Genome Sequences of Akhmeta Virus, an Early Divergent Old World Orthopoxvirus

 ,
, {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods and Materials
2.1. Virus Genome Sequences
2.2. NGS Sequencing and De Novo Assembly
2.3. Gap Closing
2.4. Phylogenetic Analysis
2.5. Genome Annotation
2.6. Tandem Repeat Analysis in the Inverse Terminal Repetitions (ITR) of AKMV Genome
2.7. Genome and Protein Comparison
2.8. Determination and Comparison of Host Range Factors
2.9. Investigation of a Recombination Event in the AKMV_VANI10 Genome
2.10. Analysis of dN/dS
3. Results
3.1. AKMV Genome Assembly and ITR
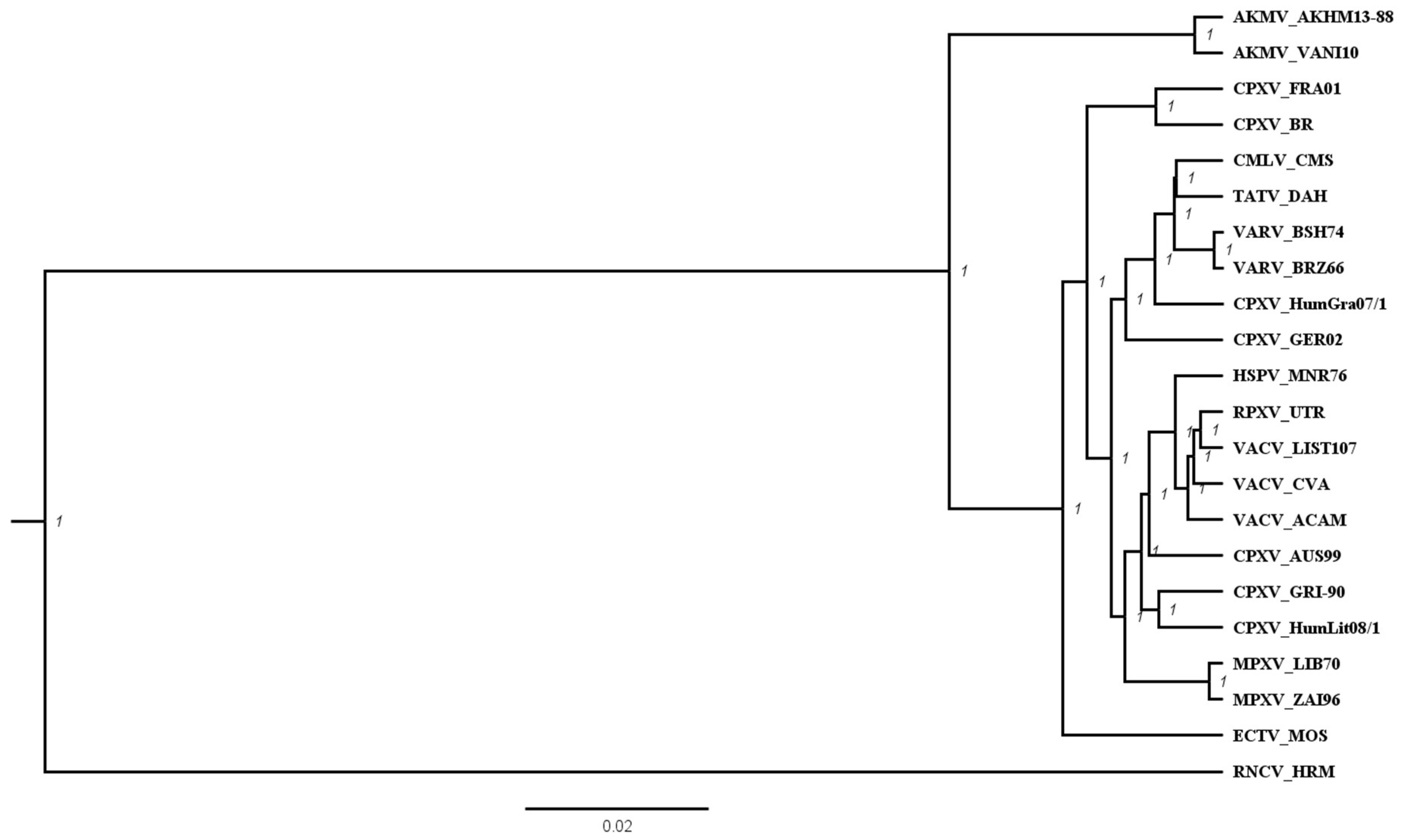
3.2. Phylogenetic Analysis of AKMV with Other OPXVs
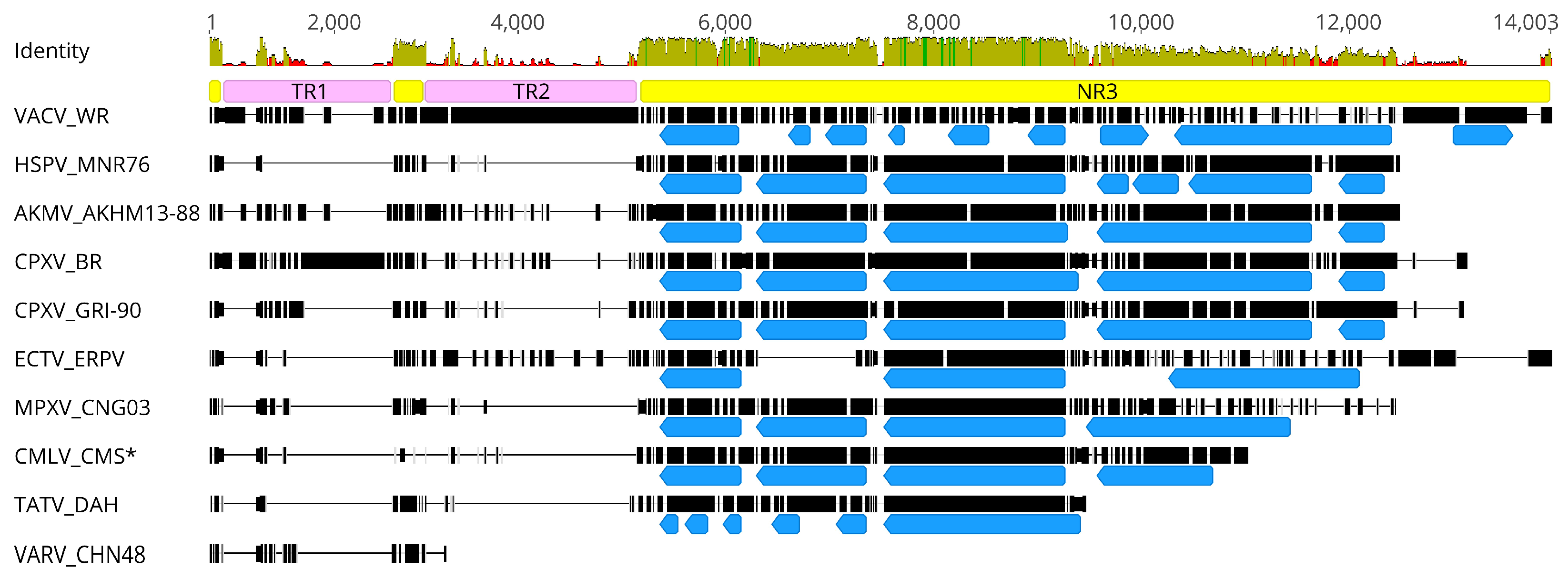
3.3. Genome Characteristics of AKMV
3.4. A Closer Look at Genes Differing between AKMV and CPXV_BR
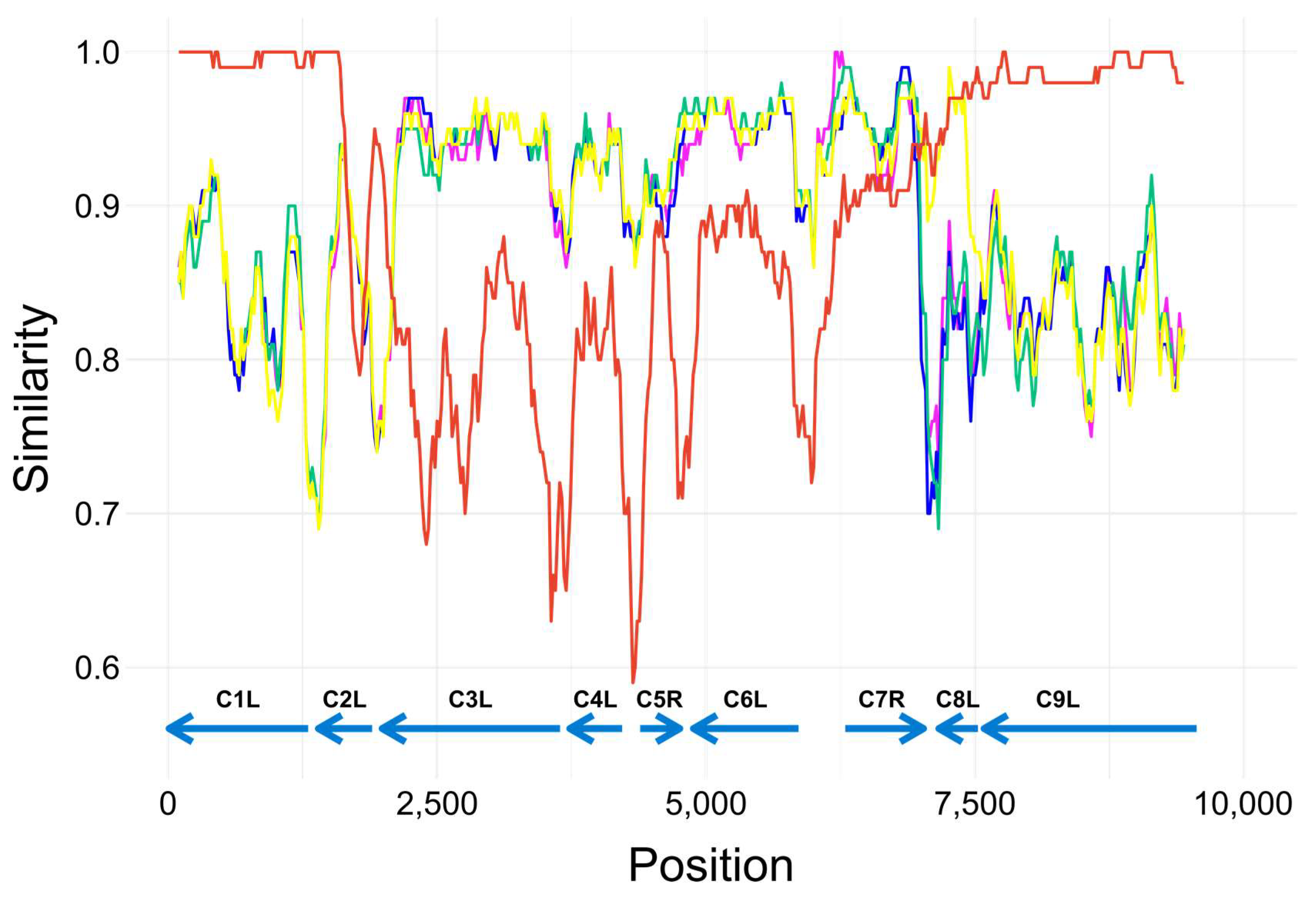
3.5. Investigation of a Recombination Event in AKMV_VANI10 Genome
4. Discussion
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Damon, I.K. Poxviruses. Fileds Virol. 2007, 2, 2943–2975. [Google Scholar]
- Mahalingam, S.; Damon, I.K.; Lidbury, B.A. 25 years since the eradication of smallpox: Why poxvirus research is still relevant. Trends Immunol. 2004, 25, 636–639. [Google Scholar] [CrossRef] [PubMed]
- Dabrowski, P.W.; Radonic, A.; Kurth, A.; Nitsche, A. Genome-wide comparison of cowpox viruses reveals a new clade related to Variola virus. PLoS ONE 2013, 8, e79953. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.G.; Damon, I.K. Outbreaks of human monkeypox after cessation of smallpox vaccination. Trends Immunol. 2012, 20, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Prevention, Multistate outbreak of monkeypox—Illinois, Indiana, and Wisconsin, 2003. MMWR Morb. Mortal. Wkly. Rep. 2003, 52, 537–540. [Google Scholar]
- Silva, D.C.; Moreira-Silva, E.A.; Gomes Jde, A.; Fonseca, F.G.; Correa-Oliveira, R. Clinical signs, diagnosis, and case reports of Vaccinia virus infections. Braz. J. Infect. Dis. 2010, 14, 129–134. [Google Scholar] [CrossRef] [PubMed]
