Inhibition of Human Immunodeficiency Virus Type 1 Entry by a Keggin Polyoxometalate

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
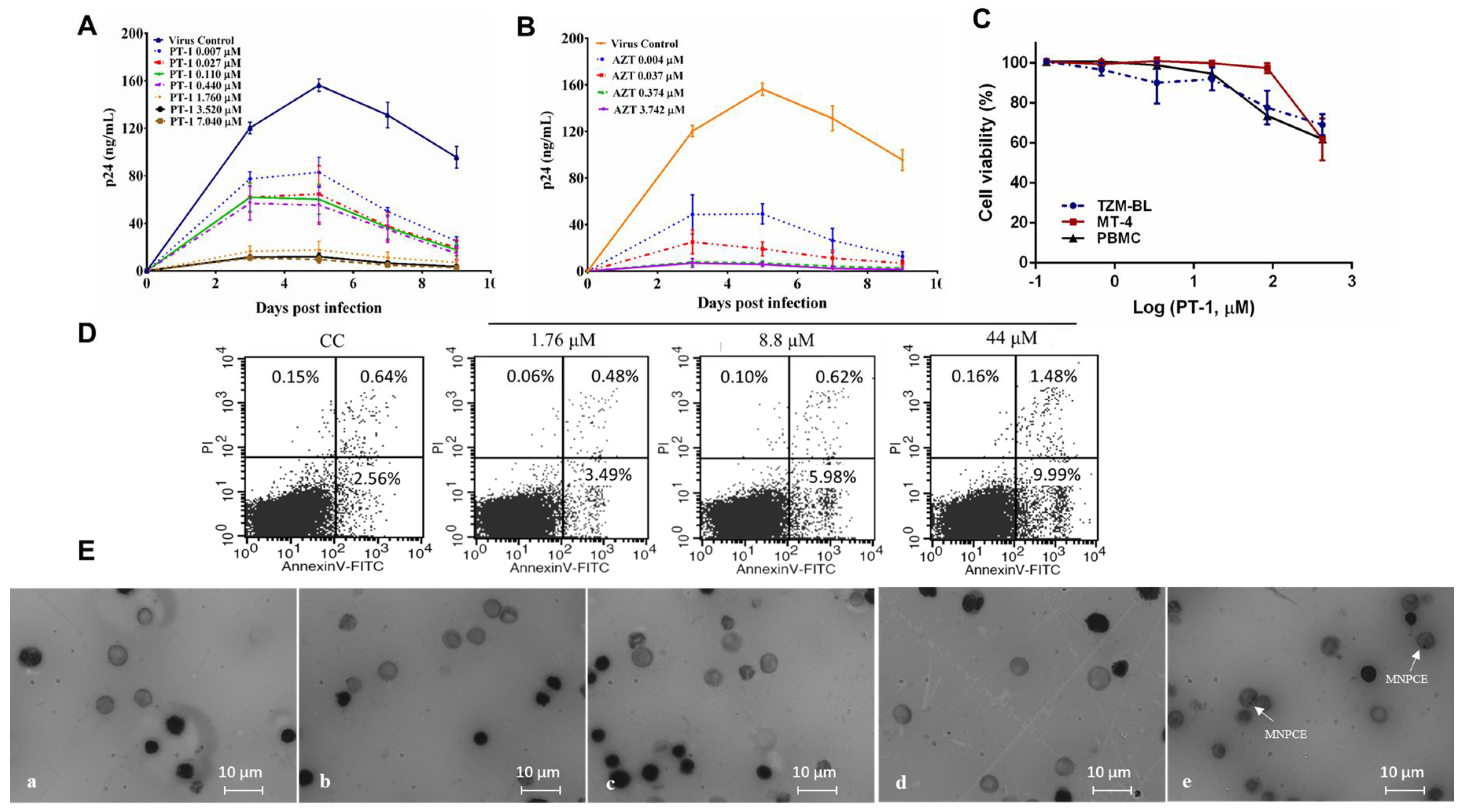
2.1. Cells, Viruses and Compounds
2.2. Cell Viability
2.3. In Vivo Mouse Micronucleus Test
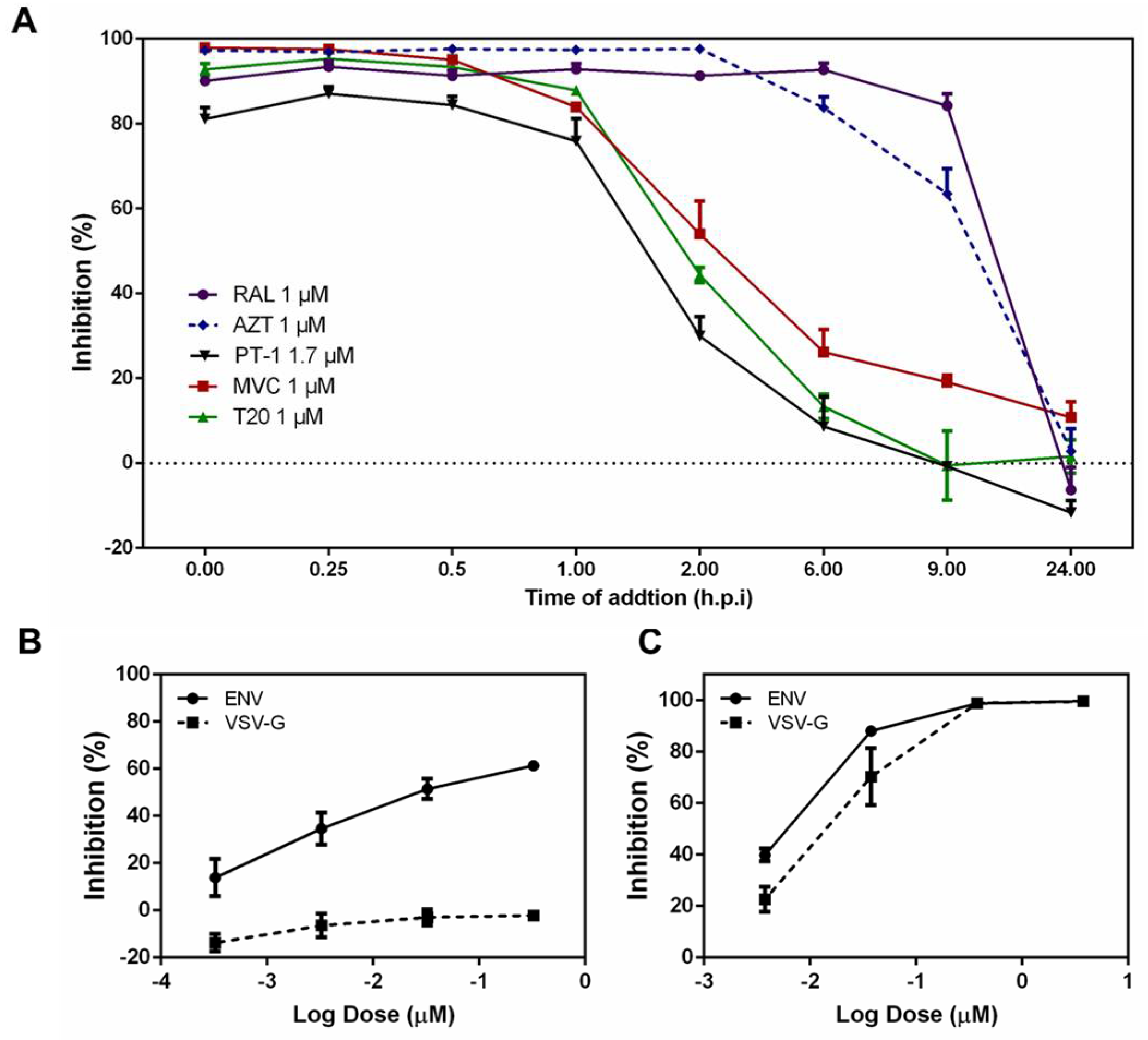
2.4. Single-Round Replication Assay
2.5. Wide-Type HIV-1 Replication Assay
2.6. Inhibition of HIV-1 Replication in Peripheral Blood Mononuclear Cells
2.7. Time-of-Addition Assay
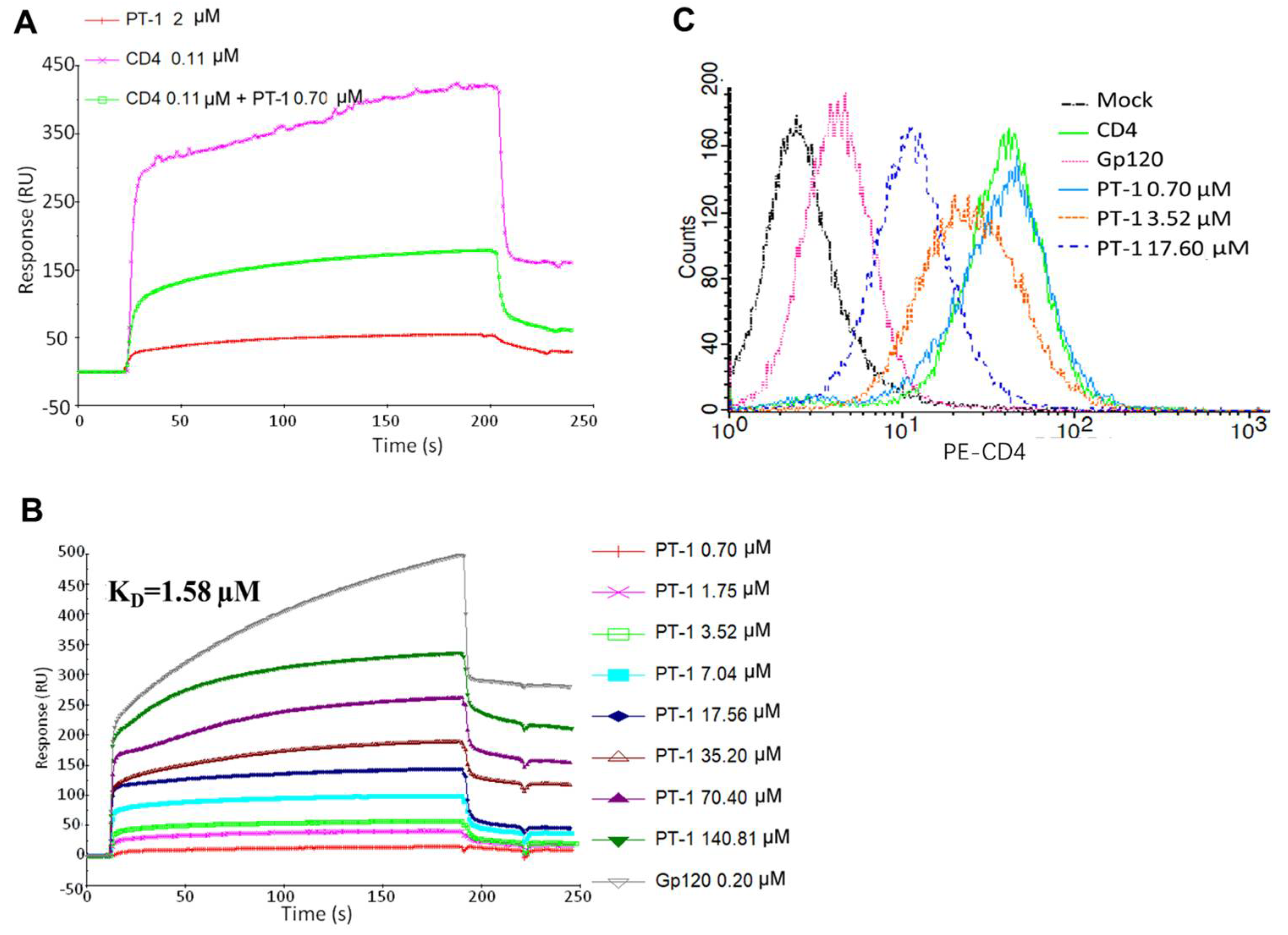
2.8. Flow Cytometry Assay
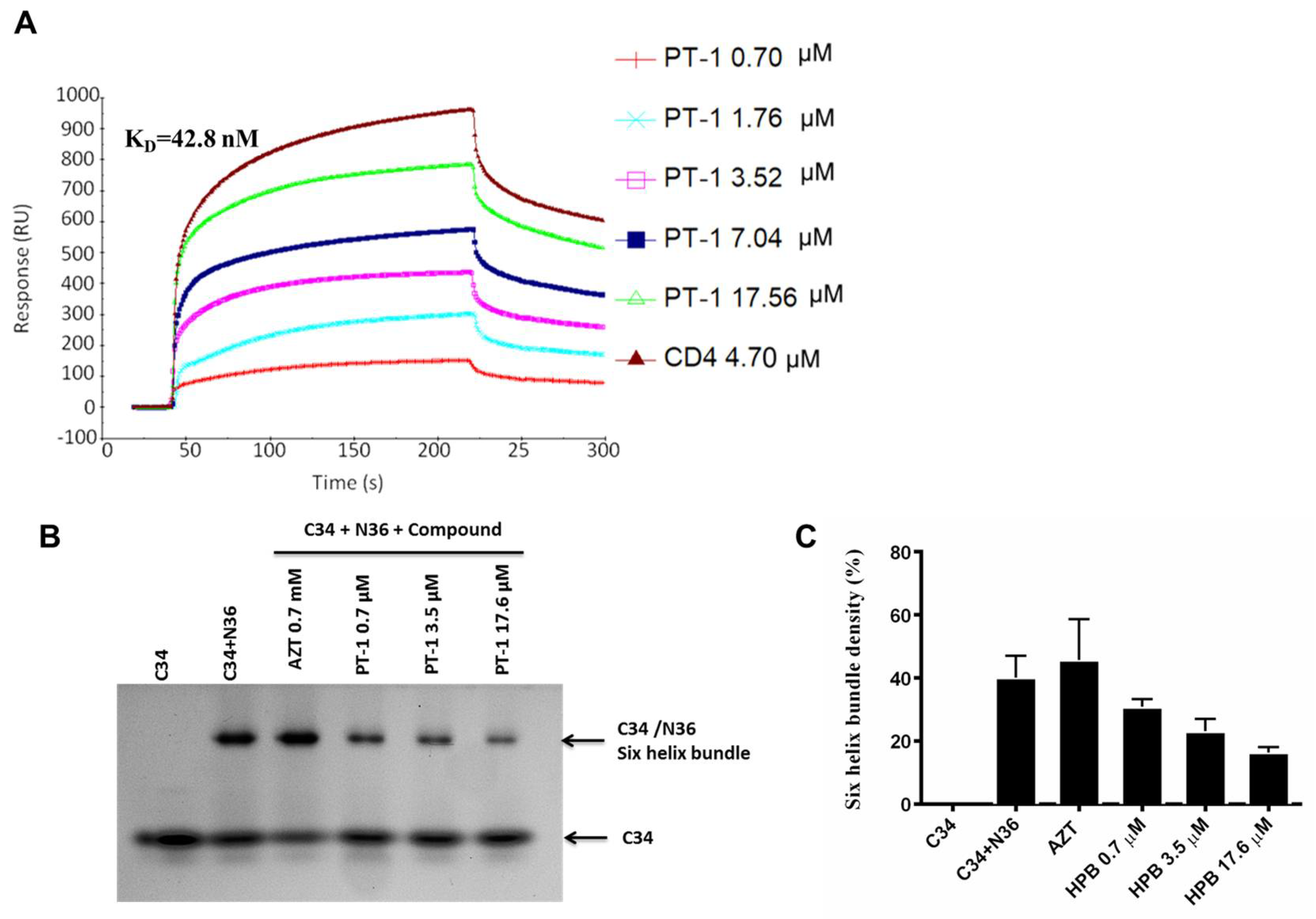
2.9. Native Polyacrylamide Gel Electrophoresis
2.10. Surface Plasmon Resonance Binding Assay
2.11. Reverse Transcriptase Assay
2.12. Protease Assay
2.13. HIV-1 Integrase 3′ Processing Assay
2.14. Statistical Analysis
3. Results
3.1. PT-1 Exhibited Potent Antiviral Activity and Low Toxicity
3.2. PT-1 Inhibited HIV-1 Replication at Early Stage
3.3. PT-1 Inhibited gp41 Core Formation and gp120-CD4 Interaction
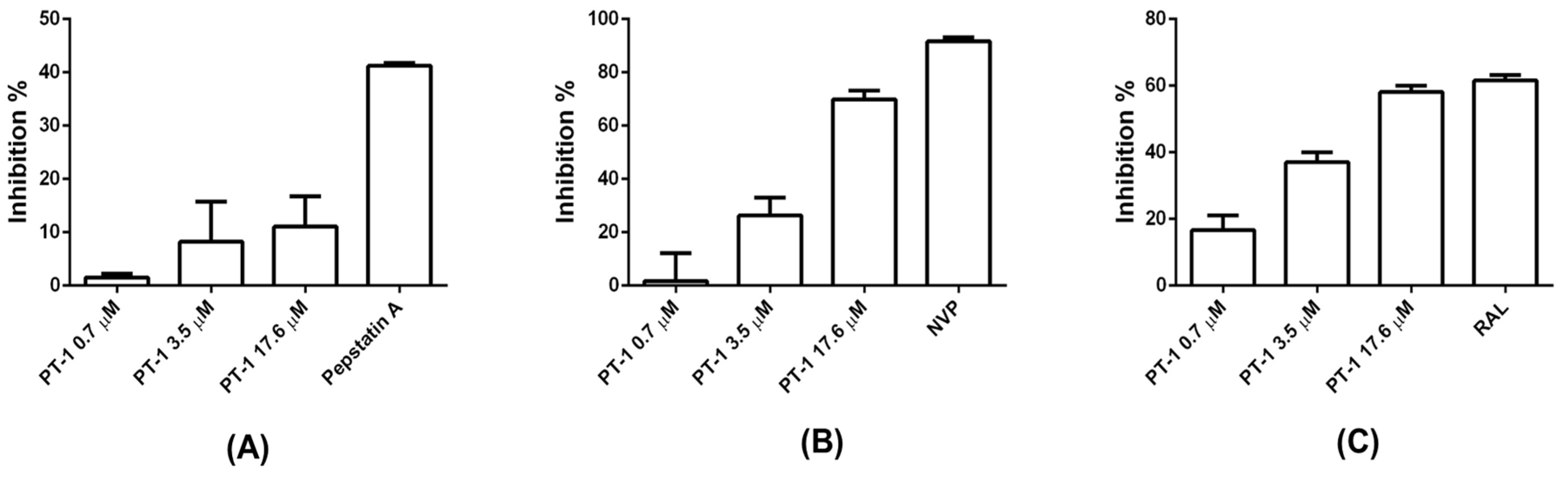
3.4. PT-1 Showed Modest Inhibitory Activity on Viral Reverse Transcriptase and Integrase
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bello, K.J.; Mesner, O.; O’Bryan, T.A.; Won, S.H.; Lalani, T.; Ganesan, A.; Agan, B.K.; Okulicz, J.F. Factors associated with 10 years of continuous viral load suppression on HAART. BMC Infect. Dis. 2016, 16, 351. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lu, L.; Li, W.; Jiang, S. Small-molecule HIV-1 entry inhibitors targeting gp120 and gp41: A patent review (2010–2015). Expert Opin. Ther. Pat. 2017, 27, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Zarr, M.; Siliciano, R. Stoichiometric parameters of HIV-1 entry. Virology 2015, 474, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Brandenberg, O.F.; Magnus, C.; Regoes, R.R.; Trkola, A. The HIV-1 entry process: A stoichiometric view. Trends Microbiol. 