Epigenetic Modulation of CD8+ T Cell Function in Lentivirus Infections: A Review


Abstract
1. Introduction
2. Animal Models of HIV Infection to Study CD8+ T Cell Dysfunction
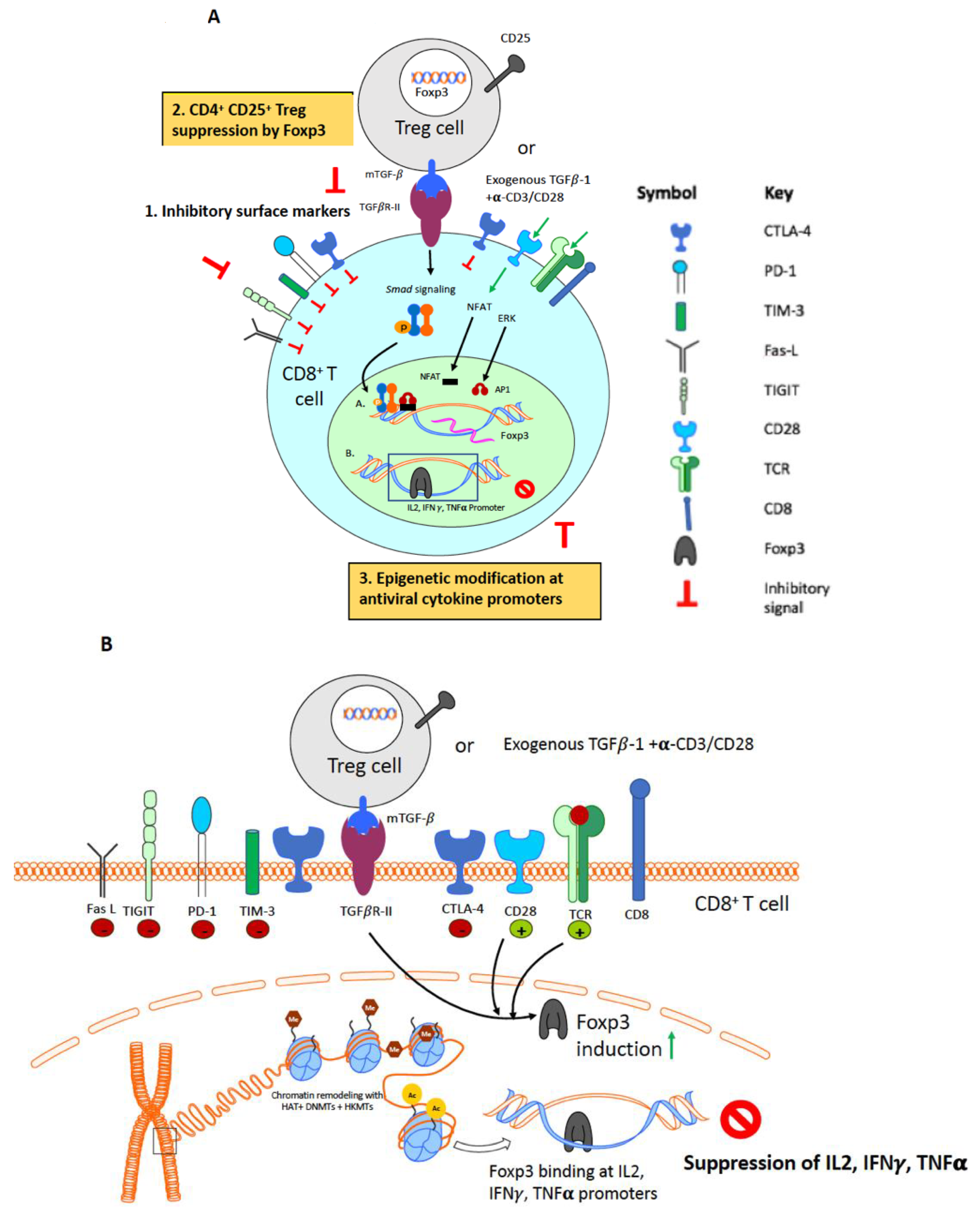
3. CD8+ T Cell Suppression by CD4+CD25+ T Regulatory Cells
4. Epigenetic Mechanisms Regulating Gene Transcription: A General Overview
5. Epigenetics in FIV and SIV
6. Epigenetics in HIV
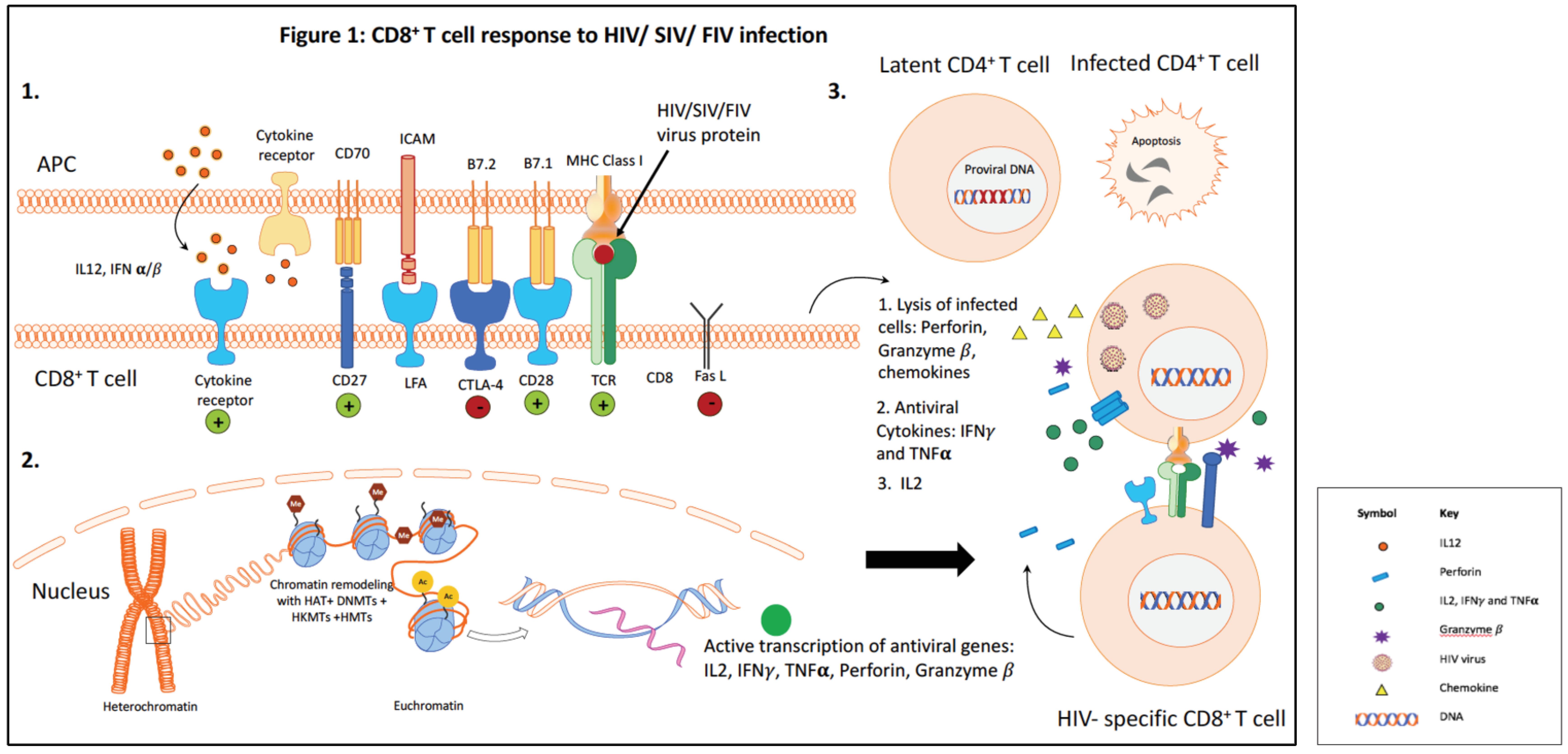
7. Epigenetic Modulation of Immune Cells as a Result of HIV Infection
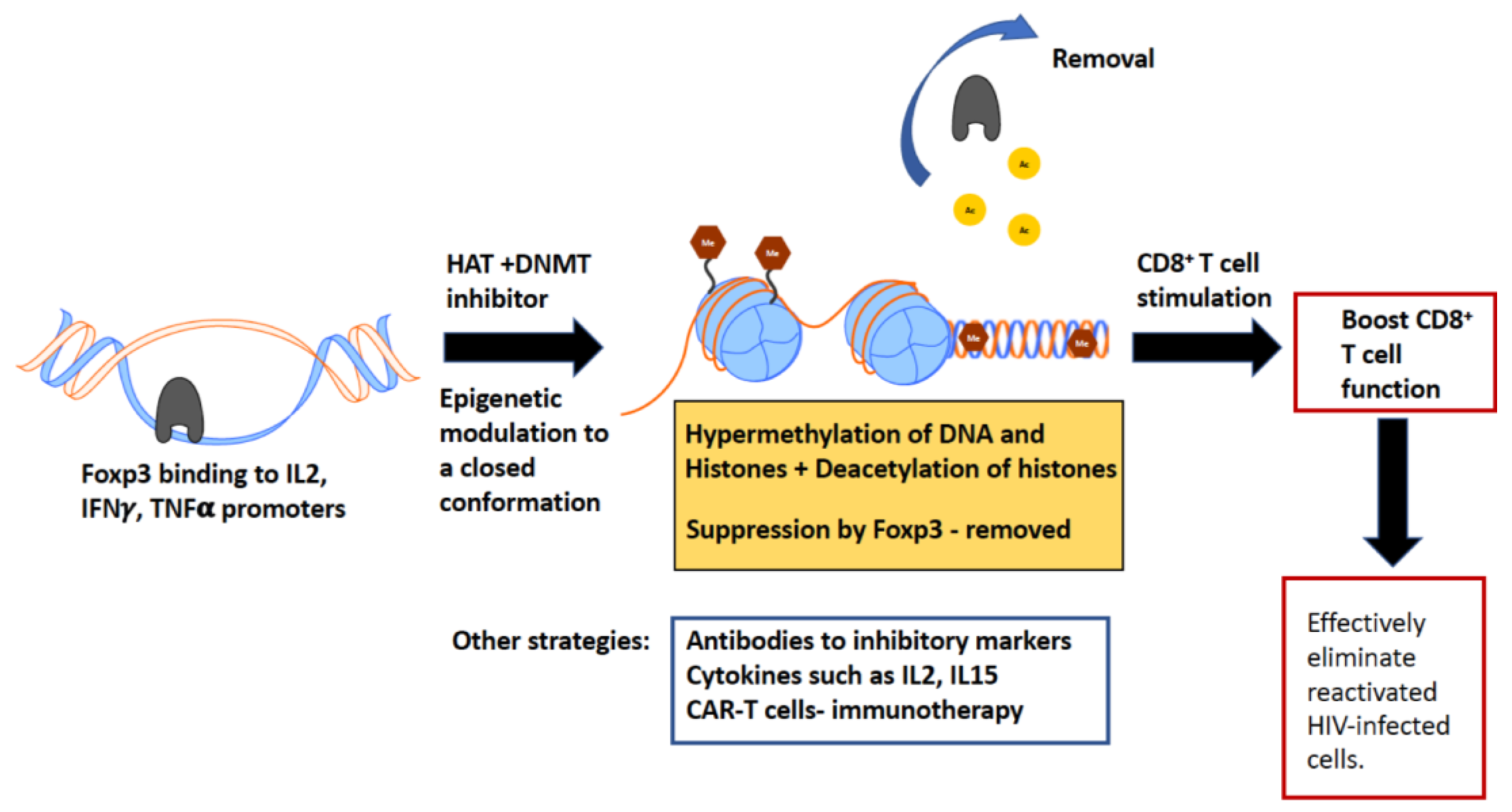
8. Current Strategies to Boost CD8+ T Cell Function
9. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Gulzar, N.; Copeland, K.F. CD8+ T-cells: Function and response to HIV infection. Curr. HIV Res. 2004, 2, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Migueles, S.A.; Connors, M. The Role of CD4+ and CD8+ T Cells in Controlling HIV Infection. Curr. Infect. Dis. Rep. 2002, 4, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.S.; Shaw, G.M.; McMichael, A.J.; Haynes, B.F. Acute HIV-1 Infection. N. Engl. J. Med. 2011, 364, 1943–1954. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Lecuroux, C.; Saez-Cirion, A.; Pancino, G.; Girault, I.; Versmisse, P.; Boufassa, F.; Taulera, O.; Sinet, M.; Lambotte, O.; et al. Potential role for HIV-specific CD38−/HLA-DR+ CD8+ T cells in viral suppression and cytotoxicity in HIV controllers. PLoS ONE 2014, 9, e101920. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Nixon, D.F.; Donahoe, S.M.; Gillespie, G.M.; Dong, T.; King, A.; Ogg, G.S.; Spiegel, H.M.; Conlon, C.; Spina, C.A.; et al. HIV-specific CD8+ T cells produce antiviral cytokines but are impaired in cytolytic function. J. Exp. Med. 2000, 192, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Goepfert, P.A.; Bansal, A.; Edwards, B.H.; Ritter, G.D., Jr.; Tellez, I.; McPherson, S.A.; Sabbaj, S.; Mulligan, M.J. A significant number of human immunodeficiency virus epitope-specific cytotoxic T lymphocytes detected by tetramer binding do not produce gamma interferon. J. Virol. 2000, 74, 10249–10255. [Google Scholar] [CrossRef] [PubMed]
- Chun, T.W.; Carruth, L.; Finzi, D.; Shen, X.; DiGiuseppe, J.A.; Taylor, H.; Hermankova, M.; Chadwick, K.; Margolick, J.; Quinn, T.C.; et al. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 1997, 387, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Pierson, T.; McArthur, J.; Siliciano, R.F. Reservoirs for HIV-1: Mechanisms for viral persistence in the presence of antiviral immune responses and antiretroviral therapy. Annu. Rev. Immunol. 2000, 18, 665–708. [Google Scholar] [CrossRef] [PubMed]
- Cartwright, E.K.; Spicer, L.; Smith, S.A.; Lee, D.; Fast, R.; Paganini, S.; Lawson, B.O.; Nega, M.; Easley, K.; Schmitz, J.E.; et al. CD8+ Lymphocytes Are Required for Maintaining Viral Suppression in SIV-Infected Macaques Treated with Short-Term Antiretroviral Therapy. Immunity 2016, 45, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. HIV: Shock and kill. Nature 2012, 487, 439–440. [Google Scholar] [CrossRef] [PubMed]
- Pandrea, I.; Gaufin, T.; Gautam, R.; Kristoff, J.; Mandell, D.; Montefiori, D.; Keele, B.F.; Ribeiro, R.M.; Veazey, R.S.; Apetrei, C. Functional cure of SIVagm infection in rhesus macaques results in complete recovery of CD4+ T cells and is reverted by CD8+ cell depletion. PLoS Pathog. 2011, 7, e1002170. [Google Scholar] [CrossRef] [PubMed]
- Schietinger, A.; Greenberg, P.D. Tolerance and exhaustion: Defining mechanisms of T cell dysfunction. Trends Immunol. 2014, 35, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J. T cell exhaustion. Nat. Immunol. 2011, 12, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Wherry, E.J.; Kurachi, M. Molecular and cellular insights into T cell exhaustion. Nat. Rev. Immunol. 2015, 15, 486–499. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, S.D.; Shin, H.; Haining, W.N.; Zou, T.; Workman, C.J.; Polley, A.; Betts, M.R.; Freeman, G.J.; Vignali, D.A.; Wherry, E.J. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 2009, 10, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Ndhlovu, L.C.; Barbour, J.D.; Sheth, P.M.; Jha, A.R.; Long, B.R.; Wong, J.C.; Satkunarajah, M.; Schweneker, M.; Chapman, J.M.; et al. Tim-3 expression defines a novel population of dysfunctional T cells with highly elevated frequencies in progressive HIV-1 infection. J. Exp. Med. 2008, 205, 2763–2779. [Google Scholar] [CrossRef] [PubMed]
- Velu, V.; Titanji, K.; Zhu, B.; Husain, S.; Pladevega, A.; Lai, L.; Vanderford, T.H.; Chennareddi, L.; Silvestri, G.; Freeman, G.J.; et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 2009, 458, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Vigano, S.; Negron, J.; Ouyang, Z.; Rosenberg, E.S.; Walker, B.D.; Lichterfeld, M.; Yu, X.G. Prolonged Antiretroviral Therapy Preserves HIV-1-Specific CD8 T Cells with Stem Cell-Like Properties. J. Virol. 2015, 89, 7829–7840. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.G.; Kitchen, S.G.; Kitchen, C.M.; Scripture-Adams, D.D.; Zack, J.A. Generation of HIV latency during thymopoiesis. Nat. Med. 2001, 7, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Brooks, D.G.; Hamer, D.H.; Arlen, P.A.; Gao, L.; Bristol, G.; Kitchen, C.M.; Berger, E.A.; Zack, J.A. Molecular characterization, reactivation, and depletion of latent HIV. Immunity 2003, 19, 413–423. [Google Scholar] [CrossRef]
- Policicchio, B.B.; Pandrea, I.; Apetrei, C. Animal Models for HIV Cure Research. Front. Immunol. 2016, 7, 12. [Google Scholar] [CrossRef] [PubMed]