- Usme-Ciro, J.A.; Paredes, A.; Walteros, D.M.; Tolosa-Perez, E.N.; Laiton-Donato, K.; Pinzon, M.D.; Petersen, B.W.; Gallardo-Romero, N.F.; Li, Y.; Wilkins, K.; et al. Detection and Molecular Characterization of Zoonotic Poxviruses Circulating in the Amazon Region of Colombia, 2014. Emerg. Infect. Dis. 2017, 23, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Kroon, E.G.; Mota, B.E.; Abrahao, J.S.; da Fonseca, F.G.; de Souza Trindade, G. Zoonotic Brazilian Vaccinia virus: From field to therapy. Antivir. Res. 2011, 92, 150–163. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Hosamani, M.; Balamurugan, V.; Bhanuprakash, V.; Rasool, T.J.; Yadav, M.P. Buffalopox: An emerging and re-emerging zoonosis. Anim. Health Res. Rev. Conf. Res. Work. Anim. Dis. 2007, 8, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Duraffour, S.; Meyer, H.; Andrei, G.; Snoeck, R. Camelpox virus. Antivir. Res. 2011, 92, 167–186. [Google Scholar] [CrossRef] [PubMed]
- Khalafalla, A.I.; Abdelazim, F. Human and Dromedary Camel Infection with Camelpox Virus in Eastern Sudan. Vector-Borne Zoonotic Dis. 2017, 17, 281–284. [Google Scholar] [CrossRef] [PubMed]
- Damle, A.S.; Gaikwad, A.A.; Patwardhan, N.S.; Duthade, M.M.; Sheikh, N.S.; Deshmukh, D.G. Outbreak of human buffalopox infection. J. Glob. Infect. Dis. 2011, 3, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Ducournau, C.; Ferrier-Rembert, A.; Ferraris, O.; Joffre, A.; Favier, A.L.; Flusin, O.; van Cauteren, D.; Kecir, K.; Auburtin, B.; Vedy, S.; et al. Concomitant human infections with 2 cowpox virus strains in related cases, France, 2011. Emerg. Infect. Dis. 2013, 19, 1996. [Google Scholar] [CrossRef] [PubMed]
- McCollum, A.M.; Damon, I.K. Human monkeypox. Clin. Infect. Dis. 2014, 58, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.G.; Emerson, G.L.; Pukuta, E.; Karhemere, S.; Muyembe, J.J.; Bikindou, A.; McCollum, A.M.; Moses, C.; Wilkins, K.; Zhao, H.; et al. Detection of human monkeypox in the Republic of the Congo following intensive community education. Am. J. Trop. Med. Hyg. 2013, 88, 982–985. [Google Scholar] [CrossRef] [PubMed]
