Swine Influenza Virus Induces RIPK1/DRP1-Mediated Interleukin-1 Beta Production



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Antibodies and Reagents
2.3. Plasmid Construction
2.4. Infection and Treatment of PAMs
2.5. RNA Interference and Real-Time PCR
2.6. NLRP3 Inflammasome Reconstitution Assay
2.7. Co-Immunoprecipitation (Co-IP)
2.8. Porcine IL-1β ELISA
2.9. Western Blotting
2.10. Confocal Microscopy
2.11. Statistical Analysis
3. Results
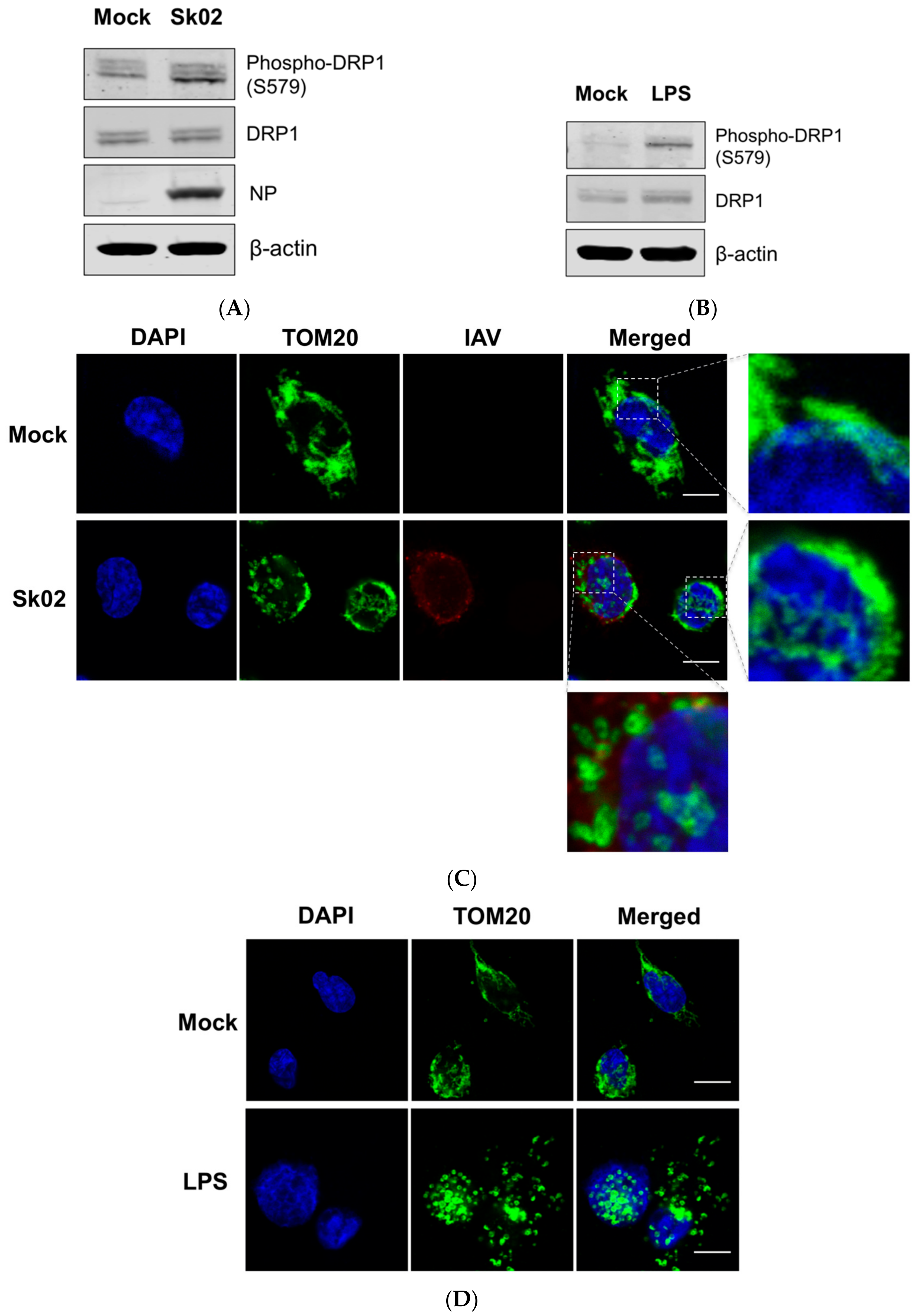
3.1. SIV Infection Induces DRP1 Phosphorylation and Mitochondrial Fission
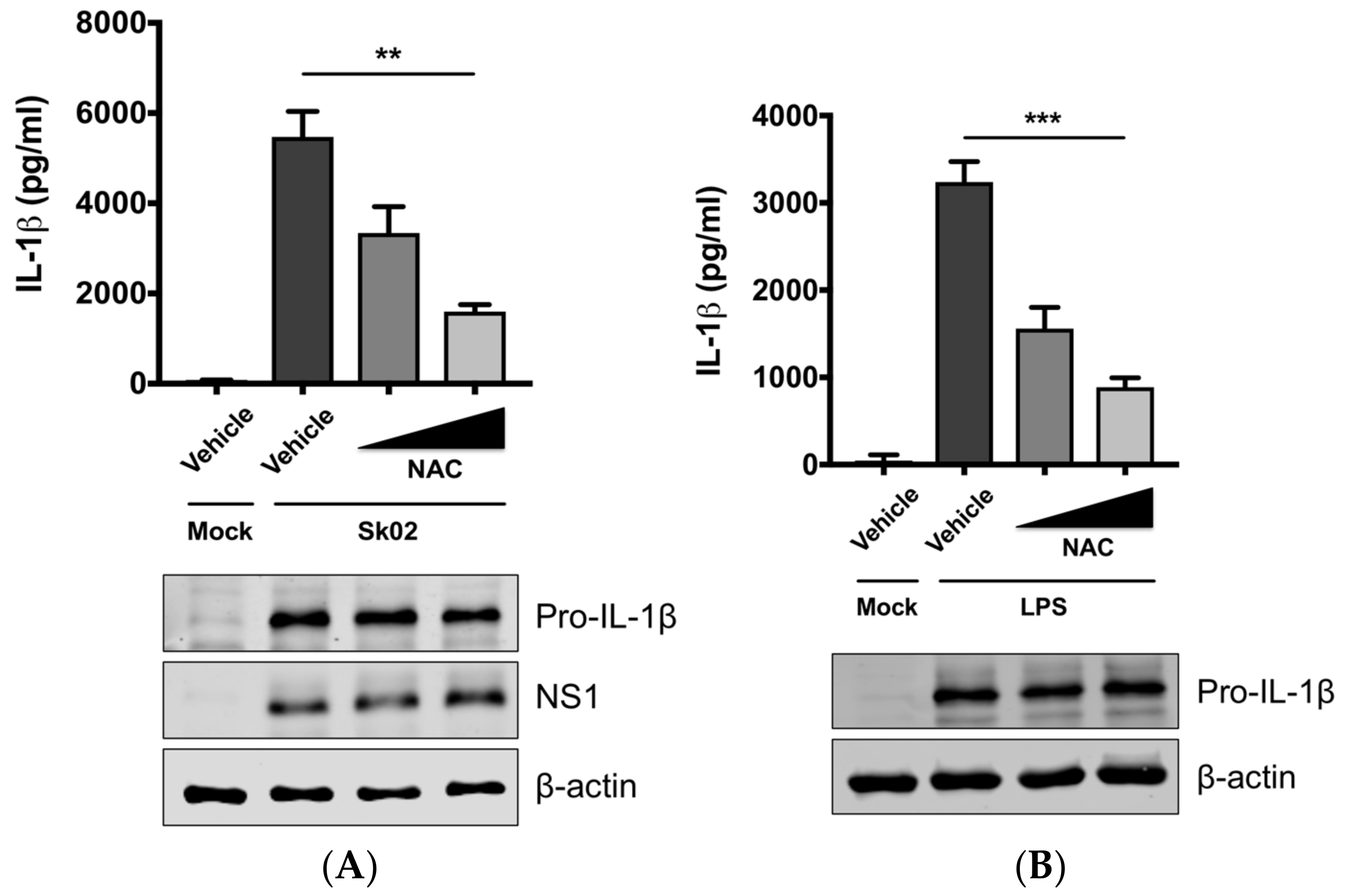
3.2. SIV-Induced Porcine IL-1β Production Is Dependent on ROS Production
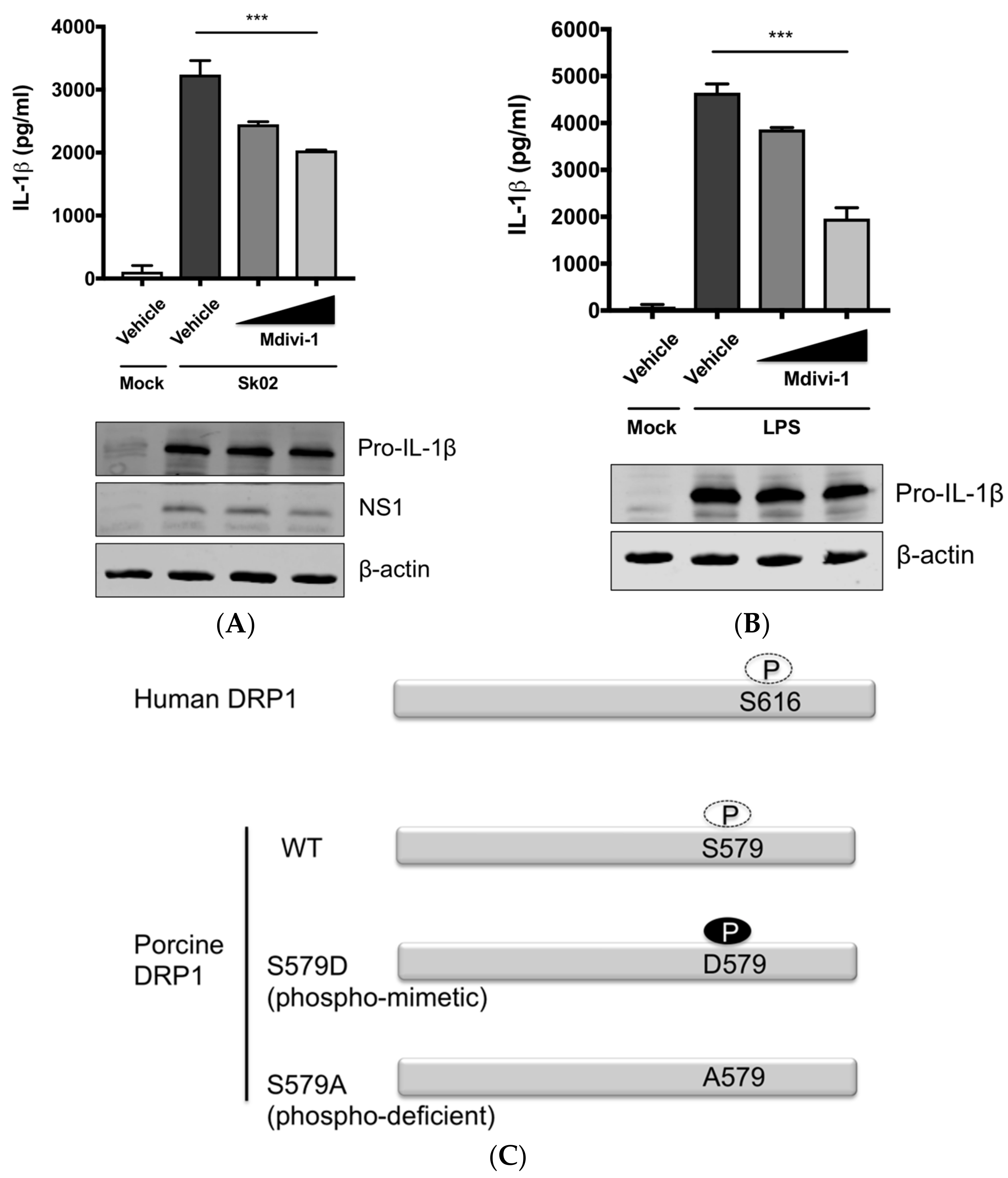
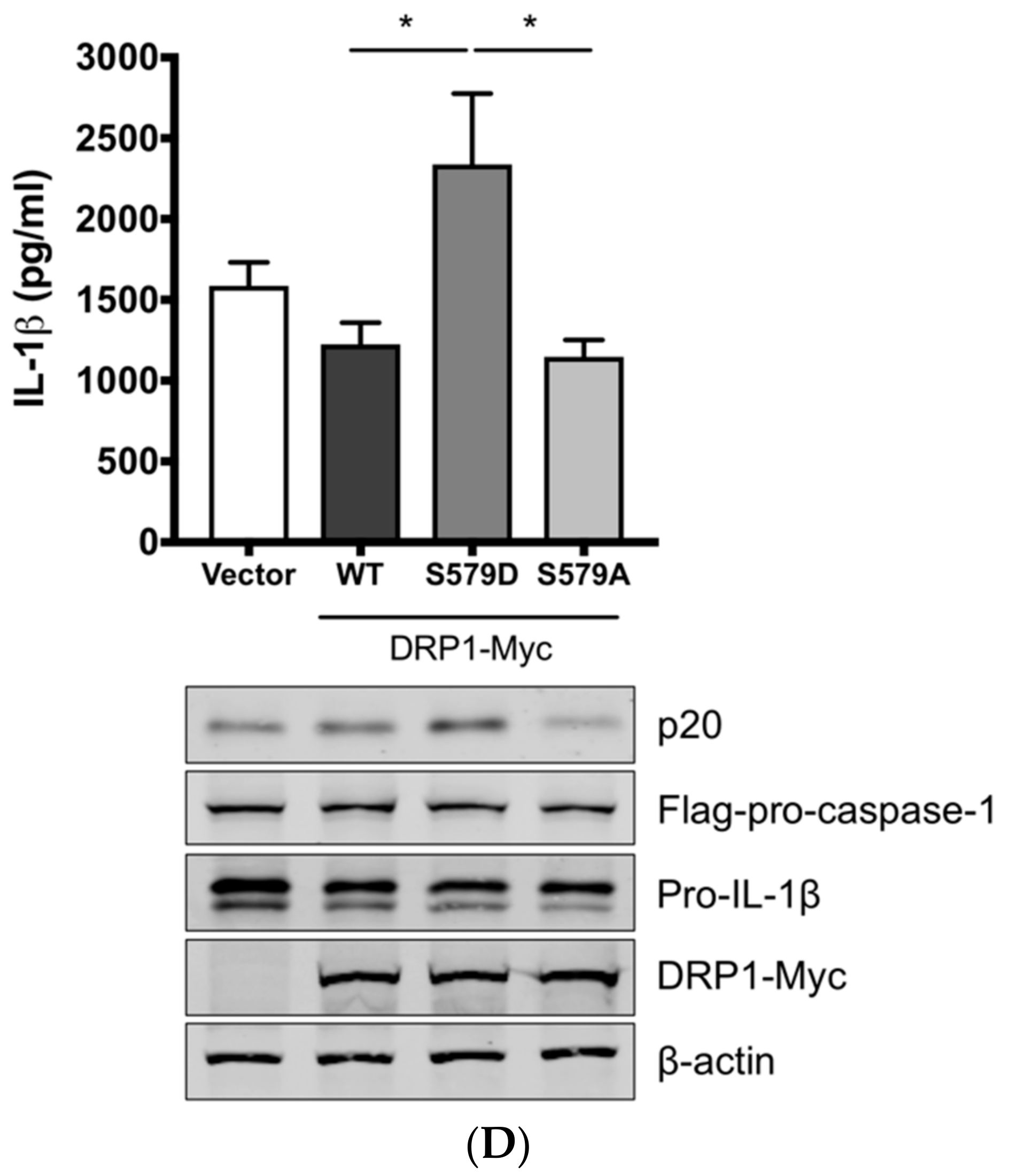
3.3. SIV-Induced Porcine IL-1β Production Is Mediated by DRP1
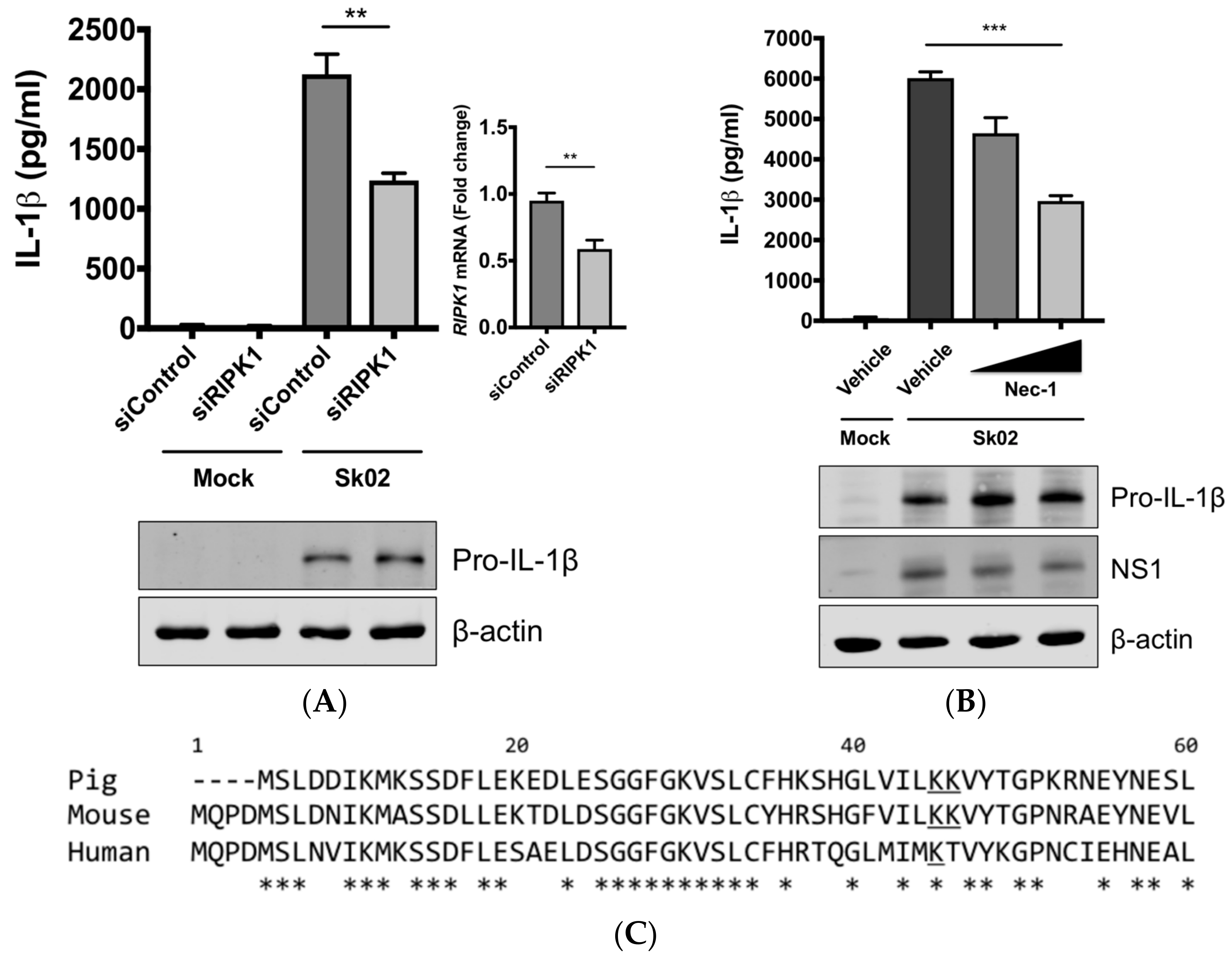
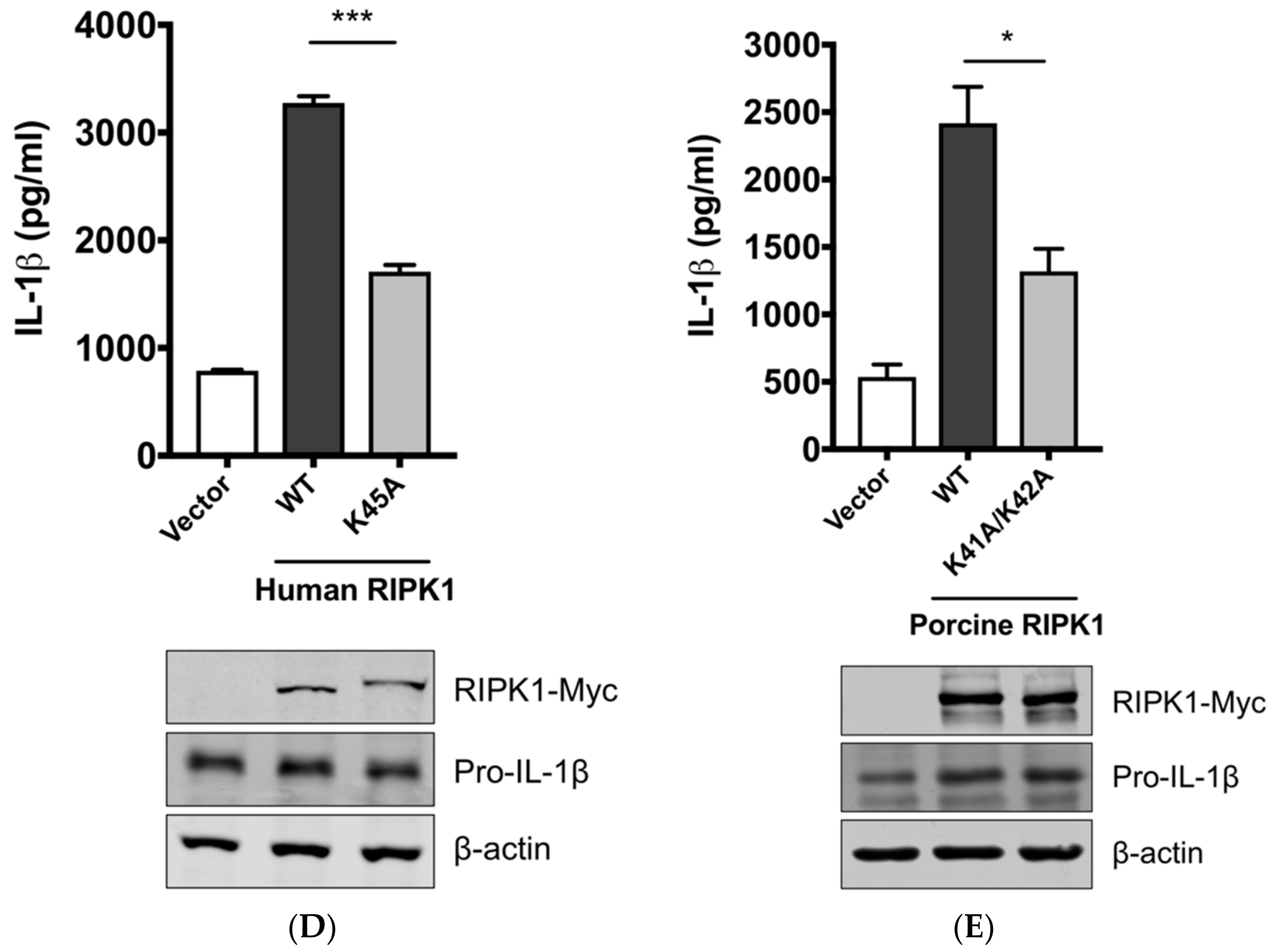
3.4. RIPK1 Kinase Activity Is Critical for SIV-Induced IL-1β Production in PAMs
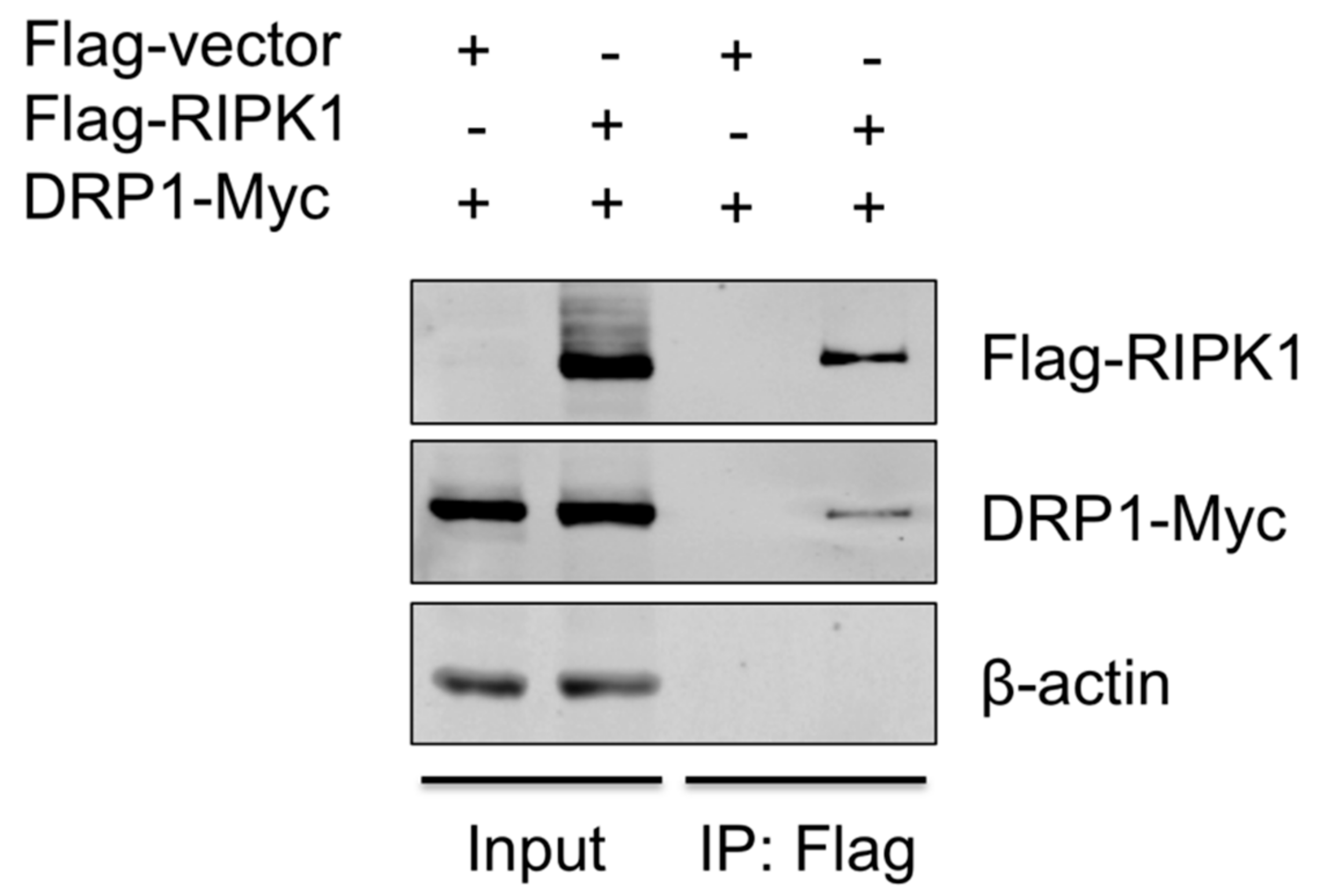
3.5. RIPK1 Interacts with DRP1
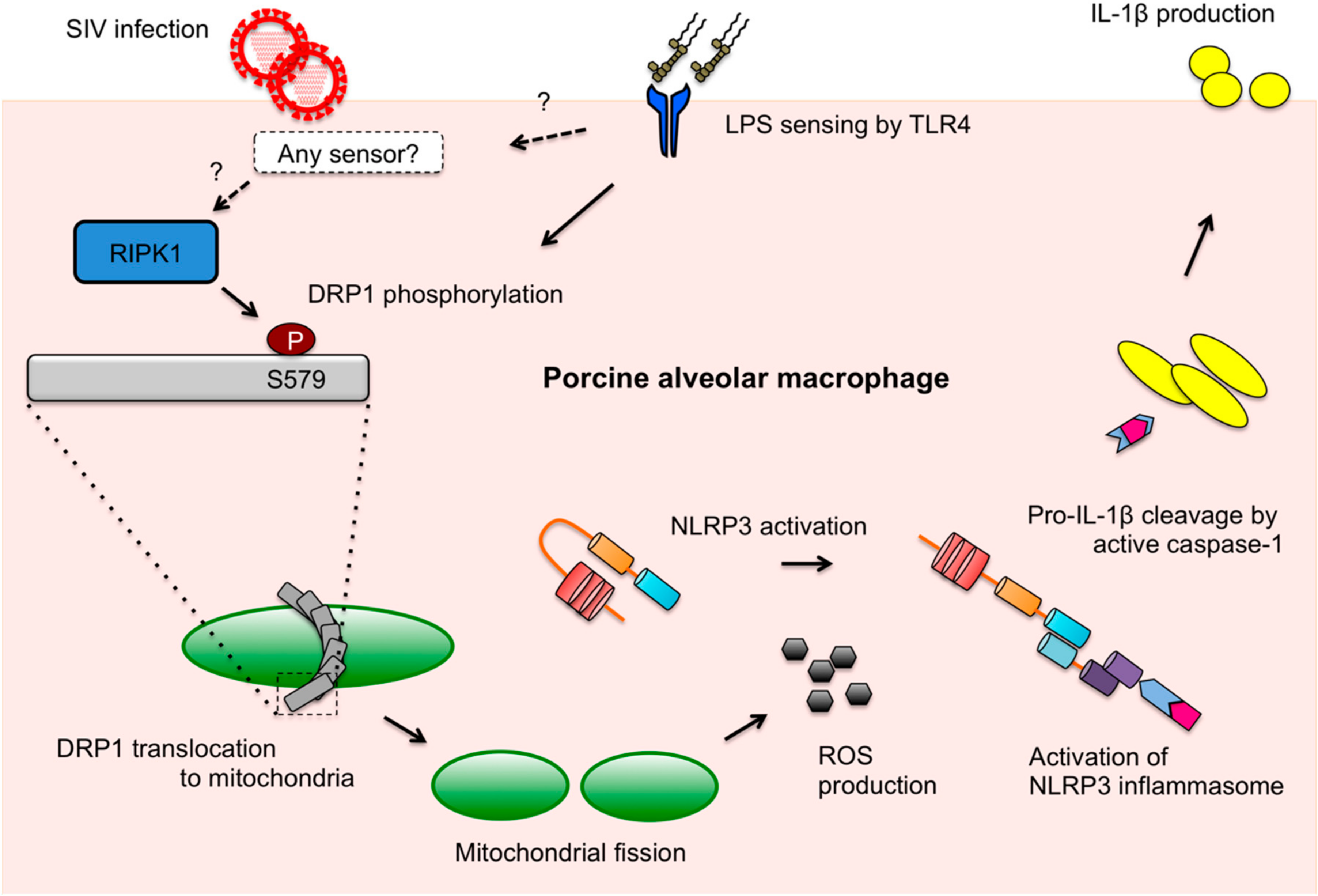
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Lee, H.K.; Ogura, Y.; Flavell, R.; Iwasaki, A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009, 206, 79–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, P.G.; Dash, P.; Aldridge, J.R.; Ellebedy, A.H.; Reynolds, C.; Funk, A.J.; Martin, W.J.; Lamkanfi, M.; Webby, R.J.; Boyd, K.L.; et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity 2009, 30, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Sarvestani, S.T.; McAuley, J.L. The role of the NLRP3 inflammasome in regulation of antiviral responses to influenza A virus infection. Antivir. Res. 2017, 148, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, T.; Kanneganti, T.D. Regulation and functions of NLRP3 inflammasome during influenza virus infection. Mol. Immunol. 