- Burkhard, M.J.; Dean, G.A. Transmission and immunopathogenesis of FIV in cats as a model for HIV. Curr. HIV Res. 2003, 1, 15–29. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.H.; Lin, Y.C.; Fink, E.; Grant, C.K. Feline immunodeficiency virus (FIV) as a model for study of lentivirus infections: Parallels with HIV. Curr. HIV Res. 2010, 8, 73–80. [Google Scholar] [CrossRef] [PubMed]
- McDonnel, S.J.; Sparger, E.E.; Murphy, B.G. Feline immunodeficiency virus latency. Retrovirology 2013, 10, 69. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, N.C.; Yamamoto, J.K.; Ishida, T.; Hansen, H. Feline immunodeficiency virus infection. Vet. Immunol. Immunopathol. 1989, 21, 111–129. [Google Scholar] [CrossRef]
- Yamamoto, J.K.; Hansen, H.; Ho, E.W.; Morishita, T.Y.; Okuda, T.; Sawa, T.R.; Nakamura, R.M.; Pedersen, N.C. Epidemiologic and clinical aspects of feline immunodeficiency virus infection in cats from the continental United States and Canada and possible mode of transmission. J. Am. Vet. Med. Assoc. 1989, 194, 213–220. [Google Scholar] [PubMed]
- Magden, E.; Quackenbush, S.L.; VandeWoude, S. FIV associated neoplasms—A mini-review. Vet. Immunol. Immunopathol. 2011, 143, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Phipps, A.J.; Hayes, K.A.; Buck, W.R.; Podell, M.; Mathes, L.E. Neurophysiologic and immunologic abnormalities associated with feline immunodeficiency virus molecular clone FIV-PPR DNA inoculation. J. Acquir. Immune Defic. Syndr. 2000, 23, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Podell, M.; Hayes, K.; Oglesbee, M.; Mathes, L. Progressive encephalopathy associated with CD4/CD8 inversion in adult FIV-infected cats. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1997, 15, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Podell, M.; Maruyama, K.; Smith, M.; Hayes, K.A.; Buck, W.R.; Ruehlmann, D.S.; Mathes, L.E. Frontal lobe neuronal injury correlates to altered function in FIV-infected cats. J. Acquir. Immune Defic. Syndr. 1999, 22, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.K.; Sanou, M.P.; Abbott, J.R.; Coleman, J.K. Feline immunodeficiency virus model for designing HIV/AIDS vaccines. Curr. HIV Res. 2010, 8, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Kuffer, M.; Balzarini, J.; Naesens, L.; Goldberg, M.; Erfle, V.; Goebel, F.D.; De Clercq, E.; Jindrich, J.; Holy, A.; et al. Efficacy of the acyclic nucleoside phosphonates (S)-9-(3-fluoro-2-phosphonylmethoxypropyl)adenine (FPMPA) and 9-(2-phosphonylmethoxyethyl)adenine (PMEA) against feline immunodeficiency virus. J. Acquir. Immune Defic. Syndr. Hum. Retrovirol. 1998, 17, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Siebelink, K.H.; Chu, I.H.; Rimmelzwaan, G.F.; Weijer, K.; van Herwijnen, R.; Knell, P.; Egberink, H.F.; Bosch, M.L.; Osterhaus, A.D. Feline immunodeficiency virus (FIV) infection in the cat as a model for HIV infection in man: FIV-induced impairment of immune function. AIDS Res. Hum. Retrovir. 1990, 6, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Arai, M.; Earl, D.D.; Yamamoto, J.K. Is AZT/3TC therapy effective against FIV infection or immunopathogenesis? Vet. Immunol. Immunopathol. 2002, 85, 189–204. [Google Scholar] [CrossRef]
- Fogle, J.E.; Tompkins, W.A.; Campbell, B.; Sumner, D.; Tompkins, M.B. Fozivudine tidoxil as single-agent therapy decreases plasma and cell-associated viremia during acute feline immunodeficiency virus infection. J. Vet. Intern. Med. 2011, 25, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, K.; Donath, A.; Beer, B.; Egberink, H.F.; Horzinek, M.C.; Lutz, H.; Hoffmann-Fezer, G.; Thum, I.; Thefeld, S. Use of two virustatica (AZT, PMEA) in the treatment of FIV and of FeLV seropositive cats with clinical symptoms. Vet. Immunol. Immunopathol. 1992, 35, 167–175. [Google Scholar] [CrossRef]
- Smyth, N.R.; McCracken, C.; Gaskell, R.M.; Cameron, J.M.; Coates, J.A.; Gaskell, C.J.; Hart, C.A.; Bennett, M. Susceptibility in cell culture of feline immunodeficiency virus to eighteen antiviral agents. J. Antimicrob. Chemother. 1994, 34, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Savarino, A.; Pistello, M.; D’Ostilio, D.; Zabogli, E.; Taglia, F.; Mancini, F.; Ferro, S.; Matteucci, D.; De Luca, L.; Barreca, M.L.; et al. Human immunodeficiency virus integrase inhibitors efficiently suppress feline immunodeficiency virus replication in vitro and provide a rationale to redesign antiretroviral treatment for feline AIDS. Retrovirology 2007, 4, 79. [Google Scholar] [CrossRef] [PubMed]
- Wongsrikeao, P.; Saenz, D.; Rinkoski, T.; Otoi, T.; Poeschla, E. Antiviral restriction factor transgenesis in the domestic cat. Nat. Methods 2011, 8, 853–859. [Google Scholar] [CrossRef] [PubMed]
- Colin, L.; Van Lint, C. Molecular control of HIV-1 postintegration latency: Implications for the development of new therapeutic strategies. Retrovirology 2009, 6, 111. [Google Scholar] [CrossRef] [PubMed]
- McDonnel, S.J.; Sparger, E.E.; Luciw, P.A.; Murphy, B.G. Transcriptional regulation of latent feline immunodeficiency virus in peripheral CD4+ T-lymphocytes. Viruses 2012, 4, 878–888. [Google Scholar] [CrossRef] [PubMed]
- McDonnel, S.J.; Liepnieks, M.L.; Murphy, B.G. Treatment of chronically FIV-infected cats with suberoylanilide hydroxamic acid. Antivir. Res. 2014, 108, 74–78. [Google Scholar] [CrossRef] [PubMed]
- Bienzle, D. FIV in cats—A useful model of HIV in people? Vet. Immunol. Immunopathol. 2014, 159, 171–179. [Google Scholar] [CrossRef] [PubMed]