2015, 23, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Melikyan, G.B. HIV entry: A game of hide-and-fuse? Curr. Opin. Virol. 2014, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gomara, M.J.; Haro, I. Updating the use of synthetic peptides as inhibitors of HIV-1 entry. Curr. Med. Chem. 2014, 21, 1188–1200. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Latinovic, O.S.; Barbault, F.; de Leeuw, E.P. A novel small-molecule inhibitor of HIV-1 entry. Drug Des. Dev. Ther. 2015, 9, 5469–5478. [Google Scholar] [CrossRef]
- Wu, C.H.; Wang, C.J.; Chang, C.P.; Cheng, Y.C.; Song, J.S.; Jan, J.J.; Chou, M.C.; Ke, Y.Y.; Ma, J.; Wong, Y.C.; et al. Function-oriented development of CXCR4 antagonists as selective human immunodeficiency virus (HIV)-1 entry inhibitors. J. Med. Chem. 2015, 58, 1452–1465. [Google Scholar] [CrossRef] [PubMed]
- Haqqani, A.A.; Tilton, J.C. Entry inhibitors and their use in the treatment of HIV-1 infection. Antivir. Res. 2013, 98, 158–170. [Google Scholar] [CrossRef] [PubMed]
- Henrich, T.J.; Kuritzkes, D.R. HIV-1 entry inhibitors: Recent development and clinical use. Curr. Opin. Virol. 2013, 3, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Xiang, Y.; Wang, J.; Qi, Y.; Li, J.; Niu, J.; Zhong, J. Inhibition of hepatitis C virus infection by polyoxometalates. Antivir. Res. 2013, 100, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Shao, C.; Guan, W.; Su, Z.; Sun, J. Studies on the interactions of Ti-containing polyoxometalates (POMs) with SARS-CoV 3CLpro by molecular modeling. J. Inorg. Biochem. 2007, 101, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.N.; Shi, S.; Mei, W.J.; Tan, C.P.; Chen, L.M.; Liu, J.; Zheng, W.J.; Ji, L.N. In vitro and in vivo investigations on the antiviral activity of a series of mixed-valence rare earth borotungstate heteropoly blues. Eur. J. Med. Chem. 2008, 43, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Shigeta, S.; Mori, S.; Yamase, T.; Yamamoto, N.; Yamamoto, N. Anti-RNA virus activity of polyoxometalates. Biomed. Pharmacother. 