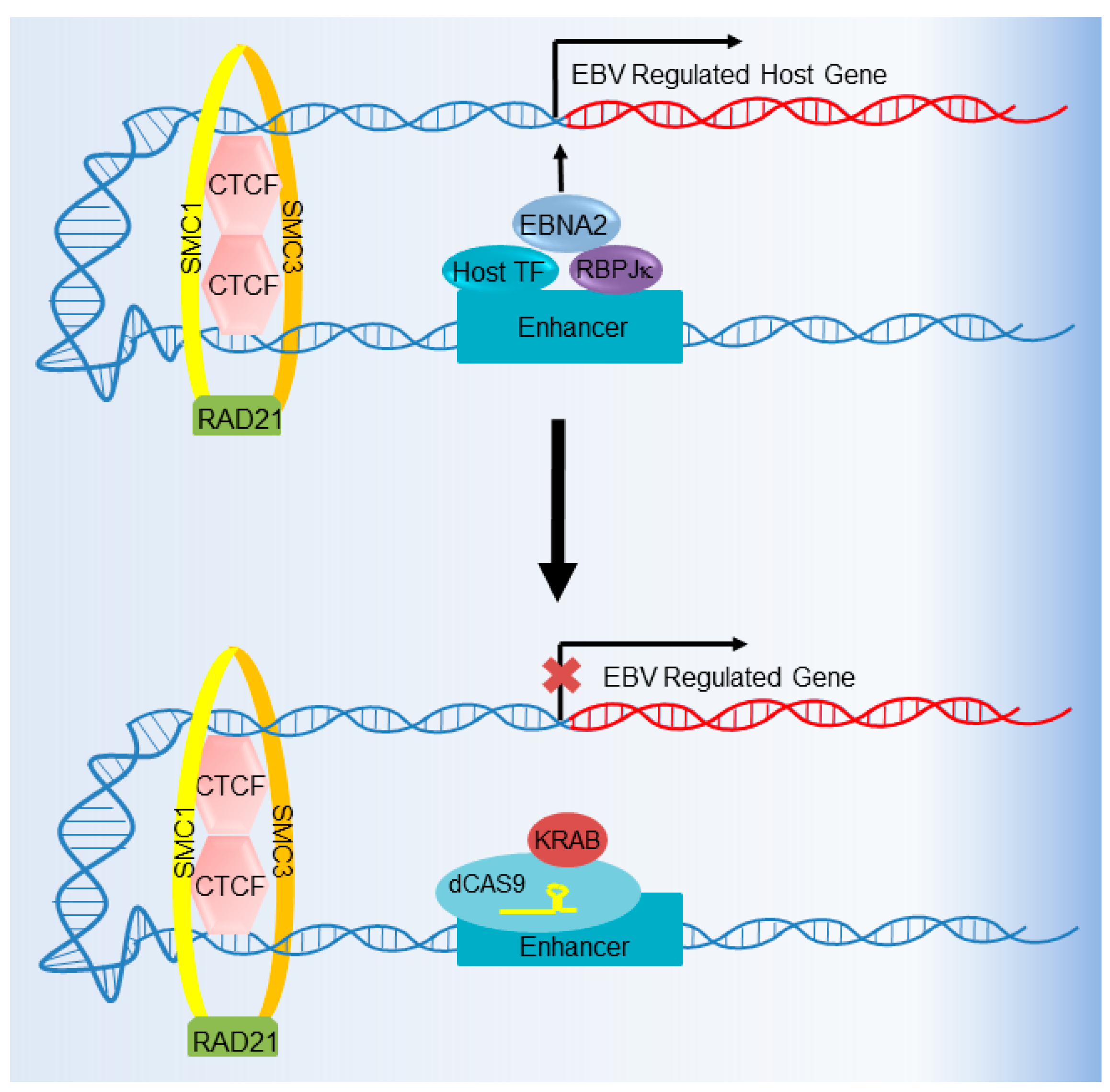
6. CRISPR Interference and CRISPR Activation
7. Future Directions
Acknowledgments
Conflicts of Interest
References
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Bhaya, D.; Davison, M.; Barrangou, R. Crispr-Cas systems in bacteria and archaea: Versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011, 45, 273–297. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Diez-Villasenor, C.; Garcia-Martinez, J.; Soria, E. Intervening sequences of regularly spaced prokaryotic repeats derive from foreign genetic elements. J. Mol. Evol. 2005, 60, 174–182. [Google Scholar] [CrossRef] [PubMed]
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. Crispr RNA maturation by trans-encoded small RNA and host factor RNAse iii. Nature 2011, 471, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Grishin, N.V.; Shabalina, S.A.; Wolf, Y.I.; Koonin, E.V. A putative rna-interference-based immune system in prokaryotes: Computational analysis of the predicted enzymatic machinery, functional analogies with eukaryotic RNAi, and hypothetical mechanisms of action. Biol. Direct 2006, 1, 7. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided Crispr CAas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Choi, J.; Bailey, S. Cut site selection by the two nuclease domains of the Cas9 RNA-guided endonuclease. J. Biol. Chem. 2014, 289, 13284–13294. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of Crispr-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G. Am I ready for Crispr? A user’s guide to genetic screens. Nat. Rev. Genet. 2017. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Zhang, F. High-throughput functional genomics using Crispr-Cas9. Nat. Rev. Genet. 2015, 16, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wei, J.J.; Sabatini, D.M.; Lander, E.S. Genetic screens in human cells using the Crispr-Cas9 system. Science 2014, 343, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Najm, F.J.; Strand, C.; Donovan, K.F.; Hegde, M.; Sanson, K.R.; Vaimberg, E.W.; Sullender, M.E.; Hartenian, E.; Kalani, Z.; Fusi, N.; et al. Orthologous Crispr-Cas9 enzymes for combinatorial genetic screens. Nat. Biotechnol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Friedland, A.E.; Baral, R.; Singhal, P.; Loveluck, K.; Shen, S.; Sanchez, M.; Marco, E.; Gotta, G.M.; Maeder, M.L.; Kennedy, E.M.; et al. Characterization of staphylococcus aureus Cas9: A smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol. 2015, 16, 257. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Topkar, V.V.; Zheng, Z.; Joung, J.K. Broadening the targeting range of staphylococcus aureus Crispr-Cas9 by modifying pam recognition. Nat. Biotechnol. 2015, 33, 1293–1298. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using Crispr/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.Y.; Pannunzio, N.R.; Adachi, N.; Lieber, M.R. Non-homologous DNA end joining and alternative pathways to double-strand break repair. Nat. Rev. Mol. Cell Biol. 2017, 18, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar] [PubMed]
- Ma, Y.; Pannicke, U.; Schwarz, K.; Lieber, M.R. Hairpin opening and overhang processing by an artemis/DNA-dependent protein kinase complex in nonhomologous end joining and v(d)j recombination. Cell 2002, 108, 781–794. [Google Scholar] [CrossRef]
- Moon, A.F.; Pryor, J.M.; Ramsden, D.A.; Kunkel, T.A.; Bebenek, K.; Pedersen, L.C. Sustained active site rigidity during synthesis by human DNA polymerase mu. Nat. Struct. Mol. Biol. 2014, 21, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Betermier, M.; Bertrand, P.; Lopez, B.S. Is non-homologous end-joining really an inherently error-prone process? PLoS Genet. 2014, 10, e1004086. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.H.; Adikaram, P.; Pandey, M.; Genis, A.; Simonds, W.F. Optimization of genome editing through Crispr-Cas9 engineering. Bioengineered 2016, 7, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Shalem, O.; Sanjana, N.E.; Hartenian, E.; Shi, X.; Scott, D.A.; Mikkelson, T.S.; Heckl, D.; Ebert, B.L.; Root, D.E.; Doench, J.G.; et al. Genome-scale Crispr-Cas9 knockout screening in human cells. Science 2014, 343, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Liu, F.; Gu, P.; Jin, H.; Chang, Y.; Wang, Q.; Liang, Q.; Qi, Q. A Crispr-Cas9 assisted non-homologous end-joining strategy for one-step engineering of bacterial genome. Sci. Rep. 2016, 6, 37895. [Google Scholar] [CrossRef] [PubMed]