- Gemeniano, M.C.; Sawai, E.T.; Sparger, E.E. Feline immunodeficiency virus Orf-A localizes to the nucleus and induces cell cycle arrest. Virology 2004, 325, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Dean, G.A.; Reubel, G.H.; Moore, P.F.; Pedersen, N.C. Proviral burden and infection kinetics of feline immunodeficiency virus in lymphocyte subsets of blood and lymph node. J. Virol. 1996, 70, 5165–5169. [Google Scholar] [PubMed]
- Shimojima, M.; Miyazawa, T.; Ikeda, Y.; McMonagle, E.L.; Haining, H.; Akashi, H.; Takeuchi, Y.; Hosie, M.J.; Willett, B.J. Use of CD134 as a primary receptor by the feline immunodeficiency virus. Science 2004, 303, 1192–1195. [Google Scholar] [CrossRef] [PubMed]
- Moir, S.; Malaspina, A.; Li, Y.; Chun, T.W.; Lowe, T.; Adelsberger, J.; Baseler, M.; Ehler, L.A.; Liu, S.; Davey, R.T., Jr.; et al. B cells of HIV-1-infected patients bind virions through CD21-complement interactions and transmit infectious virus to activated T cells. J. Exp. Med. 2000, 192, 637–646. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Moir, S.; Kulik, L.; Malaspina, A.; Donoghue, E.T.; Miller, N.J.; Wang, W.; Chun, T.W.; Fauci, A.S.; Holers, V.M. Role for CD21 in the establishment of an extracellular HIV reservoir in lymphoid tissues. J. Immunol. 2007, 178, 6968–6974. [Google Scholar] [CrossRef] [PubMed]
- Eckstrand, C.D.; Sparger, E.E.; Pitt, K.A.; Murphy, B.G. Peripheral and central immune cell reservoirs in tissues from asymptomatic cats chronically infected with feline immunodeficiency virus. PLoS ONE 2017, 12, e0175327. [Google Scholar] [CrossRef] [PubMed]
- Gardner, M.B.; Luciw, P.A. Macaque models of human infectious disease. ILAR J. 2008, 49, 220–255. [Google Scholar] [CrossRef] [PubMed]
- Chahroudi, A.; Bosinger, S.E.; Vanderford, T.H.; Paiardini, M.; Silvestri, G. Natural SIV hosts: Showing AIDS the door. Science 2012, 335, 1188–1193. [Google Scholar] [CrossRef] [PubMed]
- Hatziioannou, T.; Evans, D.T. Animal models for HIV/AIDS research. Nat. Rev. Microbiol. 2012, 10, 852–867. [Google Scholar] [CrossRef] [PubMed]
- Van Rompay, K.K. The use of nonhuman primate models of HIV infection for the evaluation of antiviral strategies. AIDS Res. Hum. Retrovir. 2012, 28, 16–35. [Google Scholar] [CrossRef] [PubMed]
- Van Rompay, K.K. Evaluation of antiretrovirals in animal models of HIV infection. Antivir. Res. 2010, 85, 159–175. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.C.; Emau, P.; Follis, K.E.; Beck, T.W.; Benveniste, R.E.; Bischofberger, N.; Lifson, J.D.; Morton, W.R. Effectiveness of postinoculation (R)-9-(2-phosphonylmethoxypropyl) adenine treatment for prevention of persistent simian immunodeficiency virus SIVmne infection depends critically on timing of initiation and duration of treatment. J. Virol. 1998, 72, 4265–4273. [Google Scholar] [PubMed]
- Abdool Karim, Q.; Abdool Karim, S.S.; Frohlich, J.A.; Grobler, A.C.; Baxter, C.; Mansoor, L.E.; Kharsany, A.B.; Sibeko, S.; Mlisana, K.P.; Omar, Z.; et al. Effectiveness and safety of tenofovir gel, an antiretroviral microbicide, for the prevention of HIV infection in women. Science 2010, 329, 1168–1174. [Google Scholar] [CrossRef] [PubMed]
- Flynn, N.M.; Forthal, D.N.; Harro, C.D.; Judson, F.N.; Mayer, K.H.; Para, M.F.; rgp120 HIV Vaccine Study Group. Placebo-controlled phase 3 trial of a recombinant glycoprotein 120 vaccine to prevent HIV-1 infection. J. Infect. Dis. 2005, 191, 654–665. [Google Scholar] [PubMed]
- Pitisuttithum, P.; Gilbert, P.; Gurwith, M.; Heyward, W.; Martin, M.; van Griensven, F.; Hu, D.; Tappero, J.W.; Choopanya, K.; Bangkok Vaccine Evaluation, G. Randomized, double-blind, placebo-controlled efficacy trial of a bivalent recombinant glycoprotein 120 HIV-1 vaccine among injection drug users in Bangkok, Thailand. J. Infect. Dis. 2006, 194, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Rerks-Ngarm, S.; Brown, A.E.; Khamboonruang, C.; Thongcharoen, P.; Kunasol, P. HIV/AIDS preventive vaccine ‘prime-boost’ phase III trial: Foundations and initial lessons learned from Thailand. AIDS 2006, 20, 1471–1479. [Google Scholar] [CrossRef] [PubMed]
- Barouch, D.H.; Alter, G.; Broge, T.; Linde, C.; Ackerman, M.E.; Brown, E.P.; Borducchi, E.N.; Smith, K.M.; Nkolola, J.P.; Liu, J.; et al. Protective efficacy of adenovirus/protein vaccines against SIV challenges in rhesus monkeys. Science 2015, 349, 320–324. [Google Scholar] [CrossRef] [PubMed]
- Crise, B.; Li, Y.; Yuan, C.; Morcock, D.R.; Whitby, D.; Munroe, D.J.; Arthur, L.O.; Wu, X. Simian immunodeficiency virus integration preference is similar to that of human immunodeficiency virus type 1. J. Virol. 2005, 79, 12199–12204. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Sadjadpour, R.; Mattapallil, J.J.; Igarashi, T.; Lee, W.; Buckler-White, A.; Roederer, M.; Chun, T.W.; Martin, M.A. High frequencies of resting CD4+ T cells containing integrated viral DNA are found in rhesus macaques during acute lentivirus infections. Proc. Natl. Acad. Sci. USA 2009, 106, 8015–8020. [Google Scholar] [CrossRef] [PubMed]
- Barber, S.A.; Gama, L.; Dudaronek, J.M.; Voelker, T.; Tarwater, P.M.; Clements, J.E. Mechanism for the establishment of transcriptional HIV latency in the brain in a simian immunodeficiency virus-macaque model. J. Infect. Dis. 2006, 193, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Darcis, G.; Van Driessche, B.; Bouchat, S.; Kirchhoff, F.; Van Lint, C. Molecular Control of HIV and SIV Latency. Curr. Top. Microbiol. Immunol. 2017. [Google Scholar] [CrossRef]
