Viruses of Eukaryotic Algae: Diversity, Methods for Detection, and Future Directions

 , , ,
, , ,
Abstract

1. Introduction
2. Diversity of Cultured Virus-Host Systems
2.1. dsDNA Viruses Infecting Eukaryotic Algae
2.2. ssDNA Viruses Infecting Eukaryotic Algae
2.3. RNA Viruses Infecting Eukaryotic Algae
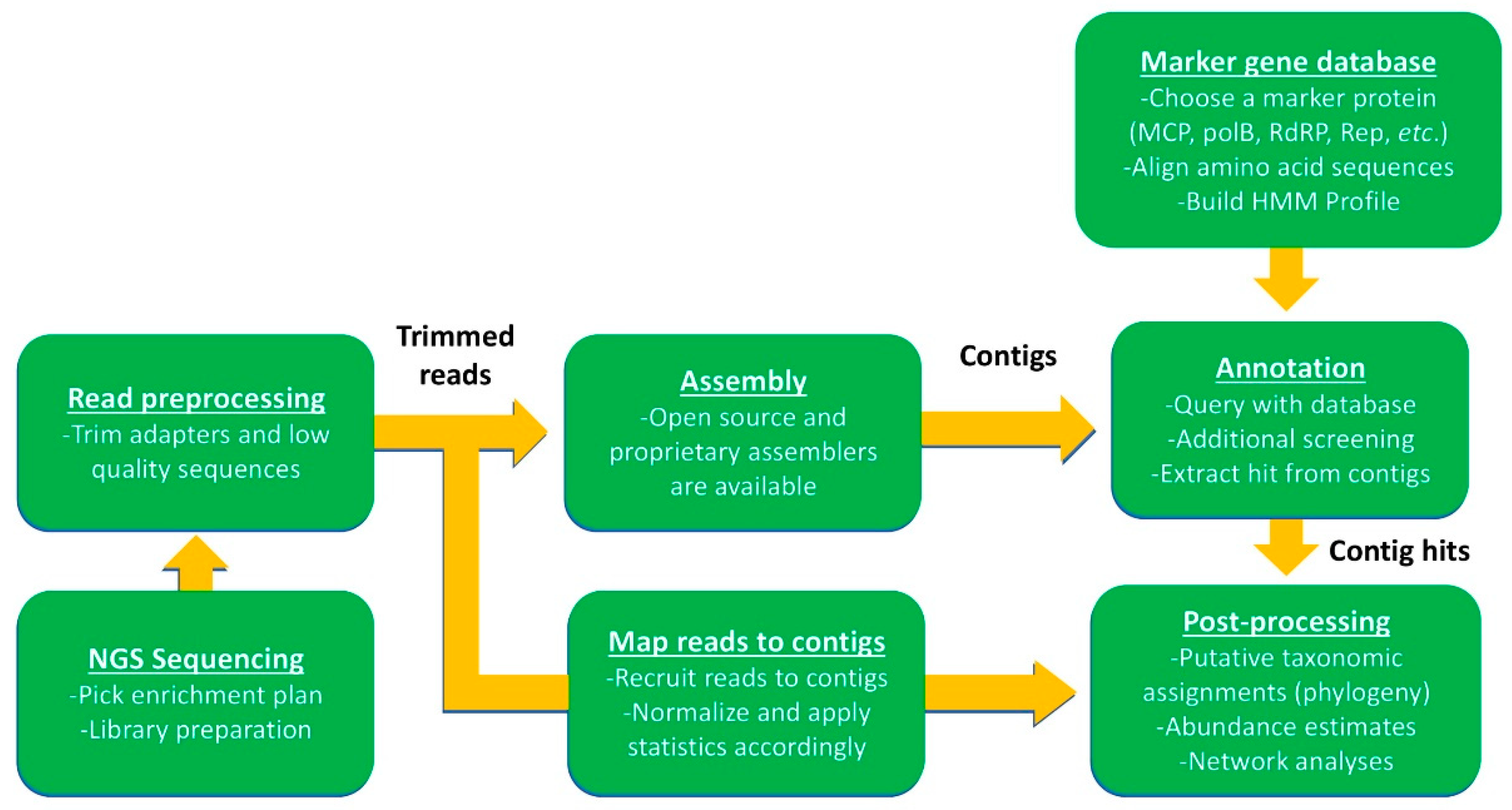
3. Culture Independent Approaches: Expanding Known Diversity
3.1. PCR Applications for Estimating Viral Diversity and Dynamics
3.2. Using Omics Approaches to Estimate Virus Diversity and Dynamics
3.3. Other Downstream Applications of Omic Assemblies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Short, S.M.; Staniewski, M.A.; Chaban, Y.V.; Long, A.M.; Wang, D. Diversity of viruses infecting eukaryotic algae. In Viruses of Microorganisms; Paul, H., Abedon, S.T., Eds.; Caister Academic Press: Poole, UK, 2018. [Google Scholar]
- Wilhelm, S.W.; Suttle, C.A. Viruses and nutrient cycles in the sea-viruses play critical roles in the structure and function of aquatic food webs. Bioscience 1999, 49, 781–788. [Google Scholar] [CrossRef]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Holligan, P.M.; Viollier, M.; Harbour, D.S.; Camus, P.; Champagne-philippe, M. Satellite and ship studies of coccolithophore production along a continental-shelf edge. Nature 1983, 304, 339–342. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; Bird, J.T.; Bonifer, K.S.; Calfee, B.C.; Chen, T.; Coy, S.R.; Gainer, P.J.; Gann, E.R.; Heatherly, H.T.; Lee, J.; et al. A student’s guide to giant viruses infecting small eukaryotes: From acanthamoeba to zooxanthellae. Viruses 2017, 9, 46. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L.; Lane, L.C.; Meints, R.H. Viruses and virus-like particles of eukaroytic algae. Microbiol. Rev. 1991, 55, 586–620. [Google Scholar] [PubMed]
- Bratbak, G.; Egge, J.K.; Heldal, M. Viral mortality of the marine alga emiliania-huxleyi (haptophyceae) and termination of algal blooms. Mar. Ecol. Prog. Ser. 1993, 93, 39–48. [Google Scholar] [CrossRef]
- Nagasaki, K.; Ando, M.; Itakura, S.; Imai, I.; Ishida, Y. Viral mortality in the final stage of heterosigma-akashiwo (raphidophyceae) red tide. J. Plankton Res. 1994, 16, 1595–1599. [Google Scholar] [CrossRef]
- Gastrich, M.D.; Anderson, O.R.; Benmayor, S.S.; Cosper, E.M. Ultrastructural analysis of viral infection in the brown-tide alga, Aureococcus anophagefferens (pelagophyceae). Phycologia 1998, 37, 300–306. [Google Scholar] [CrossRef]
- Dodds, J.A.; Cole, A. Microscopy and biology of uronema-gigas, a filamentous eukaryotic green-alga, and its associated tailed virus-like particle. Virology 1980, 100, 156–165. [Google Scholar] [CrossRef]
- Chen, F.; Suttle, C.A. Amplification of DNA-polymerase gene fragments from viruses infecting microalgae. Appl. Environ. Microbiol. 1995, 61, 1274–1278. [Google Scholar] [PubMed]
- Moniruzzaman, M.; Wurch, L.L.; Alexander, H.; Dyhrman, S.T.; Gobler, C.J.; Wilhelm, S.W. Virus-host relationships of marine single-celled eukaryotes resolved from metatranscriptomics. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L.; Burbank, D.E.; Kuczmarski, D.; Meints, R.H. Virus-infection of culturable chlorella-like algae and development of a plaque assay. Science 1983, 219, 994–996. [Google Scholar] [CrossRef] [PubMed]
- Dunigan, D.D.; Cerny, R.L.; Bauman, A.T.; Roach, J.C.; Lane, L.C.; Agarkova, I.V.; Wulser, K.; Yanai-Balser, G.M.; Gurnon, J.R.; Vitek, J.C.; et al. Paramecium bursaria chlorella virus 1 proteome reveals novel architectural and regulatory features of a giant virus. J. Virol. 2012, 86, 8821–8834. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, L.A.; Graves, M.V.; Li, X.; Feldblyum, T.; Nierman, W.C.; Van Etten, J.L. Sequence and annotation of the 369-kb NY-2a and the 345-kb AR158 viruses that infect Chlorella NC64Aa. Virology 2007, 358, 472–484. [Google Scholar] [CrossRef] [PubMed]
- Fitzgerald, L.A.; Graves, M.V.; Li, X.; Feldblyum, T.; Hartigan, J.; Van Etten, J.L. Sequence and annotation of the 314-kb MT325 and the 321-kb FR483 viruses that infect Chlorella Pbi. Virology 2007, 358, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Jeanniard, A.; Dunigan, D.D.; Gurnon, J.R.; Agarkova, I.V.; Kang, M.; Vitek, J.; Duncan, G.; McClung, O.W.; Larsen, M.; Claverie, J.M.; et al. Towards defining the chloroviruses: A genomic journey through a genus of large DNA viruses. BMC Genomics 2013, 14. [Google Scholar] [CrossRef] [PubMed]
- Quispe, C.F.; Esmael, A.; Sonderman, O.; McQuinn, M.; Agarkova, I.; Battan, M.; Duncan, G.A.; Dunigan, D.D.; Smith, T.P.L.; De Castro, C.; et al. Characterization of a new chlorovirus type with permissive and non-permissive features on phylogenetically related algal strains. Virology 2017, 500, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Yanai-Balser, G.M.; Duncan, G.A.; Eudy, J.D.; Wang, D.; Li, X.; Agarkova, I.V.; Dunigan, D.D.; Van Etten, J.L. Microarray analysis of Paramecium bursaria chlorella virus 1 transcription. J. Virol. 2010, 84, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Mozar, M.; Agarkova, I.V.; Gurnon, J.R.; Yanai-Balser, G.; Rowe, J.M.; Xia, Y.N.; Riethoven, J.J.; Dunigan, D.D.; Van Etten, J.L. Deep RNA sequencing reveals hidden features and dynamics of early gene transcription in Paramecium bursaria chlorella virus 1. PLoS ONE 2014, 9, e90989. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, R.; Strasser, P.; Vanetten, J.L. The DNA-polymerase gene from chlorella viruses PBCV-1 and NY-2A contains an intron with nuclear splicing sequences. Virology 1992, 188, 721–731. [Google Scholar] [CrossRef]
- Chen, F.; Suttle, C.A.; Short, S.M. Genetic diversity in marine algal virus communities as revealed by sequence analysis of DNA polymerase genes. Appl. Environ. Microbiol. 1996, 62, 2869–2874. [Google Scholar] [PubMed]
