A Rapid Method for Sequencing Double-Stranded RNAs Purified from Yeasts and the Identification of a Potent K1 Killer Toxin Isolated from Saccharomyces cerevisiae

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Double-Stranded RNA Extraction
2.2. Sequencing Sample Preparation
2.3. Illumina Library Preparation Using a Modified Nextera Protocol
2.4. Sequencing
2.5. Bioinformatics Analysis
2.6. Cloning of dsRNAs
2.7. Killer Toxin Assays
2.8. Verifying the Presence of DsRNA Elements in S. cerevisiae BJH001 by Reverse Transcriptase-PCR
3. Results
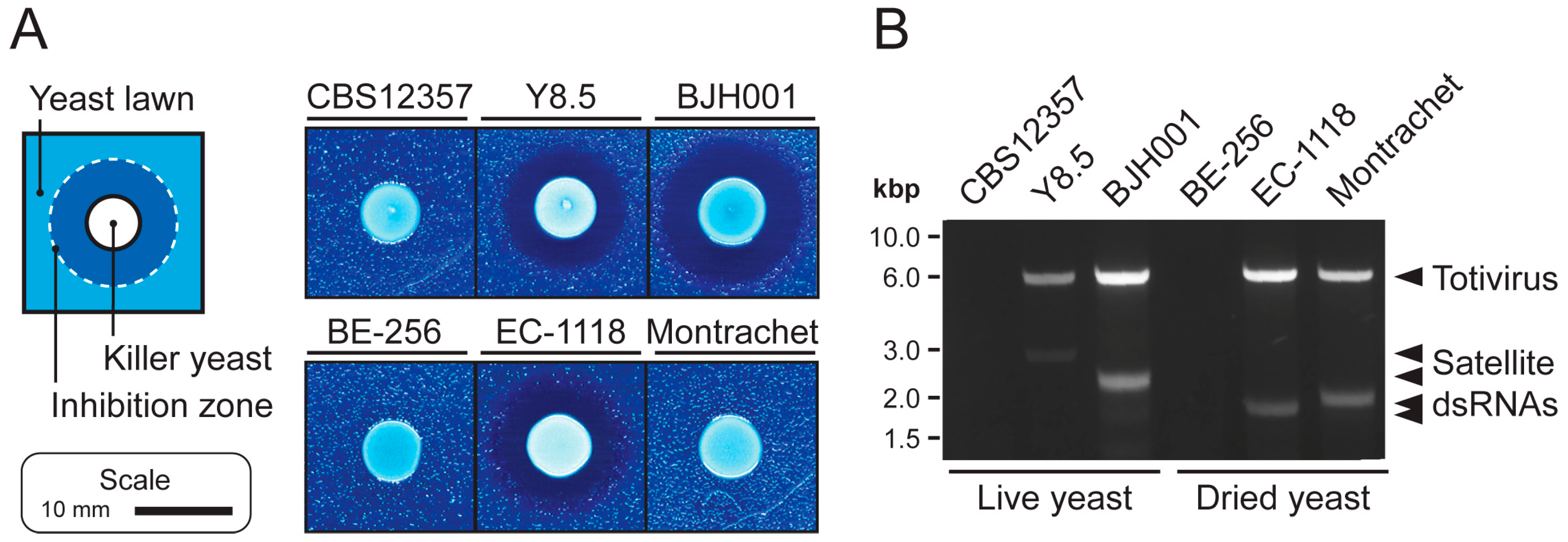
3.1. Extraction of High-Quality dsRNAs from Saccharomyces Yeasts
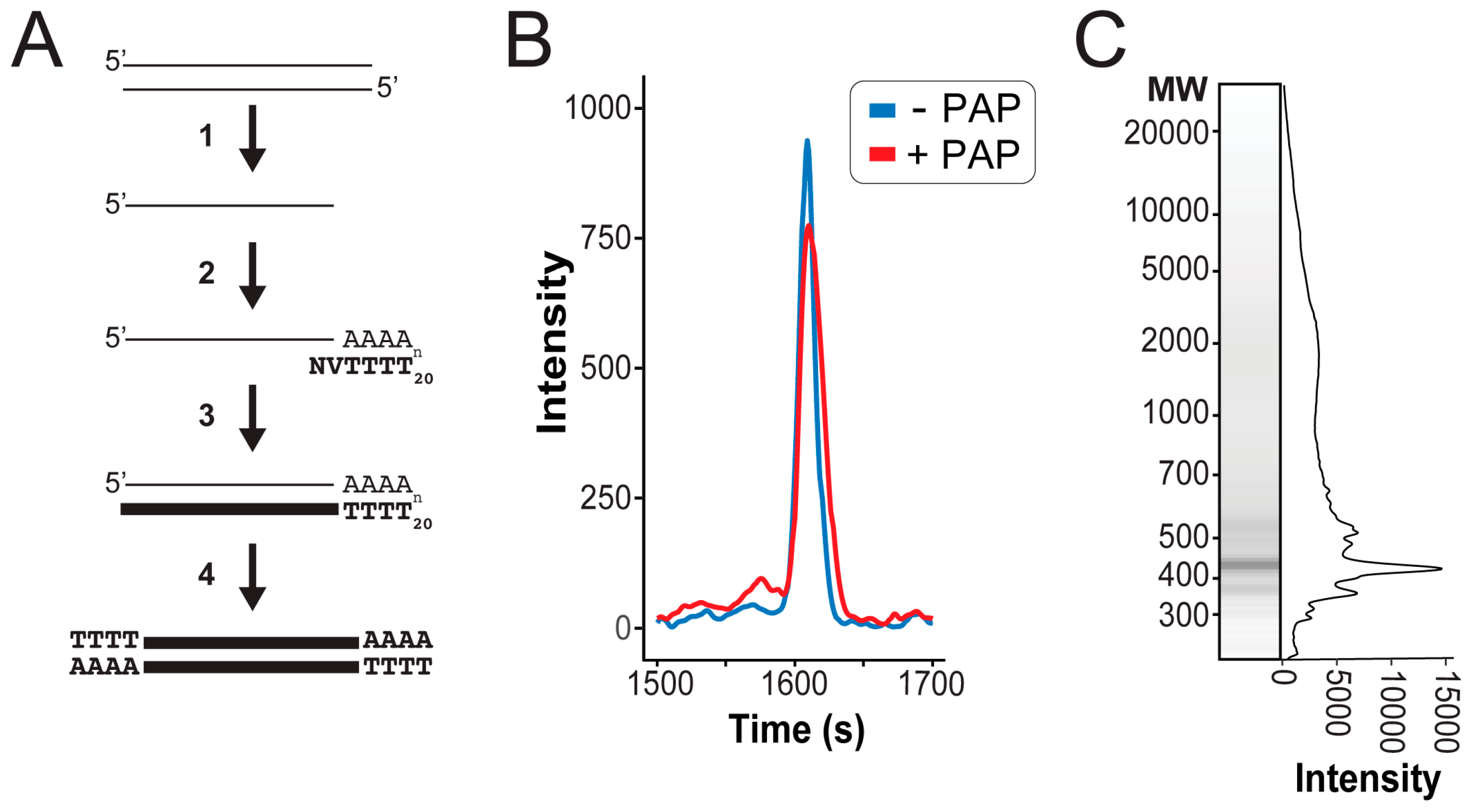
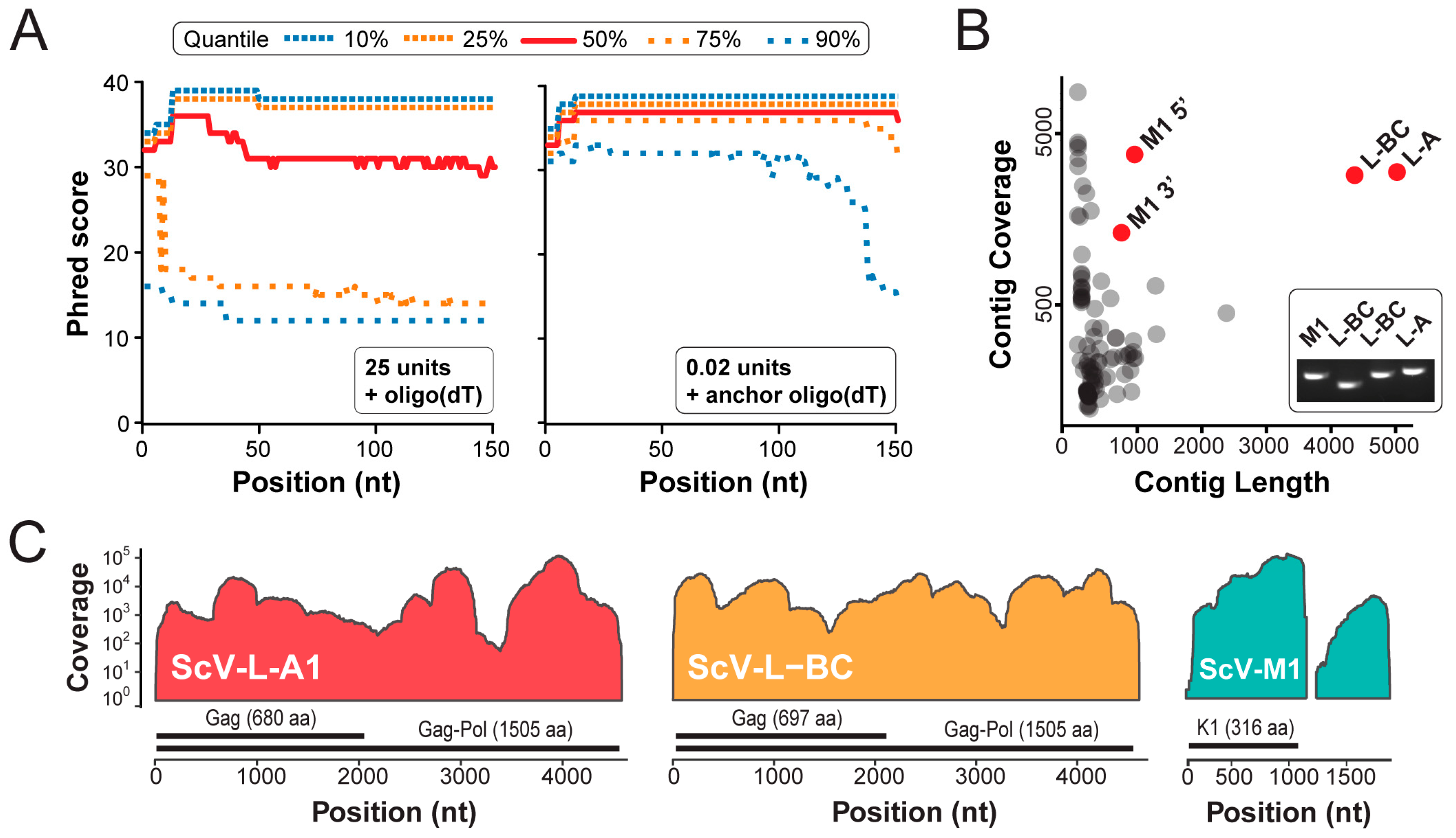