- Goulder, P.J.; Watkins, D.I. HIV and SIV CTL escape: Implications for vaccine design. Nat. Rev. Immunol. 2004, 4, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Bourry, O.; Mannioui, A.; Sellier, P.; Roucairol, C.; Durand-Gasselin, L.; Dereuddre-Bosquet, N.; Benech, H.; Roques, P.; Le Grand, R. Effect of a short-term HAART on SIV load in macaque tissues is dependent on time of initiation and antiviral diffusion. Retrovirology 2010, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Mannioui, A.; Bourry, O.; Sellier, P.; Delache, B.; Brochard, P.; Andrieu, T.; Vaslin, B.; Karlsson, I.; Roques, P.; Le Grand, R. Dynamics of viral replication in blood and lymphoid tissues during SIVmac251 infection of macaques. Retrovirology 2009, 6, 106. [Google Scholar] [CrossRef] [PubMed]
- Sellier, P.; Mannioui, A.; Bourry, O.; Dereuddre-Bosquet, N.; Delache, B.; Brochard, P.; Calvo, J.; Prevot, S.; Roques, P. Antiretroviral treatment start-time during primary SIV(mac) infection in macaques exerts a different impact on early viral replication and dissemination. PLoS ONE 2010, 5, e10570. [Google Scholar] [CrossRef] [PubMed]
- North, T.W.; Van Rompay, K.K.; Higgins, J.; Matthews, T.B.; Wadford, D.A.; Pedersen, N.C.; Schinazi, R.F. Suppression of virus load by highly active antiretroviral therapy in rhesus macaques infected with a recombinant simian immunodeficiency virus containing reverse transcriptase from human immunodeficiency virus type 1. J. Virol. 2005, 79, 7349–7354. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Y.; Tian, B.; Saifuddin, M.; Agy, M.B.; Emau, P.; Cairns, J.S.; Tsai, C.C. RT-SHIV, an infectious CCR5-tropic chimeric virus suitable for evaluating HIV reverse transcriptase inhibitors in macaque models. AIDS Res. Ther. 2009, 6, 23. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, G.Q.; Ailers, B.; Moldt, B.; Keele, B.F.; Estes, J.D.; Rodriguez, A.; Sampias, M.; Oswald, K.; Fast, R.; Trubey, C.M.; et al. Selection of unadapted, pathogenic SHIVs encoding newly transmitted HIV-1 envelope proteins. Cell Host Microbe 2014, 16, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Sakaguchi, N.; Asano, M.; Itoh, M.; Toda, M. Immunologic self-tolerance maintained by activated T cells expressing IL-2 receptor alpha-chains (CD25). Breakdown of a single mechanism of self-tolerance causes various autoimmune diseases. J. Immunol. 1995, 155, 1151–1164. [Google Scholar] [PubMed]
- Sakaguchi, S.; Wing, K.; Yamaguchi, T. Dynamics of peripheral tolerance and immune regulation mediated by Treg. Eur. J. Immunol. 2009, 39, 2331–2336. [Google Scholar] [CrossRef] [PubMed]
- Hartigan-O’Connor, D.J.; Poon, C.; Sinclair, E.; McCune, J.M. Human CD4+ regulatory T cells express lower levels of the IL-7 receptor alpha chain (CD127), allowing consistent identification and sorting of live cells. J. Immunol. Methods 2007, 319, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Apostolou, I.; von Boehmer, H. In vivo instruction of suppressor commitment in naive T cells. J. Exp. Med. 2004, 199, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Haribhai, D.; Lin, W.; Edwards, B.; Ziegelbauer, J.; Salzman, N.H.; Carlson, M.R.; Li, S.H.; Simpson, P.M.; Chatila, T.A.; Williams, C.B. A central role for induced regulatory T cells in tolerance induction in experimental colitis. J. Immunol. 2009, 182, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Kretschmer, K.; Apostolou, I.; Hawiger, D.; Khazaie, K.; Nussenzweig, M.C.; von Boehmer, H. Inducing and expanding regulatory T cell populations by foreign antigen. Nat. Immunol. 2005, 6, 1219–1227. [Google Scholar] [CrossRef] [PubMed]
- Feuerer, M.; Hill, J.A.; Kretschmer, K.; von Boehmer, H.; Mathis, D.; Benoist, C. Genomic definition of multiple ex vivo regulatory T cell subphenotypes. Proc. Natl. Acad. Sci. USA 2010, 107, 5919–5924. [Google Scholar] [CrossRef] [PubMed]
- Haribhai, D.; Williams, J.B.; Jia, S.; Nickerson, D.; Schmitt, E.G.; Edwards, B.; Ziegelbauer, J.; Yassai, M.; Li, S.H.; Relland, L.M.; et al. A requisite role for induced regulatory T cells in tolerance based on expanding antigen receptor diversity. Immunity 2011, 35, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Fantini, M.C.; Becker, C.; Monteleone, G.; Pallone, F.; Galle, P.R.; Neurath, M.F. Cutting edge: TGF-beta induces a regulatory phenotype in CD4+CD25− T cells through Foxp3 induction and down-regulation of Smad7. J. Immunol. 2004, 172, 5149–5153. [Google Scholar] [CrossRef] [PubMed]
- Sojka, D.K.; Huang, Y.H.; Fowell, D.J. Mechanisms of regulatory T-cell suppression—A diverse arsenal for a moving target. Immunology 2008, 124, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y. Regulatory T cells and infection: A dangerous necessity. Nat. Rev. Immunol. 2007, 7, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Rouse, B.T. Natural regulatory T cells in infectious disease. Nat. Immunol. 2005, 6, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Selliah, N.; Zhang, M.; White, S.; Zoltick, P.; Sawaya, B.E.; Finkel, T.H.; Cron, R.Q. FOXP3 inhibits HIV-1 infection of CD4 T-cells via inhibition of LTR transcriptional activity. Virology 2008, 381, 161–167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Holmes, D.; Jiang, Q.; Zhang, L.; Su, L. Foxp3 and Treg cells in HIV-1 infection and immuno-pathogenesis. Immunol. Res. 2008, 41, 248–266. [Google Scholar] [CrossRef] [PubMed]