2006, 60, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Dan, K.; Yamase, T. Prevention of the interaction between HVEM, herpes virus entry mediator, and gD, HSV envelope protein, by a Keggin polyoxotungstate, PM-19. Biomed. Pharmacother. 2006, 60, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Aymard, J.P.; Ferry, R.; Janot, C.; Schooneman, F.; Legras, B.; May, T.; Streiff, F. Toxicity of HPA-23 (ammonium-21-tungsto-9-antimoniate) for normal human myeloid progenitor cells (GM-CFU) in vitro. Biomed. Pharmacother. 1989, 43, 451–454. [Google Scholar] [CrossRef]
- Burgard, M.; Sansonetti, P.; Vittecoq, D.; Descamps, P.; Guetard, D.; Herson, S.; Rozenbaum, W.; Rouzioux, C. Lack of HPA-23 antiviral activity in HIV-infected patients without AIDS. AIDS 1989, 3, 665–668. [Google Scholar] [CrossRef] [PubMed]
- Hill, C.L.; Weeks, M.S.; Schinazi, R.F. Anti-HIV-1 activity, toxicity, and stability studies of representative structural families of polyoxometalates. J. Med. Chem. 1990, 33, 2767–2772. [Google Scholar] [CrossRef] [PubMed]
- Flutsch, A.; Schroeder, T.; Grutter, M.G.; Patzke, G.R. HIV-1 protease inhibition potential of functionalized polyoxometalates. Bioorg. Med. Chem. Lett. 2011, 21, 1162–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuguchi, T.; Harada, S.; Miura, T.; Ohashi, N.; Narumi, T.; Mori, H.; Irahara, Y.; Yamada, Y.; Nomura, W.; Matsushita, S.; et al. A minimally cytotoxic CD4 mimic as an HIV entry inhibitor. Bioorg. Med. Chem. Lett. 2016, 26, 397–400. [Google Scholar] [CrossRef] [PubMed]