3.2. Next Generation Sequencing of cDNAs Derived from dsRNAs
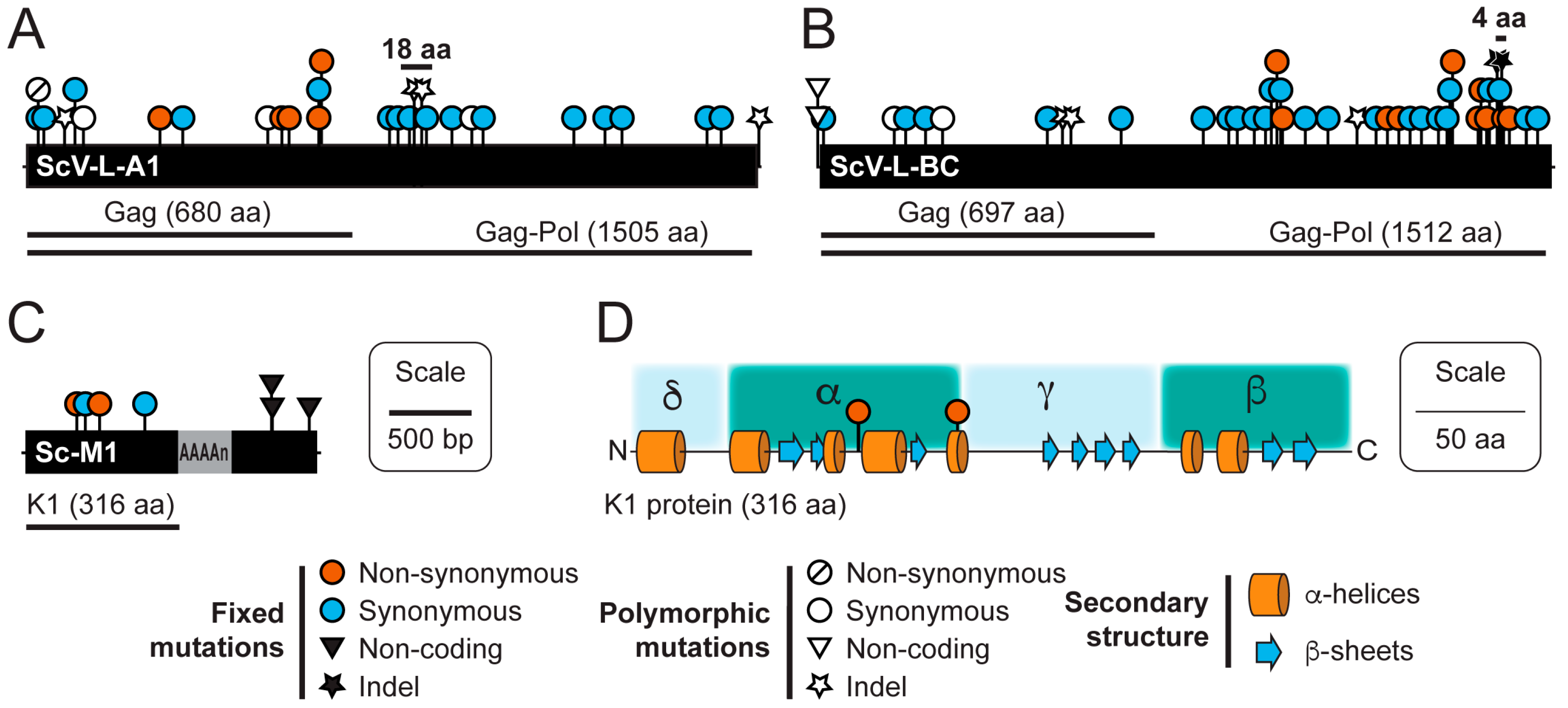
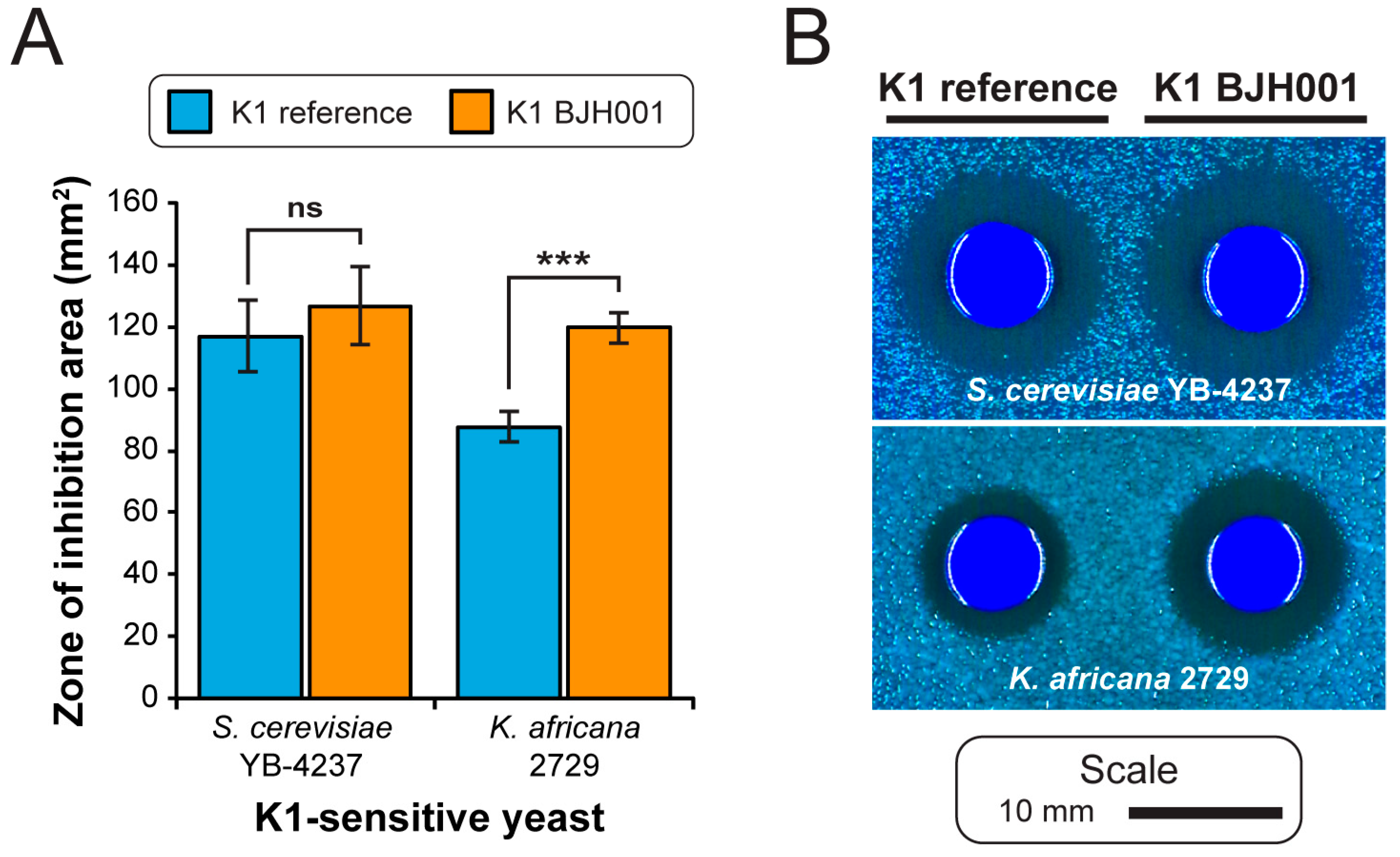
3.3. Sequence Variation in dsRNAs Identified by NGS
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ghabrial, S.A.; Suzuki, N. Viruses of plant pathogenic fungi. Annu. Rev. Phytopathol. 2009, 47, 353–384. [Google Scholar] [CrossRef] [PubMed]
- Bozarth, R.F. Mycoviruses: A new dimension in microbiology. Environ. Health Perspect. 1972, 2, 23–39. [Google Scholar] [CrossRef]
- Milgroom, M.G.; Hillman, B.I. Studies in Viral Ecology: Microbial and Botanical Host Systems, 1st ed.; Hurst, C.J., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2011; Volume 1, pp. 217–253. [Google Scholar]