- Andersson, J.; Boasso, A.; Nilsson, J.; Zhang, R.; Shire, N.J.; Lindback, S.; Shearer, G.M.; Chougnet, C.A. The prevalence of regulatory T cells in lymphoid tissue is correlated with viral load in HIV-infected patients. J. Immunol. 2005, 174, 3143–3147. [Google Scholar] [CrossRef] [PubMed]
- Kinter, A.L.; Horak, R.; Sion, M.; Riggin, L.; McNally, J.; Lin, Y.; Jackson, R.; O’Shea, A.; Roby, G.; Kovacs, C.; et al. CD25+ regulatory T cells isolated from HIV-infected individuals suppress the cytolytic and nonlytic antiviral activity of HIV-specific CD8+ T cells in vitro. AIDS Res. Hum. Retrovir. 2007, 23, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Bandera, A.; Ferrario, G.; Saresella, M.; Marventano, I.; Soria, A.; Zanini, F.; Sabbatini, F.; Airoldi, M.; Marchetti, G.; Franzetti, F.; et al. CD4+ T cell depletion, immune activation and increased production of regulatory T cells in the thymus of HIV-infected individuals. PLoS ONE 2010, 5, e10788. [Google Scholar] [CrossRef] [PubMed]
- Allers, K.; Loddenkemper, C.; Hofmann, J.; Unbehaun, A.; Kunkel, D.; Moos, V.; Kaup, F.J.; Stahl-Hennig, C.; Sauermann, U.; Epple, H.J.; et al. Gut mucosal FOXP3+ regulatory CD4+ T cells and Nonregulatory CD4+ T cells are differentially affected by simian immunodeficiency virus infection in rhesus macaques. J. Virol. 2010, 84, 3259–3269. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, C.; Ploquin, M.J.; Pandrea, I.; Faye, A.; Onanga, R.; Apetrei, C.; Poaty-Mavoungou, V.; Rouquet, P.; Estaquier, J.; Mortara, L.; et al. Antiinflammatory profiles during primary SIV infection in African green monkeys are associated with protection against AIDS. J. Clin. Investig. 2005, 115, 1082–1091. [Google Scholar] [CrossRef] [PubMed]
- Chase, A.J.; Sedaghat, A.R.; German, J.R.; Gama, L.; Zink, M.C.; Clements, J.E.; Siliciano, R.F. Severe depletion of CD4+ CD25+ regulatory T cells from the intestinal lamina propria but not peripheral blood or lymph nodes during acute simian immunodeficiency virus infection. J. Virol. 2007, 81, 12748–12757. [Google Scholar] [CrossRef] [PubMed]
- Qin, S.; Sui, Y.; Soloff, A.C.; Junecko, B.A.; Kirschner, D.E.; Murphey-Corb, M.A.; Watkins, S.C.; Tarwater, P.M.; Pease, J.E.; Barratt-Boyes, S.M.; et al. Chemokine and cytokine mediated loss of regulatory T cells in lymph nodes during pathogenic simian immunodeficiency virus infection. J. Immunol. 2008, 180, 5530–5536. [Google Scholar] [CrossRef] [PubMed]
- Vahlenkamp, T.W.; Tompkins, M.B.; Tompkins, W.A. Feline immunodeficiency virus infection phenotypically and functionally activates immunosuppressive CD4+CD25+ T regulatory cells. J. Immunol. 2004, 172, 4752–4761. [Google Scholar] [CrossRef] [PubMed]
- Estes, J.D.; Li, Q.; Reynolds, M.R.; Wietgrefe, S.; Duan, L.; Schacker, T.; Picker, L.J.; Watkins, D.I.; Lifson, J.D.; Reilly, C.; et al. Premature induction of an immunosuppressive regulatory T cell response during acute simian immunodeficiency virus infection. J. Infect. Dis. 2006, 193, 703–712. [Google Scholar] [CrossRef] [PubMed]
- Joshi, A.; Vahlenkamp, T.W.; Garg, H.; Tompkins, W.A.; Tompkins, M.B. Preferential replication of FIV in activated CD4+CD25+ T cells independent of cellular proliferation. Virology 2004, 321, 307–322. [Google Scholar] [CrossRef] [PubMed]
- Oswald-Richter, K.; Grill, S.M.; Shariat, N.; Leelawong, M.; Sundrud, M.S.; Haas, D.W.; Unutmaz, D. HIV infection of naturally occurring and genetically reprogrammed human regulatory T-cells. PLoS Biol. 2004, 2, E198. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fernandez, M.E.; Zapata, W.; Blackard, J.T.; Franchini, G.; Chougnet, C.A. Human regulatory T cells are targets for human immunodeficiency Virus (HIV) infection, and their susceptibility differs depending on the HIV type 1 strain. J. Virol. 2009, 83, 12925–12933. [Google Scholar] [CrossRef] [PubMed]
- Abel, K.; Pahar, B.; Van Rompay, K.K.; Fritts, L.; Sin, C.; Schmidt, K.; Colon, R.; McChesney, M.; Marthas, M.L. Rapid virus dissemination in infant macaques after oral simian immunodeficiency virus exposure in the presence of local innate immune responses. J. Virol. 2006, 80, 6357–6367. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mexas, A.M.; Fogle, J.E.; Tompkins, W.A.; Tompkins, M.B. CD4+CD25+ regulatory T cells are infected and activated during acute FIV infection. Vet. Immunol. Immunopathol. 2008, 126, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Zhang, L.; Wang, R.; Jeffrey, J.; Washburn, M.L.; Brouwer, D.; Barbour, S.; Kovalev, G.I.; Unutmaz, D.; Su, L. FoxP3+CD4+ regulatory T cells play an important role in acute HIV-1 infection in humanized Rag2−/−gammaC−/− mice in vivo. Blood 2008, 112, 2858–2868. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, J.; Boasso, A.; Velilla, P.A.; Zhang, R.; Vaccari, M.; Franchini, G.; Shearer, G.M.; Andersson, J.; Chougnet, C. HIV-1-driven regulatory T-cell accumulation in lymphoid tissues is associated with disease progression in HIV/AIDS. Blood 2006, 108, 3808–3817. [Google Scholar] [CrossRef] [PubMed]
- Pion, M.; Jaramillo-Ruiz, D.; Martinez, A.; Munoz-Fernandez, M.A.; Correa-Rocha, R. HIV infection of human regulatory T cells downregulates Foxp3 expression by increasing DNMT3b levels and DNA methylation in the FOXP3 gene. AIDS 2013, 27, 2019–2029. [Google Scholar] [CrossRef] [PubMed]
- Angin, M.; Sharma, S.; King, M.; Murooka, T.T.; Ghebremichael, M.; Mempel, T.R.; Walker, B.D.; Bhasin, M.K.; Addo, M.M. HIV-1 infection impairs regulatory T-cell suppressive capacity on a per-cell basis. J. Infect. Dis. 2014, 210, 899–903. [Google Scholar] [CrossRef] [PubMed]