- Witvrouw, M.; Weigold, H.; Pannecouque, C.; Schols, D.; De Clercq, E.; Holan, G. Potent anti-HIV (type 1 and type 2) activity of polyoxometalates: Structure-activity relationship and mechanism of action. J. Med. Chem. 2000, 43, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Inouye, Y.; Fujimoto, Y.; Sugiyama, M.; Yoshida, T.; Yamase, T. Structure-activity correlationship and strain specificity of polyoxometalates in anti-human immunodeficiency virus activity. Biol. Pharm. Bull. 1995, 18, 996–1000. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, N.; Schols, D.; De Clercq, E.; Debyser, Z.; Pauwels, R.; Balzarini, J.; Nakashima, H.; Baba, M.; Hosoya, M.; Snoeck, R.; et al. Mechanism of anti-human immunodeficiency virus action of polyoxometalates, a class of broad-spectrum antiviral agents. Mol. Pharmacol. 1992, 42, 1109–1117. [Google Scholar] [PubMed]
- Inouye, Y.; Tokutake, Y.; Kunihara, J.; Yoshida, T.; Yamase, T.; Nakata, A.; Nakamura, S. Suppressive effect of polyoxometalates on the cytopathogenicity of human immunodeficiency virus type 1 (HIV-1) in vitro and their inhibitory activity against HIV-1 reverse transcriptase. Chem. Pharm. Bull. 1992, 40, 805–807. [Google Scholar] [CrossRef] [PubMed]
- Take, Y.; Tokutake, Y.; Inouye, Y.; Yoshida, T.; Yamamoto, A.; Yamase, T.; Nakamura, S. Inhibition of proliferation of human immunodeficiency virus type 1 by novel heteropolyoxotungstates in vitro. Antivir. Res. 1991, 15, 113–124. [Google Scholar] [CrossRef]
- Inouye, Y.; Tokutake, Y.; Yoshida, T.; Yamamoto, A.; Yamase, T.; Nakamura, S. Antiviral activity of polyoxomolybdoeuropate PM-104 against human immunodeficiency virus type 1. Chem. Pharm. Bull. 1991, 39, 1638–1640. [Google Scholar] [CrossRef] [PubMed]