2017, 86, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Juliana, C.; Hong, S.; Datta, P.; Hwang, I.; Fernandes-Alnemri, T.; Yu, J.W.; Alnemri, E.S. The mitochondrial antiviral protein MAVS associates with NLRP3 and regulates its inflammasome activity. J. Immunol. 2013, 191, 4358–4366. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Farias, L.N.; Balaji, S.; De Rosa, M.J.; O’Donnell, C.L.; Gough, P.J.; Bertin, J.; Welsh, R.M.; Chan, F.K. The Mitochondrial Phosphatase PGAM5 Is Dispensable for Necroptosis but Promotes Inflammasome Activation in Macrophages. J. Immunol. 2016, 196, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, K.; Haspel, J.A.; Rathinam, V.A.; Lee, S.J.; Dolinay, T.; Lam, H.C.; Englert, J.A.; Rabinovitch, M.; Cernadas, M.; Kim, H.P.; et al. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol. 2011, 12, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Yoshizumi, T.; Ichinohe, T.; Sasaki, O.; Otera, H.; Kawabata, S.; Mihara, K.; Koshiba, T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Won, J.H.; Hwang, I.; Hong, S.; Lee, H.K.; Yu, J.-W. Defective mitochondrial fission augments NLRP3 inflammasome activation. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Shi, C.S.; Kehrl, J.H. Cytochrome c Negatively Regulates NLRP3 Inflammasomes. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Zemirli, N.; Morel, E.; Molino, D. Mitochondrial Dynamics in Basal and Stressful Conditions. Int. J. Mol. Sci. 2018, 19, 564. [Google Scholar] [CrossRef] [PubMed]
- Taguchi, N.; Ishihara, N.; Jofuku, A.; Oka, T.; Mihara, K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007, 282, 11521–11529. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, W.; Yan, Y.; Gong, T.; Han, J.; Tian, Z.; Zhou, R. RNA viruses promote activation of the NLRP3 inflammasome through a RIP1-RIP3-DRP1 signaling pathway. Nat. Immunol. 2014, 15, 1126–1133. [Google Scholar] [CrossRef] [PubMed]