- Fogle, J.E.; Tompkins, W.A.; Tompkins, M.B. CD4+CD25+ T regulatory cells from FIV+ cats induce a unique anergic profile in CD8+ lymphocyte targets. Retrovirology 2010, 7, 97. [Google Scholar] [CrossRef] [PubMed]
- Thorborn, G.S.; Pomeroy, L.; Ishohanni, H.; Peters, B.S.; Vyakarnam, A. Elevated effector cell sensitivity to Treg-cell suppression that is not associated with reduced Th17-cell expression distinguishes HIV+ asymptomatic subjects from progressors. Eur. J. Immunol. 2012, 42, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, S.R.; Reckling, S.K.; Egan, E.A.; Dean, G.A. In vivo depletion of CD4+CD25hi regulatory T cells is associated with improved antiviral responses in cats chronically infected with feline immunodeficiency virus. Virology 2010, 403, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Fernandez, M.E.; Presicce, P.; Chougnet, C.A. Homeostasis and function of regulatory T cells in HIV/SIV infection. J. Virol. 2012, 86, 10262–10269. [Google Scholar] [CrossRef] [PubMed]
- Elahi, S.; Dinges, W.L.; Lejarcegui, N.; Laing, K.J.; Collier, A.C.; Koelle, D.M.; McElrath, M.J.; Horton, H. Protective HIV-specific CD8+ T cells evade Treg cell suppression. Nat. Med. 2011, 17, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Nikolova, M.; Wiedemann, A.; Muhtarova, M.; Achkova, D.; Lacabaratz, C.; Levy, Y. Subset- and Antigen-Specific Effects of Treg on CD8+ T Cell Responses in Chronic HIV Infection. PLoS Pathog. 2016, 12, e1005995. [Google Scholar] [CrossRef] [PubMed]
- Fogle, J.E.; Mexas, A.M.; Tompkins, W.A.; Tompkins, M.B. CD4+CD25+ T regulatory cells inhibit CD8+ IFN-gamma production during acute and chronic FIV infection utilizing a membrane TGF-beta-dependent mechanism. AIDS Res. Hum. Retrovir. 2010, 26, 201–216. [Google Scholar] [CrossRef] [PubMed]
- Petty, C.S.; Tompkins, M.B.; Tompkins, W.A. Transforming growth factor-beta/transforming growth factor-betaRII signaling may regulate CD4+CD25+ T-regulatory cell homeostasis and suppressor function in feline AIDS lentivirus infection. J. Acquir. Immune Defic. Syndr. 2008, 47, 148–160. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.M.; Akaronu, N.; Thompson, E.M.; Hood, S.F.; Fogle, J.E. Modulating DNA methylation in activated CD8+ T cells inhibits regulatory T cell-induced binding of Foxp3 to the CD8+ T Cell IL-2 promoter. J. Immunol. 2015, 194, 990–998. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nag, M.; Tuohy, J.L.; De Paris, K.; Fogle, J.E. T Regulatory Cell Induced Foxp3 Binds the IL2, IFNγ, and TNFα Promoters in Virus-Specific CD8+ T Cells from Feline Immunodeficiency Virus Infected Cats. AIDS Res. Hum. Retrovir. 2018, 34, 269–276. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L. Histone modifications in transcriptional regulation. Curr. Opin. Genet. Dev. 2002, 12, 142–148. [Google Scholar] [CrossRef]
- Jorgensen, S.; Schotta, G.; Sorensen, C.S. Histone H4 lysine 20 methylation: Key player in epigenetic regulation of genomic integrity. Nucleic Acids Res 2013, 41, 2797–2806. [Google Scholar] [CrossRef] [PubMed]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed]
- Handy, D.E.; Castro, R.; Loscalzo, J. Epigenetic modifications: Basic mechanisms and role in cardiovascular disease. Circulation 2011, 123, 2145–2156. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.J.; Carpenter, P.B. Understanding the language of Lys36 methylation at histone H3. Nat. Rev. Mol. Cell Biol. 2012, 13, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Evertts, A.G.; Manning, A.L.; Wang, X.; Dyson, N.J.; Garcia, B.A.; Coller, H.A. H4K20 methylation regulates quiescence and chromatin compaction. Mol. Biol. Cell 2013, 24, 3025–3037. [Google Scholar] [CrossRef] [PubMed]
- Blomen, V.A.; Boonstra, J. Stable transmission of reversible modifications: Maintenance of epigenetic information through the cell cycle. Cell. Mol. Life Sci. 2011, 68, 27–44. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.H.; Zhang, Y.; Hendrich, B.; Johnson, C.A.; Turner, B.M.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D.; Bird, A. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nat. Genet. 1999, 23, 58–61. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Beard, C.; Jaenisch, R. Role for DNA methylation in genomic imprinting. Nature 1993, 366, 362–365. [Google Scholar] [CrossRef] [PubMed]
- Razin, A.; Cedar, H. DNA methylation and genomic imprinting. Cell 1994, 77, 473–476. [Google Scholar] [CrossRef]
- Siegfried, Z.; Cedar, H. DNA methylation: A molecular lock. Curr. Biol. 1997, 7, R305–R307. [Google Scholar] [CrossRef]
- Bird, A.P.; Wolffe, A.P. Methylation-induced repression—Belts, braces, and chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Robertson, K.D. DNA methylation, methyltransferases, and cancer. Oncogene 2001, 20, 3139–3155. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.; Hillman, C.; Mok, M.; Vapniarsky, N. Lentiviral latency in peripheral CD4+ T cells isolated from feline immunodeficiency virus-infected cats during the asymptomatic phase is not associated with hypermethylation of the proviral promoter. Virus Res. 2012, 169, 117–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Nag, M.; Tuohy, J.L.; Fogle, J.E. Micro-RNA 10a Is Increased in Feline T Regulatory Cells and Increases Foxp3 Protein Expression Following In Vitro Transfection. Vet. Sci. 2017, 4. [Google Scholar] [CrossRef] [PubMed]