- Inouye, Y.; Take, Y.; Tokutake, Y.; Yoshida, T.; Yamamoto, A.; Yamase, T.; Nakamura, S. Inhibition of replication of human immunodeficiency virus by a heteropolyoxotungstate (PM-19). Chem. Pharm. Bull. 1990, 38, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Sun, W.; Hu, Q.; Yan, H.; Zeng, Y. Synthesis and evaluation of pyridinium polyoxometalates as anti-HIV-1 agents. Bioorg. Med. Chem. Lett. 2017, 27, 2357–2359. [Google Scholar] [CrossRef] [PubMed]
- Domaille, P.J.; Knoth, W.H. Ti2W10PO407- and [CpFe(CO)2Sn]2W10PO385-. Preparation, properties, and structure determination by tungsten-183 NMR. Inorg. Chem. 1983, 22, 818–822. [Google Scholar] [CrossRef]
- Krishna, G.; Hayashi, M. In vivo rodent micronucleus assay: Protocol, conduct and data interpretation. Mutat. Res. 2000, 455, 155–166. [Google Scholar] [CrossRef]
- Kimpton, J.; Emerman, M. Detection of replication-competent and pseudotyped human immunodeficiency virus with a sensitive cell line on the basis of activation of an integrated beta-galactosidase gene. J. Virol. 1992, 66, 2232–2239. [Google Scholar] [PubMed]
- Baleux, F.; Loureiro-Morais, L.; Hersant, Y.; Clayette, P.; Arenzana-Seisdedos, F.; Bonnaffe, D.; Lortat-Jacob, H. A synthetic CD4-heparan sulfate glycoconjugate inhibits CCR55 and CXCR4 HIV-1 attachment and entry. Nat. Chem. Biol. 2009, 5, 743–748. [Google Scholar] [CrossRef] [PubMed]
- He, H.Q.; Ma, X.H.; Liu, B.; Zhang, X.Y.; Chen, W.Z.; Wang, C.X.; Cheng, S.H. High-throughput real-time assay based on molecular beacons for HIV-1 integrase 3′-processing reaction. Acta Pharmacol. Sin. 2007, 28, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Daelemans, D.; Pauwels, R.; De Clercq, E.; Pannecouque, C. A time-of-drug addition approach to target identification of antiviral compounds. Nat. Protoc. 2011, 6, 925–933. [Google Scholar] [CrossRef] [PubMed]
- Leydet, A.; Moullet, C.; Roque, J.P.; Witvrouw, M.; Pannecouque, C.; Andrei, G.; Snoeck, R.; Neyts, J.; Schols, D.; De Clercq, E. Polyanion inhibitors of HIV and other viruses. 7. Polyanionic compounds and polyzwitterionic compounds derived from cyclodextrins as inhibitors of HIV transmission. J. Med. Chem. 1998, 41, 4927–4932. [Google Scholar] [CrossRef] [PubMed]