- Chu, V.T.; Weber, T.; Wefers, B.; Wurst, W.; Sander, S.; Rajewsky, K.; Kuhn, R. Increasing the efficiency of homology-directed repair for Crispr-Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 2015, 33, 543–548. [Google Scholar] [CrossRef] [PubMed]
- White, M.K.; Hu, W.; Khalili, K. Gene editing approaches against viral infections and strategy to prevent occurrence of viral escape. PLoS Pathog. 2016, 12, e1005953. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Sun, L.; Gao, D.; Ding, C.; Li, Z.; Li, Y.; Cun, W.; Li, Q. High-efficiency targeted editing of large viral genomes by RNA-guided nucleases. PLoS Pathog. 2014, 10, e1004090. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Zhang, W.; Wang, J.; Al Yaghchi, C.; Ahmed, J.; Chard, L.; Lemoine, N.R.; Wang, Y. Efficiently editing the vaccinia virus genome by using the Crispr-Cas9 system. J. Virol. 2015, 89, 5176–5179. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Birsoy, K.; Hughes, N.W.; Krupczak, K.M.; Post, Y.; Wei, J.J.; Lander, E.S.; Sabatini, D.M. Identification and characterization of essential genes in the human genome. Science 2015, 350, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Dang, Y.; Wu, Y.; Jia, G.; Anaya, E.; Zhang, J.; Abraham, S.; Choi, J.G.; Shi, G.; Qi, L.; et al. A Crispr-based screen identifies genes essential for west-nile-virus-induced cell death. Cell Rep. 2015, 12, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Tzelepis, K.; Koike-Yusa, H.; De Braekeleer, E.; Li, Y.; Metzakopian, E.; Dovey, O.M.; Mupo, A.; Grinkevich, V.; Li, M.; Mazan, M.; et al. A crispr dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep. 2016, 17, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Smith, I.; Greenside, P.G.; Natoli, T.; Lahr, D.L.; Wadden, D.; Tirosh, I.; Narayan, R.; Root, D.E.; Golub, T.R.; Subramanian, A.; et al. Evaluation of RNAi and Crispr technologies by large-scale gene expression profiling in the connectivity map. PLoS Biol. 2017, 15, e2003213. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, Y.; Wu, X.; Wang, J.; Wang, Y.; Qiu, Z.; Chang, T.; Huang, H.; Lin, R.J.; Yee, J.K. Unbiased detection of off-target cleavage by Crispr-Cas9 and talens using integrase-defective lentiviral vectors. Nat. Biotechnol. 2015, 33, 175–178. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the Crispr endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Kleinstiver, B.P.; Pattanayak, V.; Prew, M.S.; Tsai, S.Q.; Nguyen, N.T.; Zheng, Z.; Joung, J.K. High-fidelity Crispr-Cas9 nucleases with no detectable genome-wide off-target effects. Nature 2016, 529, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Echeverri, C.J.; Beachy, P.A.; Baum, B.; Boutros, M.; Buchholz, F.; Chanda, S.K.; Downward, J.; Ellenberg, J.; Fraser, A.G.; Hacohen, N.; et al. Minimizing the risk of reporting false positives in large-scale RNAi screens. Nat. Methods 2006, 3, 777–779. [Google Scholar] [CrossRef] [PubMed]
- Sanjana, N.E. Genome-scale Crispr pooled screens. Anal Biochem. 2017, 532, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Lander, E.S.; Sabatini, D.M. Large-scale single guide rna library construction and use for Crispr-Cas9-based genetic screens. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Joung, J.; Engreitz, J.M.; Konermann, S.; Abudayyeh, O.O.; Verdine, V.K.; Aguet, F.; Gootenberg, J.S.; Sanjana, N.E.; Wright, J.B.; Fulco, C.P.; et al. Genome-scale activation screen identifies a lncRNA locus regulating a gene neighbourhood. Nature 2017, 548, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Hartenian, E.; Doench, J.G. Genetic screens and functional genomics using Crispr/Cas9 technology. FEBS J. 2015, 282, 1383–1393. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Koster, J.; Xu, H.; Chen, C.H.; Xiao, T.; Liu, J.S.; Brown, M.; Liu, X.S. Quality control, modeling, and visualization of Crispr screens with mageck-vispr. Genome Biol. 2015, 16, 281. [Google Scholar] [CrossRef] [PubMed]
- Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Carette, J.E. A Crispr toolbox to study virus-host interactions. Nat. Rev. Microbiol. 2017, 15, 351–364. [Google Scholar] [CrossRef] [PubMed]