- Maulik, U.; Sen, S.; Mallik, S.; Bandyopadhyay, S. Detecting TF-miRNA-gene network based modules for 5hmC and 5mC brain samples: A intra- and inter-species case-study between human and rhesus. BMC Genet. 2018, 19, 9. [Google Scholar] [CrossRef] [PubMed]
- Simon, L.; Song, K.; Vande Stouwe, C.; Hollenbach, A.; Amedee, A.; Mohan, M.; Winsauer, P.; Molina, P. Delta9-Tetrahydrocannabinol (Delta9-THC) Promotes Neuroimmune-Modulatory MicroRNA Profile in Striatum of Simian Immunodeficiency Virus (SIV)-Infected Macaques. J. Neuroimmune Pharmacol. 2016, 11, 192–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Xu, H. Potential Epigenetic Regulation in the Germinal Center Reaction of Lymphoid Tissues in HIV/SIV Infection. Front. Immunol. 2018, 9, 159. [Google Scholar] [CrossRef] [PubMed]
- Ay, E.; Banati, F.; Mezei, M.; Bakos, A.; Niller, H.H.; Buzas, K.; Minarovits, J. Epigenetics of HIV infection: Promising research areas and implications for therapy. AIDS Rev. 2013, 15, 181–188. [Google Scholar] [PubMed]
- Hensel, K.O.; Rendon, J.C.; Navas, M.C.; Rots, M.G.; Postberg, J. Virus-host interplay in hepatitis B virus infection and epigenetic treatment strategies. FEBS J. 2017, 284, 3550–3572. [Google Scholar] [CrossRef] [PubMed]
- Hundt, J.; Li, Z.; Liu, Q. Post-translational modifications of hepatitis C viral proteins and their biological significance. World J. Gastroenterol. 2013, 19, 8929–8939. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Darcis, G.; Van Lint, C.; Herbein, G. Epigenetic control of HIV-1 post integration latency: Implications for therapy. Clin. Epigenet. 2015, 7, 103. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, Y.; Shinjo, K.; Shimizu, Y.; Sano, T.; Yamao, K.; Gao, W.; Fujii, M.; Osada, H.; Sekido, Y.; Murakami, S.; et al. Hepatitis virus infection affects DNA methylation in mice with humanized livers. Gastroenterology 2014, 146, 562–572. [Google Scholar] [CrossRef] [PubMed]
- Clarke, M.A.; Wentzensen, N.; Mirabello, L.; Ghosh, A.; Wacholder, S.; Harari, A.; Lorincz, A.; Schiffman, M.; Burk, R.D. Human papillomavirus DNA methylation as a potential biomarker for cervical cancer. Cancer Epidemiol. Biomark. Prev. 2012, 21, 2125–2137. [Google Scholar] [CrossRef] [PubMed]
- Moos, W.H.; Faller, D.V.; Glavas, I.P.; Harpp, D.N.; Irwin, M.H.; Kanara, I.; Pinkert, C.A.; Powers, W.R.; Steliou, K.; Vavvas, D.G.; et al. Epigenetic Treatment of Neurodegenerative Ophthalmic Disorders: An Eye Toward the Future. Biores. Open Access 2017, 6, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Saunders, K.O.; Freel, S.A.; Overman, R.G.; Cunningham, C.K.; Tomaras, G.D. Epigenetic regulation of CD8+ T-lymphocyte mediated suppression of HIV-1 replication. Virology 2010, 405, 234–242. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shimura, M.; Toyoda, Y.; Iijima, K.; Kinomoto, M.; Tokunaga, K.; Yoda, K.; Yanagida, M.; Sata, T.; Ishizaka, Y. Epigenetic displacement of HP1 from heterochromatin by HIV-1 Vpr causes premature sister chromatid separation. J. Cell Biol. 2011, 194, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.; Reich, N.O. The early expressed HIV-1 genes regulate DNMT1 expression. Epigenetics 2008, 3, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res. Hum. Retrovir. 2009, 25, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Archin, N.M.; Sung, J.M.; Garrido, C.; Soriano-Sarabia, N.; Margolis, D.M. Eradicating HIV-1 infection: Seeking to clear a persistent pathogen. Nat. Rev. Microbiol. 2014, 12, 750–764. [Google Scholar] [CrossRef] [PubMed]
- Matalon, S.; Rasmussen, T.A.; Dinarello, C.A. Histone deacetylase inhibitors for purging HIV-1 from the latent reservoir. Mol. Med. 2011, 17, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, T.A.; Schmeltz Sogaard, O.; Brinkmann, C.; Wightman, F.; Lewin, S.R.; Melchjorsen, J.; Dinarello, C.; Ostergaard, L.; Tolstrup, M. Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Hum. Vaccin. Immunother. 2013, 9, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Kauder, S.E.; Bosque, A.; Lindqvist, A.; Planelles, V.; Verdin, E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009, 5, e1000495. [Google Scholar] [CrossRef] [PubMed]
- Maricato, J.T.; Furtado, M.N.; Takenaka, M.C.; Nunes, E.R.; Fincatti, P.; Meliso, F.M.; da Silva, I.D.; Jasiulionis, M.G.; Cecilia de Araripe Sucupira, M.; Diaz, R.S.; et al. Epigenetic modulations in activated cells early after HIV-1 infection and their possible functional consequences. PLoS ONE 2015, 10, e0119234. [Google Scholar] [CrossRef] [PubMed]
- Mikovits, J.A.; Young, H.A.; Vertino, P.; Issa, J.P.; Pitha, P.M.; Turcoski-Corrales, S.; Taub, D.D.; Petrow, C.L.; Baylin, S.B.; Ruscetti, F.W. Infection with human immunodeficiency virus type 1 upregulates DNA methyltransferase, resulting in de novo methylation of the gamma interferon (IFN-gamma) promoter and subsequent downregulation of IFN-gamma production. Mol. Cell. Biol. 1998, 18, 5166–5177. [Google Scholar] [CrossRef] [PubMed]
- Nakayama-Hosoya, K.; Ishida, T.; Youngblood, B.; Nakamura, H.; Hosoya, N.; Koga, M.; Koibuchi, T.; Iwamoto, A.; Kawana-Tachikawa, A. Epigenetic repression of interleukin 2 expression in senescent CD4+ T cells during chronic HIV type 1 infection. J. Infect. Dis. 2015, 211, 28–39. [Google Scholar] [CrossRef] [PubMed]