- Moniruzzaman, M.; LeCleir, G.R.; Brown, C.M.; Gobler, C.J.; Bidle, K.D.; Wilson, W.H.; Wilhelm, S.W. Genome of brown tide virus (AaV), the little giant of the megaviridae, elucidates ncldv genome expansion and host-virus coevolution. Virology 2014, 466, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.J.; Zhou, J.L.; Liu, T.G.; Yu, Y.X.; Pan, Y.J.; Yan, S.L.; Wang, Y.J. Four novel algal virus genomes discovered from yellowstone lake metagenomes. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Jeudy, S.; Bartoli, J.; Poirot, O.; Lescot, M.; Abergel, C.; Barbe, V.; Wommack, K.E.; Noordeloos, A.A.M.; Brussaard, C.P.D.; et al. Genome of Phaeocystis globosa virus PgV-16T highlights the common ancestry of the largest known DNA viruses infecting eukaryotes. Proc. Natl. Acad. Sci. USA 2013, 110, 10800–10805. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Koonin, E.V. Hidden evolutionary complexity of nucleo-cytoplasmic large DNA viruses of eukaryotes. Virol. J. 2012, 9. [Google Scholar] [CrossRef] [PubMed]
- Yutin, N.; Koonin, E.V. Pandoraviruses are highly derived Phycodnaviruses. Biol. Direct 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, F.; Ueki, S. Evolution and phylogeny of large DNA viruses, Mimiviridae and Phycodnaviridae including newly characterized Heterosigma akashiwo virus. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Culley, A.I.; Lang, A.S.; Suttle, C.A. High diversity of unknown picorna-like viruses in the sea. Nature 2003, 424, 1054–1057. [Google Scholar] [CrossRef] [PubMed]
- Steward, G.F.; Culley, A.I.; Mueller, J.A.; Wood-Charlson, E.M.; Belcaid, M.; Poisson, G. Are we missing half of the viruses in the ocean? ISME J. 2013, 7, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Yau, S.; Lauro, F.M.; DeMaere, M.Z.; Brown, M.V.; Thomas, T.; Raftery, M.J.; Andrews-Pfannkoch, C.; Lewis, M.; Hoffman, J.M.; Gibson, J.A.; et al. Virophage control of antarctic algal host-virus dynamics. Proc. Natl. Acad. Sci. USA 2011, 108, 6163–6168. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.W.; Zhang, W.J.; Zhou, X.W.; Wang, H.M.; Sun, G.W.; Xiao, J.Z.; Pan, Y.J.; Yan, S.L.; Wang, Y.J. Novel virophages discovered in a freshwater lake in China. Front. Microbiol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Chan, L.K.; Egan, R.; Malmstrom, R.R.; McMahon, K.D.; Sullivan, M.B. Ecogenomics of virophages and their giant virus hosts assessed through time series metagenomics. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.Z.; McCrow, J.P.; Ininbergs, K.; Dupont, C.L.; Badger, J.H.; Hoffman, J.M.; Ekman, M.; Allen, A.E.; Bergman, B.; Venter, J.C. The baltic sea virome: Diversity and transcriptional activity of DNA and RNA viruses. mSystems 2017, 2, e00125-16. [Google Scholar] [CrossRef]
- Burki, F. The eukaryotic tree of life from a global phylogenomic perspective. Cold Spring Harb. Perspect. Biol. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- De Vargas, C.; Audic, S.; Henry, N.; Decelle, J.; Mahe, F.; Logares, R.; Lara, E.; Berney, C.; Le Bescot, N.; Probert, I.; et al. Eukaryotic plankton diversity in the sunlit ocean. Science 2015, 348, 1261605. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, T.; Yamaguchi, H.; Sato, M.; Watanabe, T.; Taniuchi, Y.; Kuwata, A.; Kawachi, M. Seasonal and geographical distribution of near-surface small photosynthetic eukaryotes in the western North Pacific determined by pyrosequencing of 18s rDNA. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef] [PubMed]
- Gowing, M.M. Large viruses and infected microeukaryotes in ross sea summer pack ice habitats. Mar. Biol. 2003, 142, 1029–1040. [Google Scholar] [CrossRef]
- Honjo, T. Overview on bloom dynamics and physiological ecology of Heterosigma-akashiwo. In Toxic Phytoplankton Blooms in the Sea, Proceedings of the Fifth International Conference on Toxic Marine Phytoplankton, Newport, Rhode Island, USA, 28 October–1 November 1991; Smayda, T.J., Shimizu, Y., Eds.; Elsevier: Philadelphia, PA, USA, 1993; Volume 3, pp. 33–41. [Google Scholar]
- Karosiene, J.; Kasperoviciene, J.; Koreiviene, J.; Savadova, K.; Vitonyte, I. Factors promoting persistence of the bloom-forming Gonyostomum semen in temperate lakes. Limnologica 2016, 60, 51–58. [Google Scholar] [CrossRef]
- Leon-Munoz, J.; Urbina, M.A.; Garreaud, R.; Iriarte, J.L. Hydroclimatic conditions trigger record harmful algal bloom in western Patagonia (summer 2016). Sci. Rep. 2018, 8, 1330. [Google Scholar] [CrossRef] [PubMed]
- Gustafson, D.E.; Stoecker, D.K.; Johnson, M.D.; Van Heukelem, W.F.; Sneider, K. Cryptophyte algae are robbed of their organelles by the marine ciliate Mesodinium rubrum. Nature 2000, 405, 1049–1052. [Google Scholar] [CrossRef] [PubMed]
- Vermont, A.I.; Martinez, J.M.; Waller, J.D.; Gilg, I.C.; Leavitt, A.H.; Floge, S.A.; Archer, S.D.; Wilson, W.H.; Fields, D.M. Virus infection of Emiliania huxleyi deters grazing by the copepod Acartia tonsa. J. Plankton Res. 2016, 38, 1194–1205. [Google Scholar] [CrossRef]
- Evans, C.; Wilson, W.H. Preferential grazing of Oxyrrhis marina on virus-infected Emiliania huxleyi. Limnol. Oceanogr. 2008, 53, 2035–2040. [Google Scholar] [CrossRef]
- Schvarcz, C.R.; Steward, G.F. A giant virus infecting green algae encodes key fermentation genes. Virology 2018, 518, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Pagarete, A.; Grebert, T.; Stepanova, O.; Sandaa, R.A.; Bratbak, G. Tsv-N1: A novel DNA algal virus that infects Tetraselmis striata. Viruses 2015, 7, 3937–3953. [Google Scholar] [CrossRef] [PubMed]
- Derelle, E.; Monier, A.; Cooke, R.; Worden, A.Z.; Grimsley, N.H.; Moreau, H. Diversity of viruses infecting the green microalga Oostreococcus lucimarinus. J. Virol. 2015, 89, 5812–5821. [Google Scholar] [CrossRef] [PubMed]
- Weynberg, K.D.; Allen, M.J.; Gilg, I.C.; Scanlan, D.J.; Wilson, W.H. Genome sequence of Ostreococcus tauri virus OtV-2 throws light on the role of picoeukaryote niche separation in the ocean. J. Virol. 2011, 85, 4520–4529. [Google Scholar] [CrossRef] [PubMed]