- Perreira, J.M.; Meraner, P.; Brass, A.L. Functional genomic strategies for elucidating human-virus interactions: Will Crispr knockout RNAi and haploid cells? Adv. Virus Res. 2016, 94, 1–51. [Google Scholar] [PubMed]
- Zhang, R.; Miner, J.J.; Gorman, M.J.; Rausch, K.; Ramage, H.; White, J.P.; Zuiani, A.; Zhang, P.; Fernandez, E.; Zhang, Q.; et al. A crispr screen defines a signal peptide processing pathway required by flaviviruses. Nature 2016, 535, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Savidis, G.; McDougall, W.M.; Meraner, P.; Perreira, J.M.; Portmann, J.M.; Trincucci, G.; John, S.P.; Aker, A.M.; Renzette, N.; Robbins, D.R.; et al. Identification of zika virus and dengue virus dependency factors using functional genomics. Cell Rep. 2016, 16, 232–246. [Google Scholar] [CrossRef] [PubMed]
- Marceau, C.D.; Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Brewer, S.M.; Fuchs, G.; Swaminathan, K.; Mata, M.A.; Elias, J.E.; Sarnow, P.; et al. Genetic dissection of flaviviridae host factors through genome-scale Crispr screens. Nature 2016, 535, 159–163. [Google Scholar] [CrossRef] [PubMed]
- Orchard, R.C.; Wilen, C.B.; Doench, J.G.; Baldridge, M.T.; McCune, B.T.; Lee, Y.C.; Lee, S.; Pruett-Miller, S.M.; Nelson, C.A.; Fremont, D.H.; et al. Discovery of a proteinaceous cellular receptor for a norovirus. Science 2016, 353, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Haga, K.; Fujimoto, A.; Takai-Todaka, R.; Miki, M.; Doan, Y.H.; Murakami, K.; Yokoyama, M.; Murata, K.; Nakanishi, A.; Katayama, K. Functional receptor molecules cd300lf and cd300ld within the cd300 family enable murine noroviruses to infect cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6248–E6255. [Google Scholar] [CrossRef] [PubMed]
- Park, R.J.; Wang, T.; Koundakjian, D.; Hultquist, J.F.; Lamothe-Molina, P.; Monel, B.; Schumann, K.; Yu, H.; Krupzcak, K.M.; Garcia-Beltran, W.; et al. A genome-wide Crispr screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017, 49, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Seibert, C.; Cadene, M.; Sanfiz, A.; Chait, B.T.; Sakmar, T.P. Tyrosine sulfation of CCR5 n-terminal peptide by tyrosylprotein sulfotransferases 1 and 2 follows a discrete pattern and temporal sequence. Proc. Natl. Acad. Sci. USA 2002, 99, 11031–11036. [Google Scholar] [CrossRef] [PubMed]
- Parnas, O.; Jovanovic, M.; Eisenhaure, T.M.; Herbst, R.H.; Dixit, A.; Ye, C.J.; Przybylski, D.; Platt, R.J.; Tirosh, I.; Sanjana, N.E.; et al. A genome-wide Crispr screen in primary immune cells to dissect regulatory networks. Cell 2015, 162, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I.; Fauci, A.S.; Varmus, H.; Nabel, G.J. Epstein-barr virus: An important vaccine target for cancer prevention. Sci. Transl. Med. 2011, 3, 107fs7. [Google Scholar] [CrossRef] [PubMed]
- Longnecker, R.; Kieff, E.; Cohen, J.I. Epstein-barr virus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott, Williams and Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1898–1959. [Google Scholar]
- Jha, H.C.; Pei, Y.; Robertson, E.S. Epstein-barr virus: Diseases linked to infection and transformation. Front. Microbiol. 2016, 7, 1602. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Rickinson, A.B.; Bell, A.I. Epstein-barr virus-associated lymphomas. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, B.A.; Luftig, M.A. Recent advances in understanding epstein-barr virus. F1000Research 2017, 6, 386. [Google Scholar] [CrossRef] [PubMed]
- Young, L.S.; Yap, L.F.; Murray, P.G. Epstein-barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E. Gammaherpesviruses and lymphoproliferative disorders. Annu. Rev. Pathol. 2014, 9, 349–372. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.B.; Root, D.E. Resources for the design of Crispr gene editing experiments. Genome Biol. 2015, 16, 260. [Google Scholar] [CrossRef] [PubMed]
- Doench, J.G.; Hartenian, E.; Graham, D.B.; Tothova, Z.; Hegde, M.; Smith, I.; Sullender, M.; Ebert, B.L.; Xavier, R.J.; Root, D.E. Rational design of highly active sgRNAs for Crispr-Cas9-mediated gene inactivation. Nat. Biotechnol. 2014, 32, 1262–1267. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Wang, L.W.; Walsh, M.J.; Trudeau, S.J.; Gerdt, C.; Zhao, B.; Gewurz, B.E. Crispr/Cas9-mediated genome editing in epstein-barr virus-transformed lymphoblastoid B-cell lines. Curr. Protoc. Mol. Biol. 2018, 121, 31.12.1–31.12.23. [Google Scholar] [PubMed]