- Koirala, J.; Adamski, A.; Koch, L.; Stueber, D.; El-Azizi, M.; Khardori, N.M.; Ghassemi, M.; Novak, R.M. Interferon-gamma receptors in HIV-1 infection. AIDS Res. Hum. Retrovir. 2008, 24, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.; Hale, J.S.; Ahmed, R. T-cell memory differentiation: Insights from transcriptional signatures and epigenetics. Immunology 2013, 139, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Youngblood, B.; Noto, A.; Porichis, F.; Akondy, R.S.; Ndhlovu, Z.M.; Austin, J.W.; Bordi, R.; Procopio, F.A.; Miura, T.; Allen, T.M.; et al. Cutting edge: Prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J. Immunol. 2013, 191, 540–544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Zhou, X.; DiSpirito, J.R.; Wang, C.; Wang, Y.; Shen, H. Epigenetic manipulation restores functions of defective CD8+ T cells from chronic viral infection. Mol. Ther. 2014, 22, 1698–1706. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Hameed, E.A.; Ji, H.; Sherman, K.E.; Shata, M.T. Epigenetic modification of FOXP3 in patients with chronic HIV infection. J. Acquir. Immune Defic. Syndr. 2014, 65, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Kepp, O.; Menger, L.; Vacchelli, E.; Adjemian, S.; Martins, I.; Ma, Y.; Sukkurwala, A.Q.; Michaud, M.; Galluzzi, L.; Zitvogel, L.; et al. Anticancer activity of cardiac glycosides: At the frontier between cell-autonomous and immunological effects. Oncoimmunology 2012, 1, 1640–1642. [Google Scholar] [CrossRef] [PubMed]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.B.; Mueller, S.; O’Connor, R.; Rimpel, K.; Sloan, D.D.; Karel, D.; Wong, H.C.; Jeng, E.K.; Thomas, A.S.; Whitney, J.B.; et al. A Subset of Latency-Reversing Agents Expose HIV-Infected Resting CD4+ T-Cells to Recognition by Cytotoxic T-Lymphocytes. PLoS Pathog. 2016, 12, e1005545. [Google Scholar] [CrossRef] [PubMed]
- Rhode, P.R.; Egan, J.O.; Xu, W.; Hong, H.; Webb, G.M.; Chen, X.; Liu, B.; Zhu, X.; Wen, J.; You, L.; et al. Comparison of the Superagonist Complex, ALT-803, to IL15 as Cancer Immunotherapeutics in Animal Models. Cancer Immunol. Res. 2016, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Zarogoulidis, P.; Lampaki, S.; Yarmus, L.; Kioumis, I.; Pitsiou, G.; Katsikogiannis, N.; Hohenforst-Schmidt, W.; Li, Q.; Huang, H.; Sakkas, A.; et al. Interleukin-7 and interleukin-15 for cancer. J. Cancer 2014, 5, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Trickett, A.E.; Kwan, Y.L.; Cameron, B.; Dwyer, J.M. Ex vivo expansion of functional T lymphocytes from HIV-infected individuals. J. Immunol. Methods 2002, 262, 71–83. [Google Scholar] [CrossRef]
- Butler, M.O.; Imataki, O.; Yamashita, Y.; Tanaka, M.; Ansen, S.; Berezovskaya, A.; Metzler, G.; Milstein, M.I.; Mooney, M.M.; Murray, A.P.; et al. Ex vivo expansion of human CD8+ T cells using autologous CD4+ T cell help. PLoS ONE 2012, 7, e30229. [Google Scholar] [CrossRef] [PubMed]
- Hale, M.; Mesojednik, T.; Romano Ibarra, G.S.; Sahni, J.; Bernard, A.; Sommer, K.; Scharenberg, A.M.; Rawlings, D.J.; Wagner, T.A. Engineering HIV-Resistant, Anti-HIV Chimeric Antigen Receptor T Cells. Mol. Ther. 2017, 25, 570–579. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.; Raghavan, A.; Nandiwada, S.L.; Curtsinger, J.M.; Bohjanen, P.R.; Mueller, D.L.; Mescher, M.F. Gene regulation and chromatin remodeling by IL-12 and type I IFN in programming for CD8 T cell effector function and memory. J. Immunol. 2009, 183, 1695–1704. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Histone/Position/Modification | Location | Effect | Enzyme |
|---|---|---|---|
| H3K4me2 | Gene activation | Set1, MLL, Set7/9, SMYD3, LSD1, JAR1D1A | |
| H3K4me3 | 5′ End of transcriptionally active genes | Gene activation | Set1, MLL, Set7/9, SMYD3, JAR1D1A |
| H3K9me | Euchromatin | Gene silencing | G9a; Suv91, StB1, PRD14, CLL8, GLP, Suv39h1, Suv39h2 |
| H3K9me2 | Euchromatin | Gene silencing | G9a; Suv91, StB1, PRD14, CLL8, GLP, Suv39h1, Suv39h2, JMJD2A |
| H3K9me3 | Promoters and heterochromatin, Gene coding region | Gene silencing Gene activation | G9a; Suv91, StB1, PRD14, CLL8, GLP, Suv39h1, Suv39h2, JMJD2A |
| H3K27me1 | Heterochromatin | Gene activation | |
| H3K27me2/3 | Inactive-X chromosome, homeotic genes | Gene silencing | EZH2 |
| H3K36me | Promoter | Not well characterized | JHDM1A |
| H3K36me2 | Near double strand breaks, for repair | Gene silencing | NSD1, JMJD2A, JHDM1A |
| H3K36me3 | 3′ End of active genes. Marks exons. | Gene activation | JMJD2A |
| H3K79me2 | Gene activation | Dot1L | |
| H3K79me3 | Gene activation | Dot1L | |
| H4K20me1 | Cell cycle regulation, present at active promoters | Gene activation | SET8/PR-Set7 |
| H4K20me2 | Heterochromatin, marks origin for replication, DNA damage response | Gene silencing | NSD1, Suv4-20h1, Suv4-20H2, Set8/PR-SET7 |
| H4K20me3 | Heterochromatin, at promoters | Gene silencing | NSD1, Suv4-20h1, Suv4-20H2, Set8/PR-SET7 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nag, M.; De Paris, K.; E. Fogle, J. Epigenetic Modulation of CD8+ T Cell Function in Lentivirus Infections: A Review. Viruses 2018, 10, 227. https://doi.org/10.3390/v10050227
Nag M, De Paris K, E. Fogle J. Epigenetic Modulation of CD8+ T Cell Function in Lentivirus Infections: A Review. Viruses. 2018; 10(5):227. https://doi.org/10.3390/v10050227
Chicago/Turabian StyleNag, Mukta, Kristina De Paris, and Jonathan E. Fogle. 2018. "Epigenetic Modulation of CD8+ T Cell Function in Lentivirus Infections: A Review" Viruses 10, no. 5: 227. https://doi.org/10.3390/v10050227
APA StyleNag, M., De Paris, K., & E. Fogle, J. (2018). Epigenetic Modulation of CD8+ T Cell Function in Lentivirus Infections: A Review. Viruses, 10(5), 227. https://doi.org/10.3390/v10050227