- Moreau, H.; Piganeau, G.; Desdevises, Y.; Cooke, R.; Derelle, E.; Grimsley, N. Marine prasinovirus genomes show low evolutionary divergence and acquisition of protein metabolism genes by horizontal gene transfer. J. Virol. 2010, 84, 12555–12563. [Google Scholar] [CrossRef] [PubMed]
- Martinez, J.M.; Boere, A.; Gilg, L.; van Lent, J.W.M.; Witte, H.J.; van Bleijswijk, J.D.L.; Brussaard, C.P.D. New lipid envelope-containing dsdna virus isolates infecting Micromonas pusilla reveal a separate phylogenetic group. Aquat. Microb. Ecol. 2015, 74, 17–28. [Google Scholar] [CrossRef]
- Finke, J.F.; Winget, D.M.; Chan, A.M.; Suttle, C.A. Variation in the genetic repertoire of viruses infecting Micromonas pusilla reflects horizontal gene transfer and links to their environmental distribution. Viruses 2017, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Brussaard, C.P.D.; Noordeloos, A.A.M.; Sandaa, R.A.; Heldal, M.; Bratbak, G. Discovery of a dsRNA virus infecting the marine photosynthetic protist Micromonas pusilla. Virology 2004, 319, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Maat, D.S.; Biggs, T.; Evans, C.; van Bleijswijk, J.D.L.; van der Wel, N.N.; Dutilh, B.E.; Brussaard, C.P.D. Characterization and temperature dependence of Arctic Micromonas polaris viruses. Viruses 2017, 9. [Google Scholar] [CrossRef] [PubMed]
- Sandaa, R.A.; Heldal, M.; Castberg, T.; Thyrhaug, R.; Bratbak, G. Isolation and characterization of two viruses with large genome size infecting Chrysochromulina ericina (prymnesiophyceae) and Pyramimonas orientalis (prasinophyceae). Virology 2001, 290, 272–280. [Google Scholar] [CrossRef] [PubMed]
- Rousvoal, S.; Bouyer, B.; Lopez-Cristoffanini, C.; Boyen, C.; Collen, J. Mutant swarms of a totivirus-like entities are present in the red macroalga Chondrus crispus and have been partially transferred to the nuclear genome. J. Phycol. 2016, 52, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Ogata, H.; Toyoda, K.; Tomaru, Y.; Nakayama, N.; Shirai, Y.; Claverie, J.M.; Nagasaki, K. Remarkable sequence similarity between the dinoflagellate-infecting marine girus and the terrestrial pathogen african swine fever virus. Virol. J. 2009, 6, 178. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, Y.; Katanozaka, N.; Nishida, K.; Shirai, Y.; Tarutani, K.; Yamaguchi, M.; Nagasaki, K. Isolation and characterization of two distinct types of HcRNAV, a single-stranded RNA virus infecting the bivalve-killing microalga Heterocapsa circularisquama. Aquat. Microb. Ecol. 2004, 34, 207–218. [Google Scholar] [CrossRef]
- Kim, J.; Kim, C.H.; Takano, Y.; Jang, I.K.; Kim, S.W.; Choi, T.J. Isolation and physiological characterization of a new algicidal virus infecting the harmful dinoflagellate Heterocapsa pygmaea. Plant Pathol. J. 2012, 28, 433–438. [Google Scholar] [CrossRef]
- Onji, M.; Nakano, S.; Suzuki, S. Virus-like particles suppress growth of the red-tide-forming marine dinoflagellate Gymnodinium mikimotoi. Mar. Biotechnol. 2003, 5, 435–442. [Google Scholar] [PubMed]
- Bettarel, Y.; Kan, J.; Wang, K.; Williamson, K.E.; Cooney, S.; Ribblett, S.; Chen, F.; Wommack, K.E.; Coats, D.W. Isolation and preliminary characterisation of a small nuclear inclusion virus infecting the diatom Chaetoceros cf. gracilis. Aquat. Microb. Ecol. 2005, 40, 103–114. [Google Scholar] [CrossRef]
- Nagasaki, K.; Tomaru, Y.; Takao, Y.; Nishida, K.; Shirai, Y.; Suzuki, H.; Nagumo, T. Previously unknown virus infects marine diatom. Appl. Environ. Microbiol. 2005, 71, 3528–3535. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, Y.; Toyoda, K.; Suzuki, H.; Nagumo, T.; Kimura, K.; Takao, Y. New single-stranded DNA virus with a unique genomic structure that infects marine diatom Chaetoceros setoensis. Sci. Rep. 2013, 3. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, Y.; Takao, Y.; Suzuki, H.; Nagumo, T.; Nagasaki, K. Isolation and characterization of a single-stranded RNA virus infecting the bloom-forming diatom Chaetoceros socialis. Appl. Environ. Microbiol. 2009, 75, 2375–2381. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, Y.; Takao, Y.; Suzuki, H.; Nagumo, T.; Koike, K.; Nagasaki, K. Isolation and characterization of a single-stranded DNA virus infecting Chaetoceros lorenzianus Grunow. Appl. Environ. Microbiol. 2011, 77, 5285–5293. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, Y.; Shirai, Y.; Toyoda, K.; Nagasaki, K. Isolation and characterisation of a single-stranded DNA virus infecting the marine planktonic diatom Chaetoceros tenuissimus. Aquat. Microb. Ecol. 2011, 64, 175–184. [Google Scholar] [CrossRef]
- Kimura, K.; Tomarua, Y. Discovery of two novel viruses expands the diversity of single-stranded DNA and single-stranded RNA viruses infecting a cosmopolitan marine diatom. Appl. Environ. Microbiol. 2015, 81, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Shirai, Y.; Tomaru, Y.; Takao, Y.; Suzuki, H.; Nagumo, T.; Nagasaki, K. Isolation and characterization of a single-stranded RNA virus infecting the marine planktonic diatom Chaetoceros tenuissimus Meunier. Appl. Environ. Microbiol. 2008, 74, 4022–4027. [Google Scholar] [CrossRef] [PubMed]
- Kimura, K.; Tomaru, Y. Isolation and characterization of a single-stranded DNA virus infecting the marine diatom Chaetoceros sp. strain SS628-11 isolated from western Japan. PLoS ONE 2013, 8, e82013. [Google Scholar] [CrossRef] [PubMed]
- Toyoda, K.; Kimura, K.; Hata, N.; Nakayama, N.; Nagasaki, K.; Tomaru, Y. Isolation and characterization of a single-stranded DNA virus infecting the marine planktonic diatom Chaetoceros sp (strain TG07-C28). Plankton Benthos. Res. 2012, 7, 20–28. [Google Scholar] [CrossRef]
- Tomaru, Y.; Shirai, Y.; Suzuki, H.; Nagumo, T.; Nagasaki, K. Isolation and characterization of a new single-stranded DNA virus infecting the cosmopolitan marine diatom Chaetoceros dehilis. Aquat. Microb. Ecol. 2008, 50, 103–112. [Google Scholar] [CrossRef]
- Tomaru, Y.; Toyoda, K.; Kimura, K.; Takao, Y.; Sakurada, K.; Nakayama, N.; Nagasaki, K. Isolation and characterization of a single-stranded RNA virus that infects the marine planktonic diatom Chaetoceros sp. (SS08-c03). Phycol. Res. 2013, 61, 27–36. [Google Scholar] [CrossRef]