- Trindade, G.S.; Emerson, G.L.; Carroll, D.S.; Kroon, E.G.; Damon, I.K. Brazilian Vaccinia viruses and their origins. Emerg. Infect. Dis. 2007, 13, 965–972. [Google Scholar] [CrossRef] [PubMed]
- Bera, B.; Shanmugasundaram, K.; Barua, S.; Anand, T.; Riyesh, T.; Vaid, R.K.; Virmani, N.; Bansal, M.; Shukla, B.N.; Malik, P.; et al. Sequence and phylogenetic analysis of host-range (E3L, K3L, and C7L) and structural protein (B5R) genes of buffalopox virus isolates from buffalo, cattle, and human in India. Virus Genes 2012, 45, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Baxby, D.; Hill, B.J. Buffalopox virus. Vet. Rec. 1969, 85, 315–316. [Google Scholar] [CrossRef] [PubMed]
- Bera, B.C.; Shanmugasundaram, K.; Barua, S.; Venkatesan, G.; Virmani, N.; Riyesh, T.; Gulati, B.R.; Bhanuprakash, V.; Vaid, R.K.; Kakker, N.K.; et al. Zoonotic cases of camelpox infection in India. Vet. Microbiol. 2011, 152, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Zafar, A.; Swanepoel, R.; Hewson, R.; Nizam, M.; Ahmed, A.; Husain, A.; Grobbelaar, A.; Bewley, K.; Mioulet, V.; Dowsett, B.; et al. Nosocomial buffalopoxvirus infection, Karachi, Pakistan. Emerg. Infect. Dis. 2007, 13, 902–904. [Google Scholar] [CrossRef] [PubMed]
- Thomassen, H.A.; Fuller, T.; Asefi-Najafabady, S.; Shiplacoff, J.A.; Mulembakani, P.M.; Blumberg, S.; Johnston, S.C.; Kisalu, N.K.; Kinkela, T.L.; Fair, J.N.; et al. Pathogen-host associations and predicted range shifts of human monkeypox in response to climate change in central Africa. PLoS ONE 2013, 8, e66071. [Google Scholar] [CrossRef] [PubMed]
- McCollum, A.M.; Nakazawa, Y.; Ndongala, G.M.; Pukuta, E.; Karhemere, S.; Lushima, R.S.; Ilunga, B.K.; Kabamba, J.; Wilkins, K.; Gao, J.; et al. Human Monkeypox in the Kivus, a Conflict Region of the Democratic Republic of the Congo. Am. J. Trop. Med. Hyg. 2015, 93, 718–721. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, M.G.; Carroll, D.S.; Karem, K.L. Factors affecting the likelihood of monkeypox’s emergence and spread in the post-smallpox era. Curr. Opin. Virol. 2012, 2, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Di Giulio, D.B.; Eckburg, P.B. Human monkeypox: An emerging zoonosis. Lancet Infect. Dis. 2004, 4, 15–25. [Google Scholar] [CrossRef]