- Moskovitz, B.L. Clinical trial of tolerance of HPA-23 in patients with acquired immune deficiency syndrome. Antimicrob. Agents Chem. 1988, 32, 1300–1303. [Google Scholar] [CrossRef]
- Fu, L.; Gao, H.; Yan, M.; Li, S.; Li, X.; Dai, Z.; Liu, S. Polyoxometalate-based organic-inorganic hybrids as antitumor drugs. Small 2015, 11, 2938–2945. [Google Scholar] [CrossRef] [PubMed]
- Boulmier, A.; Feng, X.; Oms, O.; Mialane, P.; Riviere, E.; Shin, C.J.; Yao, J.; Kubo, T.; Furuta, T.; Oldfield, E.; et al. Anticancer activity of polyoxometalate-bisphosphonate complexes: Synthesis, characterization, in vitro and in vivo results. Inorg. Chem. 2017, 56, 7558–7565. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, Y.; Xu, K.; Qi, Y.; Zhong, J.; Zhang, K.; Li, J.; Wang, E.; Wu, Z.; Kang, Z. Broad-spectrum antiviral property of polyoxometalate localized on a cell surface. ACS Appl. Mater. Interfaces 2014, 6, 9785–9789. [Google Scholar] [CrossRef] [PubMed]
- Schoeberl, C.; Boehner, R.; Krebs, B.; Mueller, C.; Barnekow, A. A new polyoxometalate complex inhibits retrovirus encoded reverse transcriptase activity in vitro and in vivo. Int. J. Oncol. 1998, 12, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Inouye, Y.; Kanamori, T.; Fujimoto, Y.; Sugiyama, M.; Yoshida, T. Novel assay system favorable for the study of cell-to-cell transmission of HIV-1 and its application to the evaluation of anti-HIV drugs. Biol. Pharm. Bull. 1995, 18, 920–922. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, W.; Ying, T.; Wang, Y.; Feng, Y.; Dimitrov, D.S. Germlining of the HIV-1 broadly neutralizing antibody domain m36. Antivir. Res. 2015, 116, 62–66. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Feng, Y.; Gong, R.; Zhu, Z.; Wang, Y.; Zhao, Q.; Dimitrov, D.S. Engineered single human CD4 domains as potent HIV-1 inhibitors and components of vaccine immunogens. J. Virol. 2011, 85, 9395–9405. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Li, W.; Jiang, S. Peptide fusion inhibitors targeting the HIV-1 gp41: A patent review (2009–2014). Expert Opin. Ther. Pat. 2015, 25, 159–173. [Google Scholar] [CrossRef] [PubMed]
- Heredia, A.; Le, N.; Gartenhaus, R.B.; Sausville, E.; Medina-Moreno, S.; Zapata, J.C.; Davis, C.; Gallo, R.C.; Redfield, R.R. Targeting of mTOR catalytic site inhibits multiple steps of the HIV-1 lifecycle and suppresses HIV-1 viremia in humanized mice. Proc. Natl. Acad. Sci. USA 2015, 112, 9412–9417. [Google Scholar] [CrossRef] [PubMed]
- Readinger, J.A.; Schiralli, G.M.; Jiang, J.K.; Thomas, C.J.; August, A.; Henderson, A.J.; Schwartzberg, P.L. Selective targeting of ITK blocks multiple steps of HIV replication. Proc. Natl. Acad. Sci. USA 2008, 105, 6684–6689. [Google Scholar] [CrossRef] [PubMed]
- Andrae-Marobela, K.; Ghislain, F.W.; Okatch, H.; Majinda, R.R. Polyphenols: A diverse class of multi-target anti-HIV-1 agents. Curr. Drug Metab. 2013, 14, 392–413. [Google Scholar] [CrossRef] [PubMed]
- Rabi, S.A.; Laird, G.M.; Durand, C.M.; Laskey, S.; Shan, L.; Bailey, J.R.; Chioma, S.; Moore, R.D.; Siliciano, R.F. Multi-step inhibition explains HIV-1 protease inhibitor pharmacodynamics and resistance. J. Clin. Investig. 2013, 123, 3848–3860. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HIV-1 | Subtype | Cell Line | AZT | PT-1 | ||
|---|---|---|---|---|---|---|
| EC50 (nM) | EC50 (nM) | CC50 (µM) | TI | |||
| TRO.11 | B | TZM-bl | 60.92 ± 36.93 | 5.26 ± 0.26 | 995.20 ± 106.07 | 1.89 × 105 |
| SC422661.8 | B | TZM-bl | 98.34 ± 11.68 | 16.69 ± 6.85 | 5.95 × 104 | |
| REJO4541.67 | B | TZM-bl | 18.41 ± 11.23 | 1.96 ± 0.44 | 4.98 × 105 | |