- Eissler, Y.; Wang, K.; Chen, F.; Wommack, K.E.; Coats, D.W. Ultrastructural characterization of the lytic cycle of an intracellular virus infecting the diatom Chaetoceros cf wighamii (Bacillariophyceae) from Chesapeake Bay, USA. J. Phycol. 2009, 45, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, Y.; Toyoda, K.; Kimura, K.; Hata, N.; Yoshida, M.; Nagasaki, K. First evidence for the existence of pennate diatom viruses. ISME J. 2012, 6, 1445–1448. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, K.; Tomaru, Y.; Katanozaka, N.; Shirai, Y.; Nishida, K.; Itakura, S.; Yamaguchi, M. Isolation and characterization of a novel single-stranded RNA virus infecting the bloom-forming diatom Rhizosolenia setigera. Appl. Environ. Microbiol. 2004, 70, 704–711. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, C.H.; Youn, S.H.; Choi, T.J. Isolation and physiological characterization of a novel algicidal virus infecting the marine diatom Skeletonema costatum. Plant Pathol. J. 2015, 31, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yoon, S.H.; Choi, T.J. Isolation and physiological characterization of a novel virus infecting Stephanopyxis palmeriana (Bacillariophyta). Algae 2015, 30, 81–87. [Google Scholar] [CrossRef]
- Kapp, M.; Knippers, R.; Mueller, D.G. New members of a group of DNA viruses infecting brown algae. Phycol. Res. 1997, 45, 85–90. [Google Scholar] [CrossRef]
- Henry, E.C.; Meints, R.H. A persistent virus-infection in Feldmannia (Phaeophyceae). J. Phycol. 1992, 28, 517–526. [Google Scholar] [CrossRef]
- Maier, I.; Wolf, S.; Delaroque, N.; Muller, D.G.; Kawai, H. A DNA virus infecting the marine brown alga Pilayella littoralis (Ectocarpales, Phaeophyceae) in culture. Eur. J. Phycol. 1998, 33, 213–220. [Google Scholar] [CrossRef]
- Nagasaki, K.; Yamaguchi, M. Isolation of a virus infectious to the harmful bloom causing microalga Heterosigma akashiwo (Raphidophyceae). Aquat. Microb. Ecol. 1997, 13, 135–140. [Google Scholar] [CrossRef]
- Lawrence, J.E.; Brussaard, C.P.D.; Suttle, C.A. Virus-specific responses of Heterosigma akashiwo to infection. Appl. Environ. Microbiol. 2006, 72, 7829–7834. [Google Scholar] [CrossRef] [PubMed]
- Tai, V.; Lawrence, J.E.; Lang, A.S.; Chan, A.M.; Culley, A.I.; Suttle, C.A. Characterization of HaRNAV, a single-stranded RNA virus causing lysis of Heterosigma akashiwo (Raphidophyceae). J. Phycol. 2003, 39, 343–352. [Google Scholar] [CrossRef]
- Lawrence, J.E.; Chan, A.M.; Suttle, C.A. A novel virus (HaNIV) causes lysis of the toxic bloom-forming alga Heterosigma akashiwo (Raphidophyceae). J. Phycol. 2001, 37, 216–222. [Google Scholar] [CrossRef]
- Castberg, T.; Thyrhaug, R.; Larsen, A.; Sandaa, R.A.; Heldal, M.; Van Etten, J.L.; Bratbak, G. Isolation and characterization of a virus that infects Emiliania huxleyi (haptophyta). J. Phycol. 2002, 38, 767–774. [Google Scholar] [CrossRef]
- Baudoux, A.C.; Brussaard, C.P.D. Characterization of different viruses infecting the marine harmful algal bloom species Phaeocystis globosa. Virology 2005, 341, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Schroeder, D.C.; Ho, J.; Canty, M. Phylogenetic analysis of PgV-102P, a new virus from the english channel that infects phaeocystis globosa. J. Marine Biol. Assoc. UK 2006, 86, 485–490. [Google Scholar] [CrossRef]
- Jacobsen, A.; Bratbak, G.; Heldal, M. Isolation and characterization of a virus infecting Phaeocystis pouchetii (Prymnesiophyceae). J. Phycol. 1996, 32, 923–927. [Google Scholar] [CrossRef]
- Suttle, C.A.; Chan, A.M. Viruses infecting the marine Prymnesiophyte chrysochromulina spp.: Isolation, preliminary characterization, and natural-abundance. Mar. Ecol. Progr. Ser. 1995, 118, 275–282. [Google Scholar] [CrossRef]
- Mirza, S.F.; Staniewski, M.A.; Short, C.M.; Long, A.M.; Chaban, Y.V.; Short, S.M. Isolation and characterization of a virus infecting the freshwater algae Chrysochromulina parva. Virology 2015, 486, 105–115. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.V.; Bratbak, G.; Larsen, A.; Ogata, H.; Egge, E.S.; Edvardsen, B.; Eikrem, W.; Sandaa, R.A. Characterisation of three novel giant viruses reveals huge diversity among viruses infecting Prymnesiales (Haptophyta). Virology 2015, 476, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, B.A.; Vladu, I.C.; Barclay, J.E.; Schroeder, D.C.; Malin, G.; Field, R.A. Isolation and characterization of a double stranded DNA megavirus infecting the toxin-producing haptophyte Prymnesium parvum. Viruses 2017, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, K.; Kim, J.-J.; Tomaru, Y.; Takao, Y.; Nagai, S. Isolation and characterization of a novel virus infecting Teleaulax amphioxeia (Cryptophyceae). Plankton Benthos Res. 2009, 4, 122–124. [Google Scholar] [CrossRef]
- Brussaard, C.P.D.; Kempers, R.S.; Kop, A.J.; Riegman, R.; Heldal, M. Virus-like particles in a summer bloom of Emiliania huxleyi in the North Sea. Aquat. Microb. Ecol. 1996, 10, 105–113. [Google Scholar] [CrossRef]
- Moestrup, H.T.; Thomsen, H.A. An ultrastructural study of the flagellate Pyramimonas orientalis with particular emphasis on golgi apparatus activity and the flagellar apparatus. Protoplasma 1974, 81, 247–269. [Google Scholar] [CrossRef]
- La Scola, B.; Desnues, C.; Pagnier, I.; Robert, C.; Barrassi, L.; Fournous, G.; Merchat, M.; Suzan-Monti, M.; Forterre, P.; Koonin, E.; et al. The virophage as a unique parasite of the giant mimivirus. Nature 2008, 455, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Fischer, M.G.; Suttle, C.A. A virophage at the origin of large DNA transposons. Science 2011, 332, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Mojica, K.D.A.; Brussaard, C.P.D. Factors affecting virus dynamics and microbial host-virus interactions in marine environments. FEMS Microbiol. Ecol. 2014, 89, 495–515. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M. The ecology of viruses that infect eukaryotic algae. Environ. Microbiol. 2012, 14, 2253–2271. [Google Scholar] [CrossRef] [PubMed]