- Kashatus, J.A.; Nascimento, A.; Myers, L.J.; Sher, A.; Byrne, F.L.; Hoehn, K.L.; Counter, C.M.; Kashatus, D.F. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol. Cell 2015, 57, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Cereghetti, G.M.; Stangherlin, A.; Martins de Brito, O.; Chang, C.R.; Blackstone, C.; Bernardi, P.; Scorrano, L. Dephosphorylation by calcineurin regulates translocation of Drp1 to mitochondria. Proc. Natl. Acad. Sci. USA 2008, 105, 15803–15808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgass, K.; Pakay, J.; Ryan, M.T.; Palmer, C.S. Recent advances into the understanding of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhou, Z.H.; Chen, M.H.; Yang, J.; Leng, J.; Cao, G.S.; Xin, G.Z.; Liu, L.F.; Kou, J.P.; Liu, B.L.; et al. Inhibition of Mitochondrial Fission and NOX2 Expression Prevent NLRP3 Inflammasome Activation in the Endothelium: The Role of Corosolic Acid Action in the Amelioration of Endothelial Dysfunction. Antioxid. Redox Signal. 2016, 24, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Zhang, S.; Li, J.; Liu, K.; Huang, F.; Liu, B. Metformin and resveratrol inhibit Drp1-mediated mitochondrial fission and prevent ER stress-associated NLRP3 inflammasome activation in the adipose tissue of diabetic mice. Mol. Cell. Endocrinol. 2016, 434, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Wang, Z.; Guan, Q.; Qiu, F.; Li, Y.; Liu, Z.; Zhang, H.; Dong, H.; Zhang, Z. PEDF Inhibits the Activation of NLRP3 Inflammasome in Hypoxia Cardiomyocytes through PEDF Receptor/Phospholipase A2. Int. J. Mol. Sci. 2016, 17, 2064. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.J.; Syed, G.H.; Khan, M.; Chiu, W.W.; Sohail, M.A.; Gish, R.G.; Siddiqui, A. Hepatitis C virus triggers mitochondrial fission and attenuates apoptosis to promote viral persistence. Proc. Natl. Acad. Sci. USA 2014, 111, 6413–6418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Wang, J.; Zhou, A.; Khan, F.A.; Hu, L.; Zhang, S. Porcine reproductive and respiratory syndrome virus triggers mitochondrial fission and mitophagy to attenuate apoptosis. Oncotarget 2016, 7, 56002–56012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gou, H.; Zhao, M.; Xu, H.; Yuan, J.; He, W.; Zhu, M.; Ding, H.; Yi, L.; Chen, J. CSFV induced mitochondrial fission and mitophagy to inhibit apoptosis. Oncotarget 2017, 8, 39382–39400. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Patra, U.; Bhowmick, R.; Chawla-Sarkar, M. Rotaviral nonstructural protein 4 triggers dynamin-related protein 1-dependent mitochondrial fragmentation during infection. Cell Microbiol. 2018, 20. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.Y.; Liang, J.J.; Li, J.K.; Lee, Y.L.; Chang, B.L.; Su, C.-I.; Huang, W.-J.; Lai, M.M.C.; Lin, Y.-L. Dengue Virus Impairs Mitochondrial Fusion by Cleaving Mitofusins. PLoS Pathog. 2015, 11. [Google Scholar] [CrossRef] [PubMed]
- Chatel-Chaix, L.; Cortese, M.; Romero-Brey, I.; Bender, S.; Neufeldt, C.J.; Fischl, W.; Scaturro, P.; Schieber, N.; Schwab, Y.; Fischer, B.; et al. Dengue Virus Perturbs Mitochondrial Morphodynamics to Dampen Innate Immune Responses. Cell Host Microbe 2016, 20, 342–356. [Google Scholar] [CrossRef] [PubMed]
- Barbier, V.; Lang, D.; Valois, S.; Rothman, A.L.; Medin, C.L. Dengue virus induces mitochondrial elongation through impairment of Drp1-triggered mitochondrial fission. Virology 2017, 500, 149–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.; Fernandes-Alnemri, T.; Rogers, C.; Mayes, L.; Wang, Y.; Dillon, C.; Roback, L.; Kaiser, W.; Oberst, A.; Sagara, J.; et al. Caspase-8 scaffolding function and MLKL regulate NLRP3 inflammasome activation downstream of TLR3. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Liu, G.; Thulasi Raman, S.N.; Landreth, S.L.; Liu, Q.; Zhou, Y. NS1 Protein of 2009 Pandemic Influenza A Virus Inhibits Porcine NLRP3 Inflammasome-Mediated Interleukin-1 Beta Production by Suppressing ASC Ubiquitination. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S.B.; Read, R.C. Macrophage defences against respiratory tract infections. Br. Med. Bull. 2002, 61, 45–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ait-Ali, T.; Wilson, A.D.; Westcott, D.G.; Clapperton, M.; Waterfall, M.; Mellencamp, M.A.; Drew, T.W.; Bishop, S.C.; Archibald, A.L. Innate immune responses to replication of porcine reproductive and respiratory syndrome virus in isolated Swine alveolar macrophages. Viral Immunol. 2007, 20, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Park, H.S.; Pyo, H.M.; Liu, Q.; Zhou, Y. Influenza A Virus Panhandle Structure Is Directly Involved in RIG-I Activation and Interferon Induction. J. Virol. 2015, 89, 6067–6079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, Y.K.; Liu, Q.; Tikoo, S.K.; Babiuk, L.A.; Zhou, Y. Effect of the phosphatidylinositol 3-kinase/Akt pathway on influenza A. virus propagation. J. Gen. Virol. 2007, 88 (Pt 3), 942–950. [Google Scholar] [CrossRef]
- Blonska, M.; You, Y.; Geleziunas, R.; Lin, X. Restoration of NF-kappaB activation by tumor necrosis factor alpha receptor complex-targeted MEKK3 in receptor-interacting protein-deficient cells. Mol. Cell Biol. 2004, 24, 10757–10765. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.S.; Challa, S.; Moquin, D.; Genga, R.; Ray, T.D.; Guildford, M.; Chan, F.K. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell 2009, 137, 1112–1123. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3-new capabilities and interfaces. Nucleic Acids Res. 2012, 40. [Google Scholar] [CrossRef] [PubMed]