| Du422.1 | C | TZM-bl | 6.96 ± 2.73 | 484.56 ± 43.47 | 2.05 × 103 | |
| CE1176 | C | TZM-bl | 28.23 ± 1.62 | 187.52 ± 22.46 | 5.31 × 103 | |
| Q842env.d16 | A | TZM-bl | 11.45 ± 7.45 | ND | ND | |
| Q259env.w6 | A | TZM-bl | 24.77 ± 18.56 | ND | ND | |
| CNE8 | AE | TZM-bl | 30.73 ± 7.25 | ND | ND | |
| QA790.204I.ENV.A4 | AD | TZM-bl | 84.80 ± 42.73 | 62.48 ± 18.48 | 1.59 × 104 | |
| QA790.204I.ENV.C1 | AD | TZM-bl | 2.43 ± 1.53 | 0.60 ± 0.25 | 1.65 × 106 | |
| BJOX00200 | CRF07_BC | TZM-bl | 3.94 ± 0.91 | ND | ND | |
| CH119 | CRF07_BC | TZM-bl | 3.63 ± 1.01 | 237.16 ± 7.46 | 4.19 × 103 | |
| HXB2 | B | MT-4 | 128.11 ± 23.05 | 1248.32 ± 434.48 | 164.52 | |
| NL4-3 | B | MT-4 | 101.00 ± 10.10 | 468.20 ± 112.37 | 205.37 ± 63.54 | 438.63 |
| BH10 | B | MT-4 | 68.03 ± 8.91 | 1566.51 ± 325.74 | 131.10 | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Wang, J.; Zhang, W.; Li, B.; Zhu, Y.; Hu, Q.; Yang, Y.; Zhang, X.; Yan, H.; Zeng, Y. Inhibition of Human Immunodeficiency Virus Type 1 Entry by a Keggin Polyoxometalate. Viruses 2018, 10, 265. https://doi.org/10.3390/v10050265
Wang X, Wang J, Zhang W, Li B, Zhu Y, Hu Q, Yang Y, Zhang X, Yan H, Zeng Y. Inhibition of Human Immunodeficiency Virus Type 1 Entry by a Keggin Polyoxometalate. Viruses. 2018; 10(5):265. https://doi.org/10.3390/v10050265
Chicago/Turabian StyleWang, Xiaoli, Jiao Wang, Wenmei Zhang, Boye Li, Ying Zhu, Qin Hu, Yishu Yang, Xiaoguang Zhang, Hong Yan, and Yi Zeng. 2018. "Inhibition of Human Immunodeficiency Virus Type 1 Entry by a Keggin Polyoxometalate" Viruses 10, no. 5: 265. https://doi.org/10.3390/v10050265
APA StyleWang, X., Wang, J., Zhang, W., Li, B., Zhu, Y., Hu, Q., Yang, Y., Zhang, X., Yan, H., & Zeng, Y. (2018). Inhibition of Human Immunodeficiency Virus Type 1 Entry by a Keggin Polyoxometalate. Viruses, 10(5), 265. https://doi.org/10.3390/v10050265