- Vora, N.M.; Li, Y.; Geleishvili, M.; Emerson, G.L.; Khmaladze, E.; Maghlakelidze, G.; Navdarashvili, A.; Zakhashvili, K.; Kokhreidze, M.; Endeladze, M.; et al. Human infection with a zoonotic orthopoxvirus in the country of Georgia. N. Engl. J. Med. 2015, 372, 1223–1230. [Google Scholar] [CrossRef] [PubMed]
- Kinnunen, P.M.; Henttonen, H.; Hoffmann, B.; Kallio, E.R.; Korthase, C.; Laakkonen, J.; Niemimaa, J.; Palva, A.; Schlegel, M.; Ali, H.S.; et al. Orthopox virus infections in Eurasian wild rodents. Vector-Borne Zoonotic Dis. 2011, 11, 1133–1140. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ropp, S.L.; Zhao, H.; Damon, I.K.; Esposito, J.J. Orthopoxvirus pan-genomic DNA assay. J. Virol. Methods 2007, 141, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Olson, V.A.; Laue, T.; Laker, M.T.; Damon, I.K. Detection of monkeypox virus with real-time PCR assays. J. Clin. Virol. 2006, 36, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Meyer, H.; Zhao, H.; Damon, I.K. GC content-based pan-pox universal PCR assays for poxvirus detection. J. Clin. Microbiol. 2010, 48, 268–276. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kuhnert, D.; Vaughan, T.; Wu, C.H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Gubser, C.; Hue, S.; Kellam, P.; Smith, G.L. Poxvirus genomes: A phylogenetic analysis. J. Gen. Virol. 2004, 85 Pt 1, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Haller, S.L.; Peng, C.; McFadden, G.; Rothenburg, S. Poxviruses and the evolution of host range and virulence. Infect. Genet. Evol. 2014, 21, 15–40. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [PubMed]
- Mauldin, M.R.; Antwerpen, M.; Emerson, G.L.; Li, Y.; Zoeller, G.; Carroll, D.S.; Meyer, H. Cowpox virus: What’s in a Name? Viruses 2017, 9, 101. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Wong, W.S.; Nielsen, R. Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 2005, 22, 1107–1118. [Google Scholar] [CrossRef] [PubMed]