- Keck, F.; Brooks-Faulconer, T.; Lark, T.; Ravishankar, P.; Bailey, C.; Salvador-Morales, C.; Narayanan, A. Altered mitochondrial dynamics as a consequence of Venezuelan Equine encephalitis virus infection. Virulence 2017, 8, 1849–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.; Choi, H.; Min, J.S.; Park, S.J.; Kim, J.H.; Park, H.J.; Kim, B.; Chae, J.I.; Yim, M.; Lee, D.S. Mitochondrial dynamics modulate the expression of pro-inflammatory mediators in microglial cells. J. Neurochem. 2013, 127, 221–232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.L.; Shi, Y.Z.; Chen, G.G.; Wang, L.L.; Zheng, M.Z.; Jin, H.F.; Chen, Y.Y. TNF-α induces Drp1-mediated mitochondrial fragmentation during inflammatory cardiomyocyte injury. Int. J. Mol. Med. 2018, 41, 2317–2327. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Bryan, N.B.; Dorfleutner, A.; Rojanasakul, Y.; Stehlik, C. Activation of inflammasomes requires intracellular redistribution of the apoptotic speck-like protein containing a caspase recruitment domain. J. Immunol. 2009, 182, 3173–3182. [Google Scholar] [CrossRef] [PubMed]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Chen, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, T.; Peng, W.; Liu, Y.; Yan, C.; Maki, J.; Degterev, A.; Yuan, J.; Shi, Y. Structural basis of RIP1 inhibition by necrostatins. Structure 2013, 21, 493–499. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, K.; Chan, F.K. RIP3: A molecular switch for necrosis and inflammation. Genes Dev. 2013, 27, 1640–1649. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Su, S.S.; Zhao, S.; Yang, Z.; Zhong, C.Q.; Chen, X.; Cai, Q.; Yang, Z.H.; Huang, D.; Wu, R. RIP1 autophosphorylation is promoted by mitochondrial ROS and is essential for RIP3 recruitment into necrosome. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.B.; Kasparcova, V.; Hoffman, S.; Swift, B.; Dare, L.; Schaeffer, M.; Capriotti, C.; Cook, M.; Finger, J.; Hughes-Earle, A.; et al. Cutting Edge: RIP1 kinase activity is dispensable for normal development but is a key regulator of inflammation in SHARPIN-deficient mice. J. Immunol. 2014, 192, 5476–5480. [Google Scholar] [CrossRef] [PubMed]
- Shutinoski, B.; Alturki, N.A.; Rijal, D.; Bertin, J.; Gough, P.J.; Schlossmacher, M.G.; Sad, S. K45A mutation of RIPK1 results in poor necroptosis and cytokine signaling in macrophages, which impacts inflammatory responses in vivo. Cell Death Differ. 2016, 23, 1628–1637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardani, A.; Boulton, A.; Kim, T.S.; Braciale, T.J. Alveolar Macrophages Prevent Lethal Influenza Pneumonia By Inhibiting Infection Of Type-1 Alveolar Epithelial Cells. PLoS Pathog. 2017, 13. [Google Scholar] [CrossRef] [PubMed]
- Schneider, C.; Nobs, S.P.; Heer, A.K.; Kurrer, M.; Klinke, G.; van Nico, R.; Vogel, J.; Kopf, M. Alveolar macrophages are essential for protection from respiratory failure and associated morbidity following influenza virus infection. PLoS Pathog. 2014, 10, e1004053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.M.; Lee, Y.W.; Lee, K.J.; Kim, H.S.; Cho, S.W.; van Rooijen, N.; Guan, Y.; Seo, S.H. Alveolar macrophages are indispensable for controlling influenza viruses in lungs of pigs. J. Virol. 2008, 82, 4265–4274. [Google Scholar] [CrossRef] [PubMed]
- Masic, A.; Babiuk, L.A.; Zhou, Y. Reverse genetics-generated elastase-dependent swine influenza viruses are attenuated in pigs. J. Gen. Virol. 2009, 90 (Pt 2), 375–385. [Google Scholar] [CrossRef] [Green Version]
- Saleh, D.; Najjar, M.; Zelic, M.; Shah, S.; Nogusa, S.; Polykratis, A.; Paczosa, M.K.; Gough, P.J.; Bertin, J.; Whalen, M.; et al. Kinase Activities of RIPK1 and RIPK3 Can Direct IFN-β Synthesis Induced by Lipopolysaccharide. J. Immunol. 2017, 198, 4435–4447. [Google Scholar] [CrossRef] [PubMed]