- Wang, J.; Quake, S.R. RNA-guided endonuclease provides a therapeutic strategy to cure latent herpesviridae infection. Proc. Natl. Acad. Sci. USA 2014, 111, 13157–13162. [Google Scholar] [CrossRef] [PubMed]
- Van Diemen, F.R.; Kruse, E.M.; Hooykaas, M.J.; Bruggeling, C.E.; Schurch, A.C.; van Ham, P.M.; Imhof, S.M.; Nijhuis, M.; Wiertz, E.J.; Lebbink, R.J. Crispr/Cas9-mediated genome editing of herpesviruses limits productive and latent infections. PLoS Pathog. 2016, 12, e1005701. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.S.; Wang, Z.M.; Wong, N.M.; Zhang, Z.Q.; Cheng, T.F.; Lui, W.Y.; Chan, C.P.; Jin, D.Y. Suppression of epstein-barr virus DNA load in latently infected nasopharyngeal carcinoma cells by Crispr/Cas9. Virus Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T.; Furuse, Y.; Oshitani, H.; Kiyono, T. Highly efficient Crispr/Cas9-mediated cloning and functional characterization of gastric cancer-derived epstein-barr virus strains. J. Virol. 2016, 90, 4383–4393. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, M.; Holthaus, A.M.; Calderwood, M.A.; Lai, C.Y.; Krastins, B.; Sarracino, D.; Johannsen, E. The EBNA3 family of epstein-barr virus nuclear proteins associates with the usp46/usp12 deubiquitination complexes to regulate lymphoblastoid cell line growth. PLoS Pathog. 2015, 11, e1004822. [Google Scholar] [CrossRef] [PubMed]
- Greenfeld, H.; Takasaki, K.; Walsh, M.J.; Ersing, I.; Bernhardt, K.; Ma, Y.; Fu, B.; Ashbaugh, C.W.; Cabo, J.; Mollo, S.B.; et al. Traf1 coordinates polyubiquitin signaling to enhance epstein-barr virus lmp1-mediated growth and survival pathway activation. PLoS Pathog. 2015, 11, e1004890. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; Perez White, B.E.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor a2 is a functional entry receptor for epstein-barr virus. Nat. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Li, Y.; Wang, H.B.; Zhang, A.; Chen, M.L.; Fang, Z.X.; Dong, X.D.; Li, S.B.; Du, Y.; Xiong, D.; et al. Ephrin receptor a2 is an epithelial cell receptor for epstein-barr virus entry. Nat. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Jordi, M.; Marty, J.; Mordasini, V.; Lunemann, A.; McComb, S.; Bernasconi, M.; Nadal, D. Irak4 is essential for tlr9-induced suppression of epstein-barr virus bzlf1 transcription in akata burkitt’s lymphoma cells. PLoS ONE 2017, 12, e0186614. [Google Scholar] [CrossRef] [PubMed]
- Ersing, I.; Nobre, L.; Wang, L.W.; Soday, L.; Ma, Y.; Paulo, J.A.; Narita, Y.; Ashbaugh, C.W.; Jiang, C.; Grayson, N.E.; et al. A temporal proteomic map of epstein-barr virus lytic replication in b cells. Cell Rep. 2017, 19, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Walsh, M.J.; Bernhardt, K.; Ashbaugh, C.W.; Trudeau, S.J.; Ashbaugh, I.Y.; Jiang, S.; Jiang, C.; Zhao, B.; Root, D.E.; et al. Crispr/Cas9 screens reveal epstein-barr virus-transformed b cell host dependency factors. Cell Host Microbe 2017, 21, 580–591.e7. [Google Scholar] [CrossRef] [PubMed]
- Basso, K.; Dalla-Favera, R. Roles of bcl6 in normal and transformed germinal center B cells. Immunol. Rev. 2012, 247, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Schmitz, R.; Ceribelli, M.; Pittaluga, S.; Wright, G.; Staudt, L.M. Oncogenic mechanisms in burkitt lymphoma. Cold Spring Harb. Perspect. Med. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.; Banerjee, S.; Jha, H.C.; Sun, Z.; Robertson, E.S. An essential EBV latent antigen 3c binds bcl6 for targeted degradation and cell proliferation. PLoS Pathog. 2017, 13, e1006500. [Google Scholar] [CrossRef] [PubMed]