- Baroudy, B.M.; Moss, B. Sequence homologies of diverse length tandem repetitions near ends of vaccinia virus genome suggest unequal crossing over. Nucleic Acids Res. 1982, 10, 5673–5679. [Google Scholar] [CrossRef] [PubMed]
- Merchlinsky, M.; Moss, B. Nucleotide sequence required for resolution of the concatemer junction of vaccinia virus DNA. J. Virol. 1989, 63, 4354–4361. [Google Scholar] [PubMed]
- Senkevich, T.G.; Koonin, E.V.; Bugert, J.J.; Darai, G.; Moss, B. The genome of molluscum contagiosum virus: Analysis and comparison with other poxviruses. Virology 1997, 233, 19–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.; Danila, M.I.; Feng, Z.; Buller, R.M.; Wang, C.; Han, X.; Lefkowitz, E.J.; Upton, C. The genomic sequence of ectromelia virus, the causative agent of mousepox. Virology 2003, 317, 165–186. [Google Scholar] [CrossRef]
- Goebel, S.J.; Johnson, G.P.; Perkus, M.E.; Davis, S.W.; Winslow, J.P.; Paoletti, E. The complete DNA sequence of vaccinia virus. Virology 1990, 179, 247–266. [Google Scholar] [CrossRef]
- Wittek, R.; Moss, B. Tandem repeats within the inverted terminal repetition of vaccinia virus DNA. Cell 1980, 21, 277–284. [Google Scholar] [CrossRef]
- Tulman, E.R.; Delhon, G.; Afonso, C.L.; Lu, Z.; Zsak, L.; Sandybaev, N.T.; Kerembekova, U.Z.; Zaitsev, V.L.; Kutish, G.F.; Rock, D.L. Genome of horsepox virus. J. Virol. 2006, 80, 9244–9258. [Google Scholar] [CrossRef] [PubMed]
- Shchelkunov, S.N.; Totmenin, A.V.; Sandakhchiev, L.S. Analysis of the nucleotide sequence of 23.8 kbp from the left terminus of the genome of variola major virus strain India-1967. Virus Res. 1996, 40, 169–183. [Google Scholar] [CrossRef]
- Kochneva, G.; Kolosova, I.; Maksyutova, T.; Ryabchikova, E.; Shchelkunov, S. Effects of deletions of kelch-like genes on cowpox virus biological properties. Arch. Virol. 2005, 150, 1857–1870. [Google Scholar] [CrossRef] [PubMed]
- Alejo, A.; Ruiz-Arguello, M.B.; Ho, Y.; Smith, V.P.; Saraiva, M.; Alcami, A. A chemokine-binding domain in the tumor necrosis factor receptor from variola (smallpox) virus. Proc. Natl. Acad. Sci. USA 2006, 103, 5995–6000. [Google Scholar] [CrossRef] [PubMed]