- Brussaard, C.P.D.; Short, S.M.; Frederickson, C.M.; Suttle, C.A. Isolation and phylogenetic analysis of novel viruses infecting the phytoplankton Phaeocystis globosa (Prymnesiophyceae). Appl. Environ. Microbiol. 2004, 70, 3700–3705. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Aravind, L.; Koonin, E.V. Common origin of four diverse families of large eukaryotic DNA viruses. J. Virol. 2001, 75, 11720–11734. [Google Scholar] [CrossRef] [PubMed]
- Gallot-Lavallee, L.; Blanc, G.; Claverie, J.M. Comparative genomics of Chrysochromulina ericina virus and other microalga-infecting large DNA viruses highlights their intricate evolutionary relationship with the established Mimiviridae family. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.J.; Schroeder, D.C.; Holden, M.T.G.; Wilson, W.H. Evolutionary history of the Coccolithoviridae. Mol. Biol. Evol. 2006, 23, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Dunigan, D.D.; Fitzgerald, L.A.; Van Etten, J.L. Phycodnaviruses: A peek at genetic diversity. Virus Res. 2006, 117, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Nissimov, J.I.; Pagarete, A.; Ma, F.; Cody, S.; Dunigan, D.D.; Kimmance, S.A.; Allen, M.J. Coccolithoviruses: A review of cross-kingdom genomic thievery and metabolic thuggery. Viruses 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Clerissi, C.; Grimsley, N.; Ogata, H.; Hingamp, P.; Poulain, J.; Desdevises, Y. Unveiling of the diversity of Prasinoviruses (Phycodnaviridae) in marine samples by using high-throughput sequencing analyses of PCR-amplified DNA polymerase and major capsid protein genes. Appl. Environ. Microbiol. 2014, 80, 3150–3160. [Google Scholar] [CrossRef] [PubMed]
- Filee, J. Genomic comparison of closely related giant viruses supports an accordion-like model of evolution. Front. Microbiol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Filee, J.; Pouget, N.; Chandler, M. Phylogenetic evidence for extensive lateral acquisition of cellular genes by nucleocytoplasmic large DNA viruses. BMC Evol. Biol. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Malviya, S.; Scalco, E.; Audic, S.; Vincenta, F.; Veluchamy, A.; Poulain, J.; Wincker, P.; Iudicone, D.; de Vargas, C.; Bittner, L.; et al. Insights into global diatom distribution and diversity in the world’s ocean. Proc. Natl. Acad. Sci. USA 2016, 113, E1516–E1525. [Google Scholar] [CrossRef] [PubMed]
- Kazlauskas, D.; Dayaram, A.; Kraberger, S.; Goldstien, S.; Varsani, A.; Krupovic, M. Evolutionary history of ssDNA Bacilladnaviruses features horizontal acquisition of the capsid gene from ssRNA Nodaviruses. Virology 2017, 504, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Diemer, G.S.; Stedman, K.M. A novel virus genome discovered in an extreme environment suggests recombination between unrelated groups of RNA and DNA viruses. Biol. Direct 2012, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Koonin, E.V. Evolution of eukaryotic single-stranded DNA viruses of the Bidnaviridae family from genes of four other groups of widely different viruses. Sci. Rep. 2014, 4, 5347. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.Q.; Lefkowitz, E.J.; Mushegian, A.R.; Adams, M.J.; Dutilh, B.E.; Gorbalenya, A.E.; Harrach, B.; Harrison, R.L.; Junglen, S.; Knowles, N.J.; et al. Changes to taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses (2018). Arch. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Attoui, H.; Jaafar, F.M.; Belhouchet, M.; de Micco, P.; de Lamballerie, X.; Brussaard, C.P.D. Micromonas pusilla reovirus: A new member of the family reoviridae assigned to a novel proposed genus (Mimoreovirus). J. Gen. Virol. 2006, 87, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Delaroque, N.; Maier, I.; Knippers, R.; Muller, D.G. Persistent virus integration into the genome of its algal host, Ectocarpus siliculosus (Phaeophyceae). J. Gen. Virol. 1999, 80, 1367–1370. [Google Scholar] [CrossRef] [PubMed]
- Nagai, K.; Matsuyama, Y.; Uchida, T.; Yamaguchi, M.; Ishimura, M.; Nishimura, A.; Akamatsu, S.; Honjo, T. Toxicity and LD (50) levels of the red tide dinoflagellate Heterocapsa circularisquama on juvenile pearl oysters. Aquaculture 1996, 144, 149–154. [Google Scholar] [CrossRef]
- Nagasaki, K. Dinoflagellates, diatoms, and their viruses. J. Microbiol. 2008, 46, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Noble, R.T.; Fuhrman, J.A. Use of Sybr green I for rapid epifluorescence counts of marine viruses and bacteria. Aquat. Microb. Ecol. 1998, 14, 113–118. [Google Scholar] [CrossRef]
- Brussaard, C.P.D.; Marie, D.; Bratbak, G. Flow cytometric detection of viruses. J. Virol. Methods 2000, 85, 175–182. [Google Scholar] [CrossRef]
- Brussaard, C.P.D. Optimization of procedures for counting viruses by flow cytometry. Appl. Environ. Microbiol. 2004, 70, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Marie, D.; Brussaard, C.P.D.; Thyrhaug, R.; Bratbak, G.; Vaulot, D. Enumeration of marine viruses in culture and natural samples by flow cytometry. Appl. Environ. Microbiol. 1999, 65, 45–52. [Google Scholar]
- Larsen, J.B.; Larsen, A.; Bratbak, G.; Sandaa, R.A. Phylogenetic analysis of members of the Phycodnaviridae virus family, using amplified fragments of the major capsid protein gene. Appl. Environ. Microbiol. 2008, 74, 3048–3057. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Gann, E.R.; LeCleir, G.R.; Kang, Y.; Gobler, C.J.; Wilhelm, S.W. Diversity and dynamics of algal megaviridae members during a harmful brown tide caused by the pelagophyte, Aureococcus anophagefferens. FEMS Microbiol. Ecol. 2016, 92. [Google Scholar] [CrossRef] [PubMed]
- Kislyuk, A.O.; Haegeman, B.; Bergman, N.H.; Weitz, J.S. Genomic fluidity: An integrative view of gene diversity within microbial populations. BMC Genomics 2011, 12, 10. [Google Scholar] [CrossRef] [PubMed]