- Kieser, A.; Sterz, K.R. The latent membrane protein 1 (lmp1). Curr. Top. Microbiol. Immunol. 2015, 391, 119–149. [Google Scholar] [PubMed]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-barr virus lmp1 mediated oncogenicity. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Luftig, M.; Yasui, T.; Soni, V.; Kang, M.S.; Jacobson, N.; Cahir-McFarland, E.; Seed, B.; Kieff, E. Epstein-barr virus latent infection membrane protein 1 traf-binding site induces nik/ikk α-dependent noncanonical NF-κb activation. Proc. Natl. Acad. Sci. USA 2004, 101, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Cahir-McFarland, E.D.; Carter, K.; Rosenwald, A.; Giltnane, J.M.; Henrickson, S.E.; Staudt, L.M.; Kieff, E. Role of NF-κb in cell survival and transcription of latent membrane protein 1-expressing or epstein-barr virus latency III-infected cells. J. Virol. 2004, 78, 4108–4119. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.J.; Kang, M.S. Roles of traf2 and traf3 in epstein-barr virus latent membrane protein 1-induced alternative NF-κb activation. Virus Genes 2010, 41, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Eliopoulos, A.G.; Caamano, J.H.; Flavell, J.; Reynolds, G.M.; Murray, P.G.; Poyet, J.L.; Young, L.S. Epstein-barr virus-encoded latent infection membrane protein 1 regulates the processing of p100 NF-κb2 to p52 via an ikkγ/nemo-independent signalling pathway. Oncogene 2003, 22, 7557–7569. [Google Scholar] [CrossRef] [PubMed]
- Callahan, J.; Aster, J.; Sklar, J.; Kieff, E.; Robertson, E.S. Intracellular forms of human notch1 interact at distinctly different levels with rbp-jκ in human B and T cells. Leukemia 2000, 14, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.J.; Hayward, S.D. Masking of the cbf1/rbpj κ transcriptional repression domain by epstein-barr virus ebna2. Science 1995, 268, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Ling, P.D.; Rawlins, D.R.; Hayward, S.D. The epstein-barr virus immortalizing protein ebna-2 is targeted to DNA by a cellular enhancer-binding protein. Proc. Natl. Acad. Sci. USA 1993, 90, 9237–9241. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, E.; Miller, C.L.; Grossman, S.R.; Kieff, E. Ebna-2 and ebna-3c extensively and mutually exclusively associate with rbpjκ in epstein-barr virus-transformed b lymphocytes. J. Virol. 1996, 70, 4179–4183. [Google Scholar] [PubMed]
- Zhao, B.; Zou, J.; Wang, H.; Johannsen, E.; Peng, C.W.; Quackenbush, J.; Mar, J.C.; Morton, C.C.; Freedman, M.L.; Blacklow, S.C.; et al. Epstein-barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth. Proc. Natl. Acad. Sci. USA 2011, 108, 14902–14907. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.R.; Johannsen, E.; Tong, X.; Yalamanchili, R.; Kieff, E. The epstein-barr virus nuclear antigen 2 transactivator is directed to response elements by the j κ recombination signal binding protein. Proc. Natl. Acad. Sci. USA 1994, 91, 7568–7572. [Google Scholar] [CrossRef] [PubMed]
- Henkel, T.; Ling, P.D.; Hayward, S.D.; Peterson, M.G. Mediation of epstein-barr virus ebna2 transactivation by recombination signal-binding protein j κ. Science 1994, 265, 92–95. [Google Scholar] [CrossRef] [PubMed]
- Skalska, L.; White, R.E.; Franz, M.; Ruhmann, M.; Allday, M.J. Epigenetic repression of p16(ink4a) by latent epstein-barr virus requires the interaction of ebna3a and ebna3c with ctbp. PLoS Pathog. 2010, 6, e1000951. [Google Scholar] [CrossRef] [PubMed]
- Touitou, R.; Hickabottom, M.; Parker, G.; Crook, T.; Allday, M.J. Physical and functional interactions between the corepressor ctbp and the epstein-barr virus nuclear antigen ebna3c. J. Virol. 2001, 75, 7749–7755. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Tourigny, J.P.; Forte, E.; Salinas, R.E.; Dave, S.S.; Luftig, M.A. Analysis of epstein-barr virus-regulated host gene expression changes through primary b-cell outgrowth reveals delayed kinetics of latent membrane protein 1-mediated NF-κb activation. J. Virol. 2012, 86, 11096–11106. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, K.; Haar, J.; Tsai, M.H.; Poirey, R.; Feederle, R.; Delecluse, H.J. A viral microrna cluster regulates the expression of pten, p27 and of a bcl-2 homolog. PLoS Pathog. 2016, 12, e1005405. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.A.; Hernandez-Hopkins, D.; Vider, J.; Ponomarev, V.; Hyjek, E.; Schattner, E.J.; Cesarman, E. NF-κb is essential for the progression of kshv- and ebv-infected lymphomas in vivo. Blood 2006, 107, 3295–3302. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Barrera, L.A.; Ersing, I.; Willox, B.; Schmidt, S.C.; Greenfeld, H.; Zhou, H.; Mollo, S.B.; Shi, T.T.; Takasaki, K.; et al. The NF-κb genomic landscape in lymphoblastoid B cells. Cell Rep. 2014, 8, 1595–1606. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Schmidt, S.C.; Jiang, S.; Willox, B.; Bernhardt, K.; Liang, J.; Johannsen, E.C.; Kharchenko, P.; Gewurz, B.E.; Kieff, E.; et al. Epstein-barr virus oncoprotein super-enhancers control B cell growth. Cell Host Microbe 2015, 17, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhou, H.; Liang, J.; Gerdt, C.; Wang, C.; Ke, L.; Schmidt, S.C.S.; Narita, Y.; Ma, Y.; Wang, S.; et al. The epstein-barr virus regulome in lymphoblastoid cells. Cell Host Microbe 2017, 22, 561–573.e4. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Lu, J.; Cai, Q.; Saha, A.; Jha, H.C.; Dzeng, R.K.; Robertson, E.S. The ebv latent antigen 3c inhibits apoptosis through targeted regulation of interferon regulatory factors 4 and 8. PLoS Pathog. 2013, 9, e1003314. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Willox, B.; Zhou, H.; Holthaus, A.M.; Wang, A.; Shi, T.T.; Maruo, S.; Kharchenko, P.V.; Johannsen, E.C.; Kieff, E.; et al. Epstein-barr virus nuclear antigen 3c binds to batf/irf4 or spi1/irf4 composite sites and recruits sin3a to repress cdkn2a. Proc. Natl. Acad. Sci. USA 2014, 111, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.C.; Jiang, S.; Zhou, H.; Willox, B.; Holthaus, A.M.; Kharchenko, P.V.; Johannsen, E.C.; Kieff, E.; Zhao, B. Epstein-barr virus nuclear antigen 3a partially coincides with ebna3c genome-wide and is tethered to DNA through batf complexes. Proc. Natl. Acad. Sci. USA 2015, 112, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Allday, M.J. Ebv finds a polycomb-mediated, epigenetic solution to the problem of oncogenic stress responses triggered by infection. Front. Genet. 2013, 4, 212. [Google Scholar] [CrossRef] [PubMed]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two epstein-barr virus (ebv) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor bim: Clues to the pathogenesis of burkitt’s lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Parker, G.A.; Watanatanasup, E.; White, R.E.; Allday, M.J. Bim promoter directly targeted by ebna3c in polycomb-mediated repression by EBV. Nucleic Acids Res. 2012, 40, 7233–7246. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.D.; Veenstra, H.; Khasnis, S.; Gunnell, A.; Webb, H.M.; Shannon-Lowe, C.; Andrews, S.; Osborne, C.S.; West, M.J. Myc activation and bcl2l11 silencing by a tumour virus through the large-scale reconfiguration of enhancer-promoter hubs. eLife 2016, 5, e18270. [Google Scholar] [CrossRef] [PubMed]
- Paschos, K.; Bazot, Q.; Ho, G.; Parker, G.A.; Lees, J.; Barton, G.; Allday, M.J. Core binding factor (cbf) is required for epstein-barr virus ebna3 proteins to regulate target gene expression. Nucleic Acids Res. 2017, 45, 2368–2383. [Google Scholar] [CrossRef] [PubMed]
- Minamitani, T.; Ma, Y.; Zhou, H.; Kida, H.; Tsai, C.Y.; Obana, M.; Okuzaki, D.; Fujio, Y.; Kumanogoh, A.; Zhao, B.; et al. Mouse model of epstein-barr virus lmp1- and lmp2a-driven germinal center B-cell lymphoproliferative disease. Proc. Natl. Acad. Sci. USA 2017, 114, 4751–4756. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Nie, K.; Redmond, D.; Liu, Y.; Elemento, O.; Knowles, D.M.; Tam, W. Ebv-mir-bhrf1-2 targets prdm1/blimp1: Potential role in EBV lymphomagenesis. Leukemia 2016, 30, 594–604. [Google Scholar] [CrossRef] [PubMed]
- Nawandar, D.M.; Wang, A.; Makielski, K.; Lee, D.; Ma, S.; Barlow, E.; Reusch, J.; Jiang, R.; Wille, C.K.; Greenspan, D.; et al. Differentiation-dependent klf4 expression promotes lytic epstein-barr virus infection in epithelial cells. PLoS Pathog. 2015, 11, e1005195. [Google Scholar] [CrossRef] [PubMed]
- Reusch, J.A.; Nawandar, D.M.; Wright, K.L.; Kenney, S.C.; Mertz, J.E. Cellular differentiation regulator blimp1 induces epstein-barr virus lytic reactivation in epithelial and B cells by activating transcription from both the R and Z promoters. J. Virol. 2015, 89, 1731–1743. [Google Scholar] [CrossRef] [PubMed]
- Buettner, M.; Lang, A.; Tudor, C.S.; Meyer, B.; Cruchley, A.; Barros, M.H.; Farrell, P.J.; Jack, H.M.; Schuh, W.; Niedobitek, G. Lytic epstein-barr virus infection in epithelial cells but not in B-lymphocytes is dependent on blimp1. J. Gen. Virol. 2012, 93, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Sanjana, N.E.; Zheng, K.; Shalem, O.; Lee, K.; Shi, X.; Scott, D.A.; Song, J.; Pan, J.Q.; Weissleder, R.; et al. Genome-wide Crispr screen in a mouse model of tumor growth and metastasis. Cell 2015, 160, 1246–1260. [Google Scholar] [CrossRef] [PubMed]