- Loparev, V.N.; Parsons, J.M.; Knight, J.C.; Panus, J.F.; Ray, C.A.; Buller, R.M.; Pickup, D.J.; Esposito, J.J. A third distinct tumor necrosis factor receptor of orthopoxviruses. Proc. Natl. Acad. Sci. USA 1998, 95, 3786–3791. [Google Scholar] [CrossRef] [PubMed]
- Caillat, C.; Topalis, D.; Agrofoglio, L.A.; Pochet, S.; Balzarini, J.; Deville-Bonne, D.; Meyer, P. Crystal structure of poxvirus thymidylate kinase: An unexpected dimerization has implications for antiviral therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 16900–16905. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.J.; Johnston, L.H.; de Carlos, A.; Smith, G.L. Vaccinia virus encodes an active thymidylate kinase that complements a cdc8 mutant of Saccharomyces cerevisiae. J. Biol. Chem. 1991, 266, 20103–20109. [Google Scholar] [PubMed]
- Guimaraes, A.P.; de Souza, F.R.; Oliveira, A.A.; Goncalves, A.S.; de Alencastro, R.B.; Ramalho, T.C.; Franca, T.C. Design of inhibitors of thymidylate kinase from Variola virus as new selective drugs against smallpox. Eur. J. Med. Chem. 2015, 91, 72–90. [Google Scholar] [CrossRef] [PubMed]
- Luteijn, R.D.; Hoelen, H.; Kruse, E.; van Leeuwen, W.F.; Grootens, J.; Horst, D.; Koorengevel, M.; Drijfhout, J.W.; Kremmer, E.; Fruh, K.; et al. Cowpox virus protein CPXV012 eludes CTLs by blocking ATP binding to TAP. J. Immunol. 2014, 193, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Voigt, S.; Mesci, A.; Ettinger, J.; Fine, J.H.; Chen, P.; Chou, W.; Carlyle, J.R. Cytomegalovirus evasion of innate immunity by subversion of the NKR-P1B:Clr-b missing-self axis. Immunity 2007, 26, 617–627. [Google Scholar] [CrossRef] [PubMed]
- McCoy, W.H.T.; Wang, X.; Yokoyama, W.M.; Hansen, T.H.; Fremont, D.H. Structural mechanism of ER retrieval of MHC class I. by cowpox. PLoS Biol. 2012, 10, e1001432. [Google Scholar] [CrossRef] [PubMed]
- McCoy, W.H.T.; Wang, X.; Yokoyama, W.M.; Hansen, T.H.; Fremont, D.H. Cowpox virus employs a two-pronged strategy to outflank MHCI antigen presentation. Mol. Immunol. 2013, 55, 156–158. [Google Scholar] [CrossRef] [PubMed]