- Lefever, S.; Pattyn, F.; Hellemans, J.; Vandesompele, J. Single-nucleotide polymorphisms and other mismatches reduce performance of quantitative PCR assays. Clin. Chem. 2013, 59, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Gulvik, C.A.; Effler, T.C.; Wilhelm, S.W.; Buchan, A. De-metast-blast: A tool for the validation of degenerate primer sets and data mining of publicly available metagenomes. PLoS ONE 2012, 7, e50362. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M.; Suttle, C.A. Sequence analysis of marine virus communities reveals that groups of related algal viruses are widely distributed in nature. Appl. Environ. Microbiol. 2002, 68, 1290–1296. [Google Scholar] [CrossRef] [PubMed]
- Short, S.M.; Short, C.M. Diversity of algal viruses in various North American freshwater environments. Aquat. Microb. Ecol. 2008, 51, 13–21. [Google Scholar] [CrossRef]
- Clasen, J.L.; Suttle, C.A. Identification of freshwater Phycodnaviridae and their potential phytoplankton hosts, using DNA pol sequence fragments and a genetic-distance analysis. Appl. Environ. Microbiol. 2009, 75, 991–997. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.M.; Fabre, M.F.; Gobena, D.; Wilson, W.H.; Wilhelm, S.W. Application of the major capsid protein as a marker of the phylogenetic diversity of Emiliania huxleyi viruses. FEMS Microbiol. Ecol. 2011, 76, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Gilg, I.C.; Duarte, A.; Ogata, H. Development of DNA mismatch repair gene, muts, as a diagnostic marker for detection and phylogenetic analysis of algal megaviruses. Virology 2014, 466, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Culley, A.I.; Steward, G.F. New genera of RNA viruses in subtropical seawater, inferred from polymerase gene sequences. Appl. Environ. Microbiol. 2007, 73, 5937–5944. [Google Scholar] [CrossRef] [PubMed]
- Culley, A. New insight into the RNA aquatic virosphere via viromics. Virus Res. 2018, 244, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B. Viromes, not gene markers, for studying double-stranded DNA virus communities. J. Virol. 2015, 89, 2459–2461. [Google Scholar] [CrossRef] [PubMed]
- Polz, M.F.; Cavanaugh, C.M. Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 1998, 64, 3724–3730. [Google Scholar] [PubMed]
- Short, S.M.; Short, C.M. Quantitative PCR reveals transient and persistent algal viruses in Lake Ontario, Canada. Environ. Microbiol. 2009, 11, 2639–2648. [Google Scholar] [CrossRef] [PubMed]
- Short, C.M.; Rusanova, O.; Short, S.M. Quantification of virus genes provides evidence for seed-bank populations of phycodnaviruses in Lake Ontario, Canada. ISME J. 2011, 5, 810–821. [Google Scholar] [CrossRef] [PubMed]
- Baran, N.; Goldin, S.; Maidanik, I.; Lindell, D. Quantification of diverse virus populations in the environment using the polony method. Nat. Microbiol. 2018, 3, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Tadmor, A.D.; Ottesen, E.A.; Leadbetter, J.R.; Phillips, R. Probing individual environmental bacteria for viruses by using microfluidic digital PCR. Science 2011, 333, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Culley, A.I.; Mueller, J.A.; Belcaid, M.; Wood-Charlson, E.M.; Poisson, G.; Steward, G.F. The characterization of RNA viruses in tropical seawater using targeted PCR and metagenomics. mBio 2014, 5, e01210-14. [Google Scholar] [CrossRef] [PubMed]
- Simmonds, P.; Adams, M.J.; Benko, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Krupovic, M. Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 2018, 163, 2005–2015. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.J.; Lefkowitz, E.J.; King, A.M.Q.; Harrach, B.; Harrison, R.L.; Knowles, N.J.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Mushegian, A.R.; et al. Changes to taxonomy and the international code of virus classification and nomenclature ratified by the international committee on taxonomy of viruses (2017). Arch. Virol. 2017, 162, 2505–2538. [Google Scholar] [CrossRef] [PubMed]
- Wommack, K.E.; Hill, R.T.; Colwell, R.R. A simple method for the concentration of viruses from natural water samples. J. Virol. Methods 1995, 22, 57–67. [Google Scholar] [CrossRef]
- John, S.G.; Mendez, C.B.; Deng, L.; Poulos, B.; Kauffman, A.K.M.; Kern, S.; Brum, J.; Polz, M.F.; Boyle, E.A.; Sullivan, M.B. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 2011, 3, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Solonenko, N.E.; Dang, V.T.; Poulos, B.T.; Schwenk, S.M.; Goldsmith, D.B.; Coleman, M.L.; Breitbare, M.; Sullivan, M.B. Towards quantitative viromics for both double-stranded and single-stranded DNA viruses. PeerJ 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Tomita, R.; Sakamoto, M. Recombinant plant dsRNA-binding protein as an effective tool for the isolation of viral replicative form dsRNA and universal detection of RNA viruses. J. Gen. Plant Pathol. 2009, 75, 87–91. [Google Scholar] [CrossRef]
- Andrews-Pfannkoch, C.; Fadrosh, D.W.; Thorpe, J.; Williamson, S.J. Hydroxyapatite-mediated separation of double-stranded DNA, single-stranded DNA, and RNA genomes from natural viral assemblages. Appl. Environ. Microbiol. 2010, 76, 5039–5045. [Google Scholar] [CrossRef] [PubMed]
- Shatkin, A.J. Animal RNA viruses-genome structure and function. Annu. Rev. Biochem. 1974, 43, 643–665. [Google Scholar] [CrossRef] [PubMed]
- Steward, G.F.; Culley, A. Extraction and purification of nucleic acids from viruses. In Manual of Aquatic Viral Ecology; ASLO: Waco, TX, USA, 2010; pp. 154–165. [Google Scholar]