- Manguso, R.T.; Pope, H.W.; Zimmer, M.D.; Brown, F.D.; Yates, K.B.; Miller, B.C.; Collins, N.B.; Bi, K.; LaFleur, M.W.; Juneja, V.R.; et al. In vivo crispr screening identifies ptpn2 as a cancer immunotherapy target. Nature 2017, 547, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing crispr as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Kampmann, M. Crispri and crispra screens in mammalian cells for precision biology and medicine. ACS Chem. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Larson, M.H.; Morsut, L.; Liu, Z.; Brar, G.A.; Torres, S.E.; Stern-Ginossar, N.; Brandman, O.; Whitehead, E.H.; Doudna, J.A. Crispr-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 2013, 154, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Groner, A.C.; Meylan, S.; Ciuffi, A.; Zangger, N.; Ambrosini, G.; Dénervaud, N.; Bucher, P.; Trono, D. Krab–zinc finger proteins and kap1 can mediate long-range transcriptional repression through heterochromatin spreading. PLoS Genet. 2010, 6, e1000869. [Google Scholar] [CrossRef] [PubMed]
- Chavez, A.; Scheiman, J.; Vora, S.; Pruitt, B.W.; Tuttle, M.; Iyer, E.P.; Lin, S.; Kiani, S.; Guzman, C.D.; Wiegand, D.J. Highly efficient Cas9-mediated transcriptional programming. Nat. Methods 2015, 12, 326–328. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, L.A.; Horlbeck, M.A.; Adamson, B.; Villalta, J.E.; Chen, Y.; Whitehead, E.H.; Guimaraes, C.; Panning, B.; Ploegh, H.L.; Bassik, M.C. Genome-scale Crispr-mediated control of gene repression and activation. Cell 2014, 159, 647–661. [Google Scholar] [CrossRef] [PubMed]
- Tanenbaum, M.E.; Gilbert, L.A.; Qi, L.S.; Weissman, J.S.; Vale, R.D. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 2014, 159, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Perez-Pinera, P.; Kocak, D.D.; Vockley, C.M.; Adler, A.F.; Kabadi, A.M.; Polstein, L.R.; Thakore, P.I.; Glass, K.A.; Ousterout, D.G.; Leong, K.W. RNA-guided gene activation by Crispr-Cas9-based transcription factors. Nat. Methods 2013, 10, 973–976. [Google Scholar] [CrossRef] [PubMed]
- Horlbeck, M.A.; Gilbert, L.A.; Villalta, J.E.; Adamson, B.; Pak, R.A.; Chen, Y.; Fields, A.P.; Park, C.Y.; Corn, J.E.; Kampmann, M. Compact and highly active next-generation libraries for crispr-mediated gene repression and activation. eLife 2016, 5, e19760. [Google Scholar] [CrossRef] [PubMed]
- Du, D.; Qi, L.S. Crispr technology for genome activation and repression in mammalian cells. Cold Spring Harb. Protoc. 2016, 2016. [Google Scholar] [CrossRef] [PubMed]
- Horlbeck, M.A.; Witkowsky, L.B.; Guglielmi, B.; Replogle, J.M.; Gilbert, L.A.; Villalta, J.E.; Torigoe, S.E.; Tijan, R.; Weissman, J.S. Nucleosomes impede Cas9 access to DNA in vivo and in vitro. eLife 2016, 5, e12677. [Google Scholar] [CrossRef] [PubMed]
- Heaton, B.E.; Kennedy, E.M.; Dumm, R.E.; Harding, A.T.; Sacco, M.T.; Sachs, D.; Heaton, N.S. A Crispr activation screen identifies a pan-avian influenza virus inhibitory host factor. Cell Rep. 2017, 20, 1503–1512. [Google Scholar] [CrossRef] [PubMed]
- Stencel-Baerenwald, J.E.; Reiss, K.; Reiter, D.M.; Stehle, T.; Dermody, T.S. The sweet spot: Defining virus-sialic acid interactions. Nat. Rev. Microbiol. 2014, 12, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Wilks, S.; de Graaf, M.; Smith, D.J.; Burke, D.F. A review of influenza haemagglutinin receptor binding as it relates to pandemic properties. Vaccine 2012, 30, 4369–4376. [Google Scholar] [CrossRef] [PubMed]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding rnas. Cell 2009, 136, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Rinn, J.L.; Chang, H.Y. Genome regulation by long noncoding RNAs. Annu. Rev. Biochem. 2012, 81, 145–166. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, A.M.; Chang, H.Y. Long noncoding RNAs in cancer pathways. Cancer Cell 2016, 29, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.; Kraus, W.L. From discovery to function: The expanding roles of long noncoding RNAs in physiology and disease. Endocr. Rev. 2015, 36, 25–64. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, Y.; Anderson, E.M.; Birmingham, A.; Reynolds, A.; Karpilow, J.; Robinson, K.; Leake, D.; Marshall, W.S.; Khvorova, A. Off-target effects by siRNA can induce toxic phenotype. RNA 2006, 12, 1188–1196. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.L.; Linsley, P.S. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat. Rev. Drug Discov. 2010, 9, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Cullen, B.R. RNA interference in human cells is restricted to the cytoplasm. RNA 2002, 8, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.J.; Horlbeck, M.A.; Cho, S.W.; Birk, H.S.; Malatesta, M.; He, D.; Attenello, F.J.; Villalta, J.E.; Cho, M.Y.; Chen, Y. Crispri-based genome-scale identification of functional long noncoding RNA loci in human cells. Science 2017, 355. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, A.J.; Meyers, R.M.; Weir, B.A.; Vazquez, F.; Zhang, C.-Z.; Ben-David, U.; Cook, A.; Ha, G.; Harrington, W.F.; Doshi, M.B. Genomic copy number dictates a gene-independent cell response to Crispr/Cas9 targeting. Cancer Discov. 2016, 6, 914–929. [Google Scholar] [CrossRef] [PubMed]
- Mandage, R.; Telford, M.; Rodríguez, J.A.; Farré, X.; Layouni, H.; Marigorta, U.M.; Cundiff, C.; Heredia-Genestar, J.M.; Navarro, A.; Santpere, G. Genetic factors affecting EBV copy number in lymphoblastoid cell lines derived from the 1000 genome project samples. PLoS ONE 2017, 12, e0179446. [Google Scholar] [CrossRef] [PubMed]
- Delecluse, H.; Bartnizke, S.; Hammerschmidt, W.; Bullerdiek, J.; Bornkamm, G. Episomal and integrated copies of epstein-barr virus coexist in burkitt lymphoma cell lines. J. Virol. 1993, 67, 1292–1299. [Google Scholar] [PubMed]
- McClellan, M.J.; Wood, C.D.; Ojeniyi, O.; Cooper, T.J.; Kanhere, A.; Arvey, A.; Webb, H.M.; Palermo, R.D.; Harth-Hertle, M.L.; Kempkes, B. Modulation of enhancer looping and differential gene targeting by epstein-barr virus transcription factors directs cellular reprogramming. PLoS Pathog. 2013, 9, e1003636. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.W.; Trudeau, S.J.; Wang, C.; Gerdt, C.; Jiang, S.; Zhao, B.; Gewurz, B.E. Modulating gene expression in epstein-barr virus (EBV)-positive B cell lines with CRISPRa and CRISPRi. Curr. Protoc. Mol. Biol. 2018, 121, 31.13.1–31.13.18. [Google Scholar] [CrossRef] [PubMed]
- Carleton, J.B.; Berrett, K.C.; Gertz, J. Multiplex enhancer interference reveals collaborative control of gene regulation by estrogen receptor α-bound enhancers. Cell Syst. 2017, 5, 333–344.e5. [Google Scholar] [CrossRef] [PubMed]
- Costanzo, M.; VanderSluis, B.; Koch, E.N.; Baryshnikova, A.; Pons, C.; Tan, G.; Wang, W.; Usaj, M.; Hanchard, J.; Lee, S.D.; et al. A global genetic interaction network maps a wiring diagram of cellular function. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Murugan, K.; Babu, K.; Sundaresan, R.; Rajan, R.; Sashital, D.G. The revolution continues: Newly discovered systems expand the Crispr-Cas toolkit. Mol. Cell 2017, 68, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Abudayyeh, O.O.; Gootenberg, J.S.; Essletzbichler, P.; Han, S.; Joung, J.; Belanto, J.J.; Verdine, V.; Cox, D.B.; Kellner, M.J.; Regev, A. RNA targeting with Crispr–Cas13. Nature 2017, 550, 280–284. [Google Scholar] [CrossRef] [PubMed]
- Cox, D.B.; Gootenberg, J.S.; Abudayyeh, O.O.; Franklin, B.; Kellner, M.J.; Joung, J.; Zhang, F. RNA editing with Crispr-Cas13. Science 2017, 358, 1019–1027. [Google Scholar] [CrossRef] [PubMed]
- Mahas, A.; Stewart, C.N.; Mahfouz, M.M. Harnessing Crispr/Cas systems for programmable transcriptional and post-transcriptional regulation. Biotechnol. Adv. 2017. [Google Scholar] [CrossRef] [PubMed]
- Gootenberg, J.S.; Abudayyeh, O.O.; Lee, J.W.; Essletzbichler, P.; Dy, A.J.; Joung, J.; Verdine, V.; Donghia, N.; Daringer, N.M.; Freije, C.A.; et al. Nucleic acid detection with Crispr-Cas13a/c2c2. Science 2017, 356, 438–442. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gebre, M.; Nomburg, J.L.; Gewurz, B.E. CRISPR–Cas9 Genetic Analysis of Virus–Host Interactions. Viruses 2018, 10, 55. https://doi.org/10.3390/v10020055
Gebre M, Nomburg JL, Gewurz BE. CRISPR–Cas9 Genetic Analysis of Virus–Host Interactions. Viruses. 2018; 10(2):55. https://doi.org/10.3390/v10020055
Chicago/Turabian StyleGebre, Makda, Jason L. Nomburg, and Benjamin E. Gewurz. 2018. "CRISPR–Cas9 Genetic Analysis of Virus–Host Interactions" Viruses 10, no. 2: 55. https://doi.org/10.3390/v10020055
APA StyleGebre, M., Nomburg, J. L., & Gewurz, B. E. (2018). CRISPR–Cas9 Genetic Analysis of Virus–Host Interactions. Viruses, 10(2), 55. https://doi.org/10.3390/v10020055