- Bratke, K.A.; McLysaght, A.; Rothenburg, S. A survey of host range genes in poxvirus genomes. Infect. Genet. Evol. 2013, 14, 406–425. [Google Scholar] [CrossRef] [PubMed]
- Likos, A.M.; Sammons, S.A.; Olson, V.A.; Frace, A.M.; Li, Y.; Olsen-Rasmussen, M.; Davidson, W.; Galloway, R.; Khristova, M.L.; Reynolds, M.G.; et al. A tale of two clades: Monkeypox viruses. J. Gen. Virol. 2005, 86 Pt 10, 2661–2672. [Google Scholar] [CrossRef] [PubMed]
- Earl, P.L.; Moss, B.; Wyatt, L.S.; Carroll, M.W. Generation of Recombinant Vaccinia Viruses. Curr. Protoc. Mol. Biol. 1998, 43, 16–17. [Google Scholar]
- Paszkowski, P.; Noyce, R.S.; Evans, D.H. Live-Cell Imaging of Vaccinia Virus Recombination. PLoS Pathog. 2016, 12, e1005824. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gao, J.; Gigante, C.; Khmaladze, E.; Liu, P.; Tang, S.; Wilkins, K.; Zhao, K.; Davidson, W.; Nakazawa, Y.; Maghlakelidze, G.; et al. Genome Sequences of Akhmeta Virus, an Early Divergent Old World Orthopoxvirus. Viruses 2018, 10, 252. https://doi.org/10.3390/v10050252
Gao J, Gigante C, Khmaladze E, Liu P, Tang S, Wilkins K, Zhao K, Davidson W, Nakazawa Y, Maghlakelidze G, et al. Genome Sequences of Akhmeta Virus, an Early Divergent Old World Orthopoxvirus. Viruses. 2018; 10(5):252. https://doi.org/10.3390/v10050252
Chicago/Turabian StyleGao, Jinxin, Crystal Gigante, Ekaterine Khmaladze, Pengbo Liu, Shiyuyun Tang, Kimberly Wilkins, Kun Zhao, Whitni Davidson, Yoshinori Nakazawa, Giorgi Maghlakelidze, and et al. 2018. "Genome Sequences of Akhmeta Virus, an Early Divergent Old World Orthopoxvirus" Viruses 10, no. 5: 252. https://doi.org/10.3390/v10050252
APA StyleGao, J., Gigante, C., Khmaladze, E., Liu, P., Tang, S., Wilkins, K., Zhao, K., Davidson, W., Nakazawa, Y., Maghlakelidze, G., Geleishvili, M., Kokhreidze, M., Carroll, D. S., Emerson, G., & Li, Y. (2018). Genome Sequences of Akhmeta Virus, an Early Divergent Old World Orthopoxvirus. Viruses, 10(5), 252. https://doi.org/10.3390/v10050252