- Hurwitz, B.L.; Deng, L.; Poulos, B.T.; Sullivan, M.B. Evaluation of methods to concentrate and purify ocean virus communities through comparative, replicated metagenomics. Environ. Microbiol. 2013, 15, 1428–1440. [Google Scholar] [CrossRef] [PubMed]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of virus metagenomic classification methods and their biological applications. Front. Microbiol. 2018, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Skewes-Cox, P.; Sharpton, T.J.; Pollard, K.S.; DeRisi, J.L. Profile Hidden Markov Models for the detection of viruses within metagenomic sequence data. PLoS ONE 2014, 9, e105067. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- Matsen, F.A.; Kodner, R.B.; Armbrust, E.V. Pplacer: Linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinform. 2010, 11. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-tree: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Gallot-Lavallee, L.; Blanc, G. A glimpse of nucleo-cytoplasmic large DNA virus biodiversity through the eukaryotic genomicswindow. Viruses 2017, 9, 17. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Emerson, J.B.; Eloe-Fadrosh, E.A.; Sullivan, M.B. Benchmarking viromics: An in silico evaluation of metagenome-enabled estimates of viral community composition and diversity. PeerJ 2017, 5. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B.; Weitz, J.S.; Wilhelm, S.W. Viral ecology comes of age. Environ. Microbiol. Rep. 2017, 9, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Allers, E.; Moraru, C.; Duhaime, M.B.; Beneze, E.; Solonenko, N.; Barrero-Canosa, J.; Amann, R.; Sullivan, M.B. Single-cell and population level viral infection dynamics revealed by phagefish, a method to visualize intracellular and free viruses. Environ. Microbiol. 2013, 15, 2306–2318. [Google Scholar] [CrossRef] [PubMed]
- Deng, L.; Ignacio-Espinoza, J.C.; Gregory, A.C.; Poulos, B.T.; Weitz, J.S.; Hugenholtz, P.; Sullivan, M.B. Viral tagging reveals discrete populations in Synechococcus viral genome sequence space. Nature 2014, 513, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Weiss, S.; Van Treuren, W.; Lozupone, C.; Faust, K.; Friedman, J.; Deng, Y.; Xia, L.C.; Xu, Z.Z.; Ursell, L.; Alm, E.J.; et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016, 10, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Youens-Clark, K.; Roux, S.; Hurwitz, B.L.; Sullivan, M.B. Ivirus: Facilitating new insights in viral ecology with software and community data sets imbedded in a cyberinfrastructure. ISME J. 2017, 11, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Flaviani, F.; Schroeder, D.C.; Balestreri, C.; Schroeder, J.L.; Moore, K.; Paszkiewicz, K.; Pfaff, M.C.; Rybicki, E.P. A pelagic microbiome (viruses to protists) from a small cup of seawater. Viruses 2017, 9, 47. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.H.; Gilg, I.C.; Moniruzzaman, M.; Field, E.K.; Koren, S.; LeCleir, G.R.; Martinez, J.M.; Poulton, N.J.; Swan, B.K.; Stepanauskas, R.; et al. Genomic exploration of individual giant ocean viruses. ISME J. 2017, 11, 1736–1745. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Hernandez, F.; Fornas, O.; Gomez, M.L.; Bolduc, B.; de la Cruz Pena, M.J.; Martinez, J.M.; Anton, J.; Gasol, J.M.; Rosselli, R.; Rodriguez-Valera, F.; et al. Single-virus genomics reveals hidden cosmopolitan and abundant viruses. Nat. Commun. 2017, 8, 15892. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.N.; Li, Y.; Que, Q.D.; Bhattacharya, M.; Lane, L.C.; Chaney, W.G.; Vanetten, J.L. Evidence for virus-encoded glycosylation specificity. Proc. Natl. Acad. Sci. USA 1993, 90, 3840–3844. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L.; Gurnon, J.R.; Yanai-Balser, G.M.; Dunigan, D.D.; Graves, M.V. Chlorella viruses encode most, if not all, of the machinery to glycosylate their glycoproteins independent of the endoplasmic reticulum and golgi. Biochim. Biophys. Acta 2010, 1800, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Van Etten, J.L.; Schuster, A.M.; Girton, L.; Burbank, D.E.; Swinton, D.; Hattman, S. DNA methylation of viruses infecting a eukaryotic Chlorella-like green alga. Nucleic Acids Res. 1985, 13, 3471–3478. [Google Scholar] [CrossRef] [PubMed]
- Agarkova, I.V.; Dunigan, D.D.; Van Etten, J.L. Virion-associated restriction endonucleases of chloroviruses. J. Virol. 2006, 80, 8114–8123. [Google Scholar] [CrossRef] [PubMed]
- Agarkova, I.; Dunigan, D.; Gurnon, J.; Greiner, T.; Barres, J.; Thiel, G.; Van Etten, J.L. Chlorovirus-mediated membrane depolarization of Chlorella alters secondary active transport of solutes. J. Virol. 2008, 82, 12181–12190. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.M.; Jeanniard, A.; Gurnon, J.R.; Xia, Y.N.; Dunigan, D.D.; Van Etten, J.L.; Blanc, G. Global analysis of Chlorella variabilis NC64A mRNA profiles during the early phase of Paramecium bursaria chlorella virus-1 infection. PLoS ONE 2014, 9, e90988. [Google Scholar] [CrossRef] [PubMed]
- DeLong, J.P.; Al-Ameeli, Z.; Duncan, G.; Van Etten, J.L.; Dunigan, D.D. Predators catalyze an increase in chloroviruses by foraging on the symbiotic hosts of zoochlorellae. Proc. Natl. Acad. Sci. USA 2016, 113, 13780–13784. [Google Scholar] [CrossRef] [PubMed]
- DeLong, J.P.; Al-Ameeli, Z.; Lyon, S.; Van Etten, J.L.; Dunigan, D.D. Size-dependent catalysis of chlorovirus population growth by a messy feeding predator. Microb. Ecol. 2018, 75, 847–853. [Google Scholar] [CrossRef] [PubMed]
- Long, A.M.; Short, S.M. Seasonal determinations of algal virus decay rates reveal overwintering in a temperate freshwater pond. ISME J. 2016, 10, 1602–1612. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
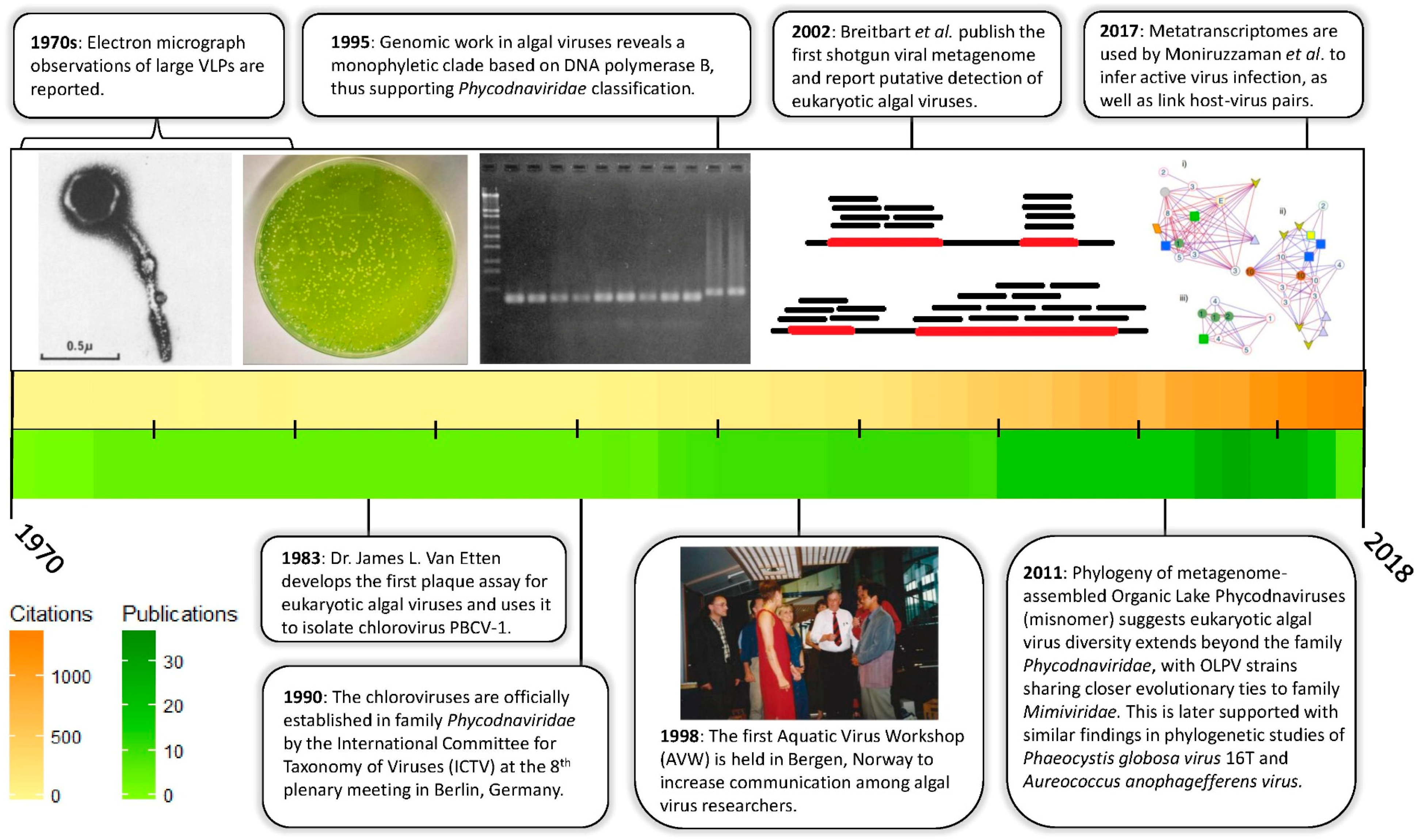{kind=link}
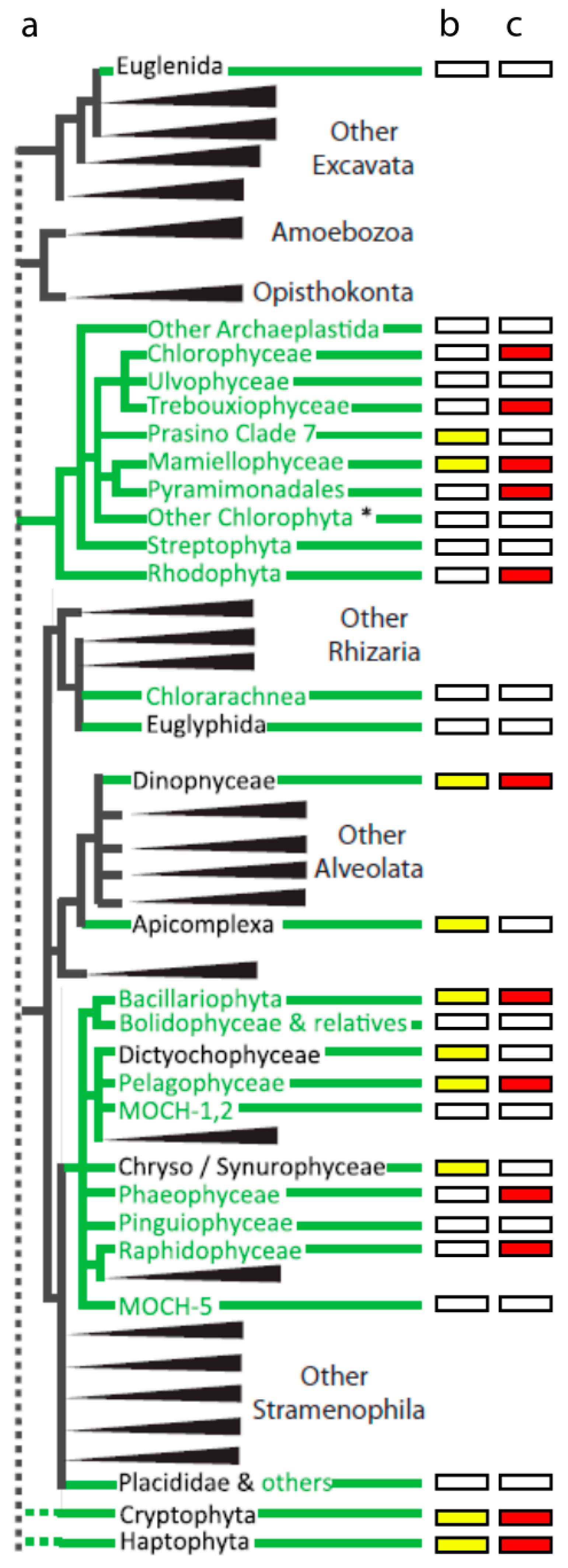{kind=link}
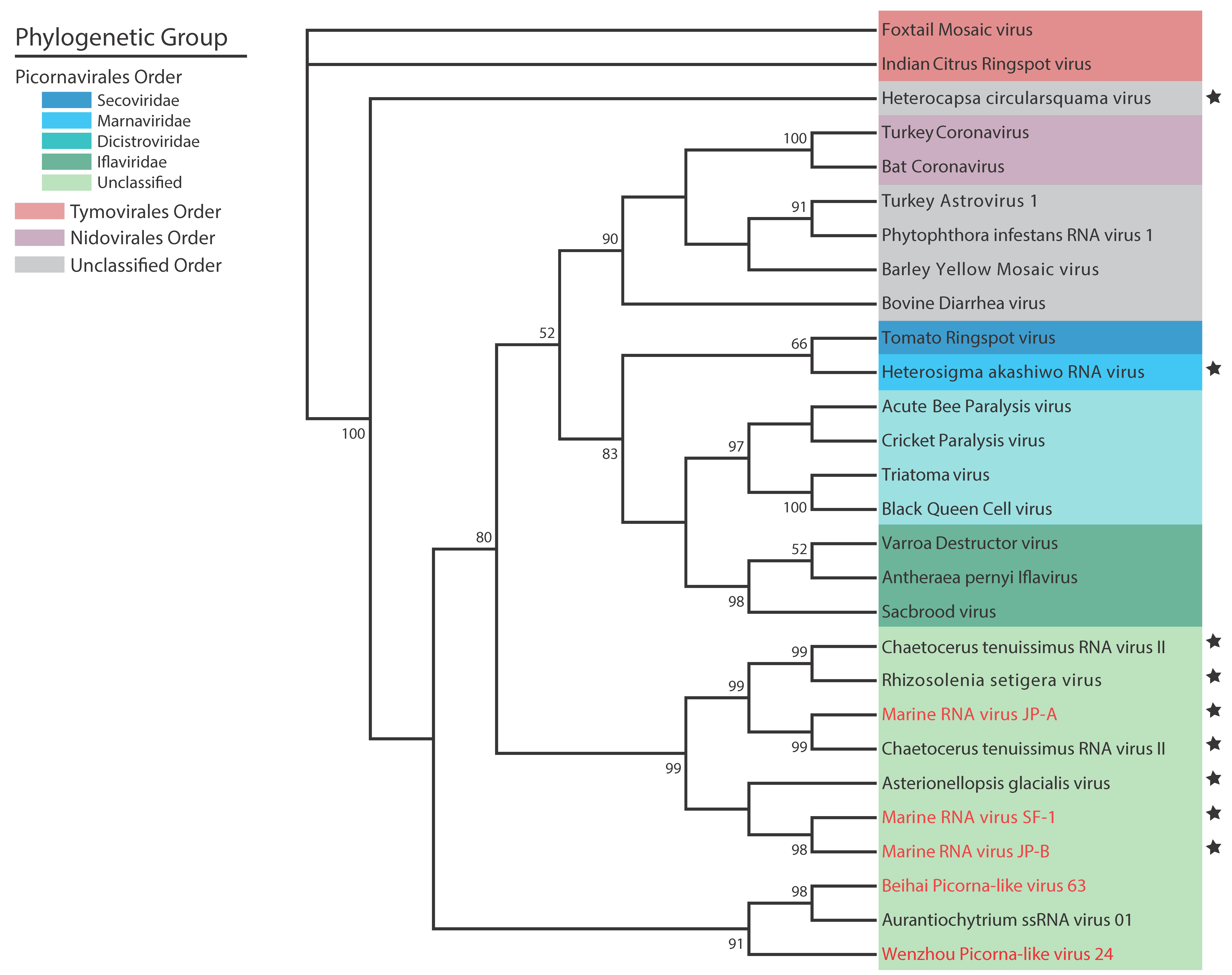{kind=link}
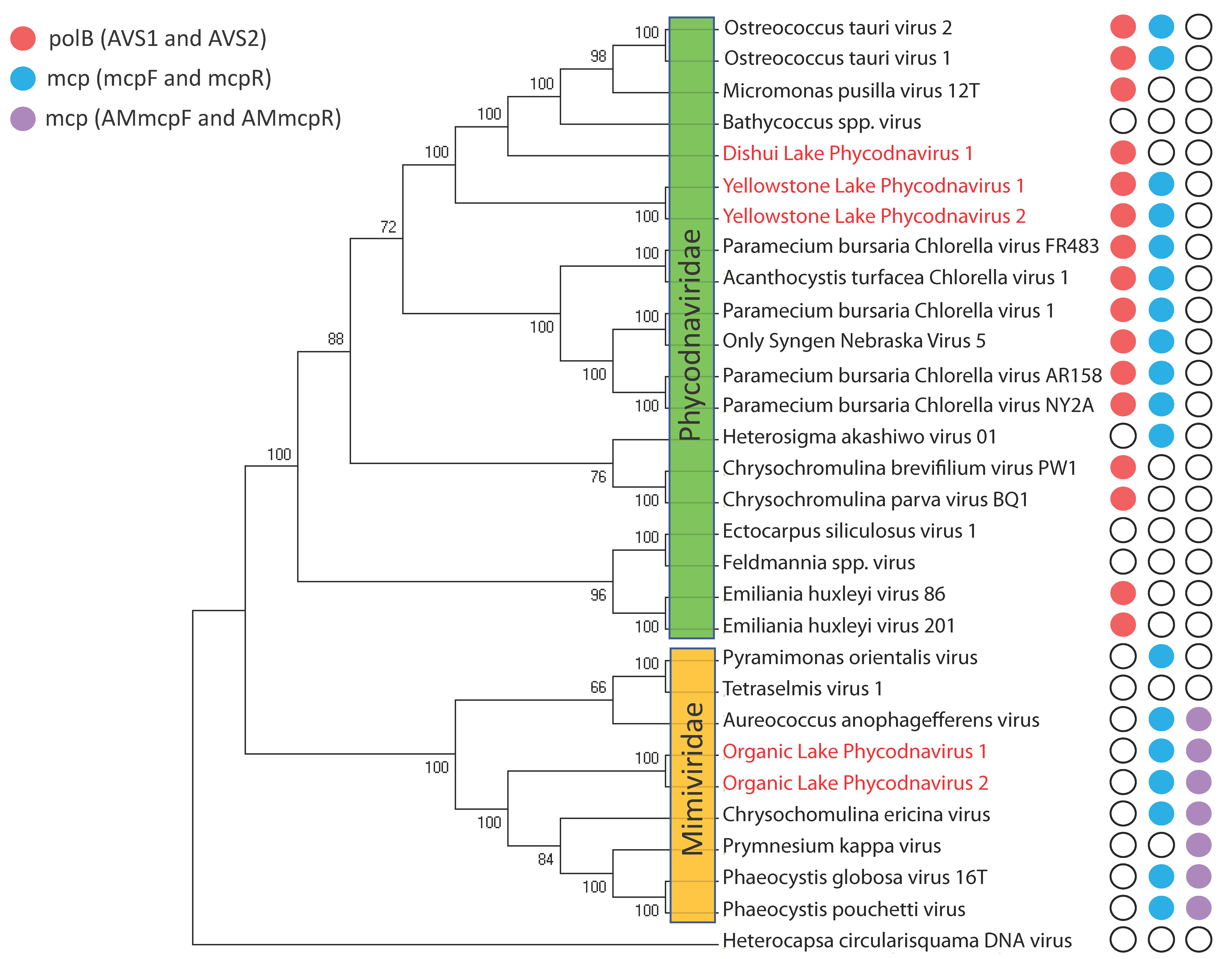{kind=link}
{kind=link}
| Host Algae | Type | Size (kbp or knt) | Code | References |
|---|---|---|---|---|
| Chlorophyceaea | ||||
| Tetraselmis spp. | dsDNA | 668 | TetV | Schvarcz et al., 2018 [45] |
| Tetraselmis striata | dsDNA | 31 | Tsv-N1 | Pagarete et al., 2015 [46] |
| Trebouxiophyceae | ||||
| Chlorella variabilis NC64A | dsDNA | 287–369 | PBCV-1 | Jeanniard et al., 2013 [17] |
| Chlorella variabilis Syngen 2-3 | dsDNA | 327 | OSy-NE5 | Quispe et al., 2017 [18] |