- Najjar, M.; Saleh, D.; Zelic, M.; Nogusa, S.; Shah, S.; Tai, A.; Finger, J.N.; Polykratis, A.; Gough, P.J.; Bertin, J.; et al. RIPK1 and RIPK3 Kinases Promote Cell-Death-Independent Inflammation by Toll-like Receptor 4. Immunity 2016, 45, 46–59. [Google Scholar] [CrossRef] [PubMed]
- Kuriakose, T.; Man, S.M.; Malireddi, R.K.; Karki, R.; Kesavardhana, S.; David, E.P.; Geoffrey, N.; Peter, V.; Thirumala-Devi, K. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci. Immunol. 2016, 1. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, H.-S. VIDO-InterVac, Saskatoon, Canada, unpublished data. 2018. [Google Scholar]
- Chang, C.R.; Blackstone, C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann. N. Y. Acad. Sci. 2010, 1201, 34–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, L.; Lin, H.; Wen, J.; Sun, Q.; Gao, Y.; Xu, X.; Wang, J.; Zhang, J.; Weng, D. The kinase receptor-interacting protein 1 is required for inflammasome activation induced by endoplasmic reticulum stress. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.-S.; Liu, G.; Liu, Q.; Zhou, Y. Swine Influenza Virus Induces RIPK1/DRP1-Mediated Interleukin-1 Beta Production. Viruses 2018, 10, 419. https://doi.org/10.3390/v10080419
Park H-S, Liu G, Liu Q, Zhou Y. Swine Influenza Virus Induces RIPK1/DRP1-Mediated Interleukin-1 Beta Production. Viruses. 2018; 10(8):419. https://doi.org/10.3390/v10080419
Chicago/Turabian StylePark, Hong-Su, Guanqun Liu, Qiang Liu, and Yan Zhou. 2018. "Swine Influenza Virus Induces RIPK1/DRP1-Mediated Interleukin-1 Beta Production" Viruses 10, no. 8: 419. https://doi.org/10.3390/v10080419
APA StylePark, H. -S., Liu, G., Liu, Q., & Zhou, Y. (2018). Swine Influenza Virus Induces RIPK1/DRP1-Mediated Interleukin-1 Beta Production. Viruses, 10(8), 419. https://doi.org/10.3390/v10080419