| Chlorella heliozoae SAG 3.83 | dsDNA | 288–327 | ATCV-1 | Jeanniard et al., 2013 [17] |
| Micratinium conductrix Pbi | dsDNA | 302–329 | CVM | Jeanniard et al., 2013 [17] |
| Mamiellophyceae | ||||
| Ostreococcus lucimarinus | dsDNA | 182–196 | OlV1 | Derelle et al., 2015 [47] |
| Ostreococcus tauri | dsDNA | 184–192 | OtV5 | Weynberg et al., 2011 [48] |
| Ostreococcus mediterraneus | dsDNA | 193 | OmV1 | Derelle et al., 2015 [47] |
| Bathycoccus sp. RCC1105 | dsDNA | 187–198 | BpV | Moreau et al., 2010 [49] |
| Micromonas pusilla CCMP1545 | dsDNA | 186–195 | MpV-02T | Martinez Martinez et al., 2015 [50] |
| Micromonas pusilla LAC38 | dsDNA | 173–205 | MpV1 | Finke et al., 2017 [51] |
| Micromonas pusilla LAC38 | dsRNA | 25.5 | MpRV | Brussaard et al., 2004 [52] |
| Micromonas polaris | dsDNA | 191–205 | MpoV | Maat et al., 2017 [53] |
| Pyramimonadales | ||||
| Pyramimonas orientalis | dsDNA | 560 | PoV | Sandaa et al., 2001 [54] |
| Rhodophyta | ||||
| Chondrus crispus | dsRNA | 6 | CcV | Rousvoal et al., 2016 [55] |
| Dinophyceaea | ||||
| Heterocapsa circularisquama | dsDNA | 356 | HcDNAV | Ogata et al., 2009 [56] |
| Heterocapsa circularisquama | ssRNA | 4.4 | HcRNAV | Tomaru et al., 2004 [57] |
| Heterocapsa pygmea | dsDNA | ND | HpygDNAV | Kim et al., 2012 [58] |
| Gymnodinium mikimotoi | ND | ND | GM6/GM7 | Onji et al., 2003 [59] |
| Bacillariophyta | ||||
| Chaetoceros cf. gracilise | ND | ND | CspNIV | Bettarel et al., 2005 [60] |
| Chaetoceros salsugineum | ssDNA | 6 | CsalDNAV* | Nagasaki et al., 2005 [61] |
| Chaetoceros setoensis | ssDNA | 5.8 | CsetDNAV* | Tomaru et al., 2013 [62] |
| Chaetoceros socialis f. radians | ssRNA | 9.4 | CsfrRNAV | Tomaru et al., 2009b [63] |
| Chaetoceros lorenzianus | ssDNA | 5.8 | ClorDNAV* | Tomaru et al., 2011 [64] |
| Chaetoceros tenuissimus | ssDNA | 5.6 | CtenDNAV-I* | Tomaru et al., 2011 [65] |
| Chaetoceros tenuissimus | ssDNA | 5.6 | CtenDNAV-II* | Kimura and Tomaru 2015 [66] |
| Chaetoceros tenuissimus | ssRNA | 9.4 | CtenRNAV | Shirai et al., 2008 [67] |
| Chaetoceros tenuissimus, Chaetoceros spp. | ssRNA | 9.6 | CtenRNAV-II | Kimura and Tomaru 2015 [66] |
| Chaetoceros spp. SS628-11 | ssDNA | 5.5 | Csp07DNAV* | Kimura et al., 2013 [68] |
| Chaetoceros spp. TG07-C28 | ssDNA | ND | Csp05DNAV | Toyoda et al., 2012 [69] |
| Chaetoceros debilis | ssDNA | ND | CdebDNAV | Tomaru et al., 2008 [70] |
| Chaetoceros sp. SS08-C03 | ssRNA | 9.4 | Csp03RNAV | Tomaru et al., 2013 [71] |
| Chaetoceros cf. wighamii | ssDNA | 7-8 | CwNIV | Eissler et al., 2009 [72] |
| Asterionellopsis glacialis | ssRNA | 9.5 | AglaRNAV | Tomaru et al., 2012 [73] |
| Thalassionema nitzschioides | ssDNA | 5.5 | TnitDNAV | Tomaru et al., 2012 [73] |
| Rhizosolenia setigera | ssRNA | 11.2 | RsetRNAV | Nagasaki et al., 2004 [74] |
| Skeletonema costatum | ND | ND | ScosV | Kim et al., 2015 [75] |
| Stephanopyxis palmeriana | ND | ND | SpalV | Kim et al., 2015 [76] |
| Pelagophyceae | ||||
| Aureococcus anophagefferens | dsDNA | 370 | AaV | Moniruzzaman et al., 2014 [23] |
| Phaeophyceae | ||||
| Ectocarpus fasciculatus | dsDNA | 340 | EfasV | Kapp et al., 1997 [77] |
| Ectocarpus siliculosus | dsDNA | 320 | EsV | Kapp et al., 1997 [77] |
| Feldmannia irregularis | dsDNA | 180 | FirrV | Kapp et al., 1997 [77] |
| Feldmannia simplex | dsDNA | 220 | FlexV | Kapp et al., 1997 [77] |
| Feldmannia species | dsDNA | 170 | FsV | Henry and Meints 1992 [78] |
| Hincksia hinckiae | dsDNA | 240 | HincV | Kapp et al., 1997 [77] |
| Myriotrichia clavaeformis | dsDNA | 320 | MclaV | Kapp et al., 1997 [77] |
| Pilayella littoralis | dsDNA | 280 | PlitV | Maier et al., 1998 [79] |
| Raphidophyceae | ||||
| Heterosigma akashiwo | dsDNA | ND | HaV | Nagasaki et al., 1997 [80] |
| Heterosigma akashiwo | dsDNA | 180 | O1s1 | Lawrence et al., 2006 [81] |
| Heterosigma akashiwo | ssRNA | 9.1 | HaRNAV | Tai et al., 2003 [82] |
| Heterosigma akashiwo | ND | ND | HaNIV | Lawrence et al., 2001 [83] |
| Haptophyta | ||||
| Emiliania huxleyi | dsDNA | 415 | EhV | Castberg et al., 2002 [84] |
| Phaeocystis globosa | dsDNA | 466 | PgV-16T (Group I) | Baudoux et al., 2005 [85] |
| Phaeocystis globosa | dsDNA | 177 | PgV-03T (Group II) | Baudoux et al., 2005 [85] |
| Phaeocystis globosa | dsDNA | 176 | PgV-102P | Wilson et al., 2006 [86] |
| Phaeocystis pouchetii | dsDNA | 485 | PpV | Jacobsen et al., 1996 [87] |
| Chrysochromulina brevifilum, Chrysochromulina strobilus | dsDNA | ND | CbV | Suttle and Chan 1995 [88] |
| Chrysochromulina ericina | dsDNA | 510 | CeV | Sandaa et al., 2001 [54] |
| Chrysochromulina parva | dsDNA | 485 | CpV | Mirza et al., 2015 [89] |
| Haptolina ericina, Prymnesium kappa | dsDNA | 530 | HeV-RF02 | Johannessen et al., 2015 [90] |
| Prymnesium kappa, Haptolina ericina | dsDNA | ND | PkV-RF01 | Johannessen et al., 2015 [90] |
| Prymnesium kappa | dsDNA | 507 | PkV-RF02 | Johannessen et al., 2015 [90] |
| Prymnesium parvum | dsDNA | ND | PpDNAV | Wagstaff et al., 2017 [91] |
| Cryptophyta | ||||
| Teleaulax amphioxeia | ND | ND | TampV | Nagasaki et al., 2009 [92] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coy, S.R.; Gann, E.R.; Pound, H.L.; Short, S.M.; Wilhelm, S.W. Viruses of Eukaryotic Algae: Diversity, Methods for Detection, and Future Directions. Viruses 2018, 10, 487. https://doi.org/10.3390/v10090487
Coy SR, Gann ER, Pound HL, Short SM, Wilhelm SW. Viruses of Eukaryotic Algae: Diversity, Methods for Detection, and Future Directions. Viruses. 2018; 10(9):487. https://doi.org/10.3390/v10090487
Chicago/Turabian StyleCoy, Samantha R., Eric R. Gann, Helena L. Pound, Steven M. Short, and Steven W. Wilhelm. 2018. "Viruses of Eukaryotic Algae: Diversity, Methods for Detection, and Future Directions" Viruses 10, no. 9: 487. https://doi.org/10.3390/v10090487
APA StyleCoy, S. R., Gann, E. R., Pound, H. L., Short, S. M., & Wilhelm, S. W. (2018). Viruses of Eukaryotic Algae: Diversity, Methods for Detection, and Future Directions. Viruses, 10(9), 487. https://doi.org/10.3390/v10090487