- Nuss, D.L. Hypovirulence: Mycoviruses at the fungal–plant interface. Nat. Rev. Microbiol. 2005, 3, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.J.; Breinig, F. Yeast viral killer toxins: Lethality and self-protection. Nat. Rev. Microbiol. 2006, 4, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Márquez, L.M.; Redman, R.S.; Rodriguez, R.J.; Roossinck, M.J. A virus in a fungus in a plant: Three-way symbiosis required for thermal tolerance. Science 2007, 315, 513–515. [Google Scholar] [CrossRef] [PubMed]
- Franklin, R.M. Purification and properties of the replicative intermediate of the RNA bacteriophage R17. Proc. Natl. Acad. Sci. USA 1966, 55, 1504–1511. [Google Scholar] [CrossRef]
- Kobayashi, K.; Tomita, R.; Sakamoto, M. Recombinant plant dsRNA-binding protein as an effective tool for the isolation of viral replicative form dsRNA and universal detection of RNA viruses. J. Gen. Plant Pathol. 2009, 75, 87–91. [Google Scholar] [CrossRef]
- Cashdollar, L.W.; Esparza, J.; Hudson, G.R.; Chmelo, R.; Lee, P.W.; Joklik, W.K. Cloning the double-stranded RNA genes of reovirus: Sequence of the cloned S2 gene. Proc. Natl. Acad. Sci. USA 1982, 79, 7644–7648. [Google Scholar] [CrossRef]
- Fried, H.M.; Fink, G.R. Electron microscopic heteroduplex analysis of “killer” double-stranded RNA species from yeast. Proc. Natl. Acad. Sci. USA 1978, 75, 4224–4228. [Google Scholar] [CrossRef]
- Lambden, P.R.; Cooke, S.J.; Caul, E.O.; Clarke, I.N. Cloning of noncultivatable human rotavirus by single primer amplification. J. Virol. 1992, 66, 1817–1822. [Google Scholar]
- Ramìrez, M.; Velázquez, R.; Maqueda, M.; López-Piñeiro, A.; Ribas, J.C. A new wine Torulaspora delbrueckii killer strain with broad antifungal activity and its toxin-encoding double-stranded RNA virus. Front. Microbiol. 2015, 6, 403–412. [Google Scholar] [CrossRef] [PubMed]
- Ramìrez, M.; Velázquez, R.; López-Piñeiro, A.; Naranjo, B.; Roig, F.; Llorens, C. New Insights into the Genome Organization of Yeast Killer Viruses Based on “Atypical” Killer Strains Characterized by High-Throughput Sequencing. Toxins 2017, 9, 292. [Google Scholar] [CrossRef] [PubMed]
- Brizzard, B.L.; de Kloet, S.R. Reverse transcription of yeast double-stranded RNA and ribosomal RNA using synthetic oligonucleotide primers. Biochim. Biophys. Acta 1983, 739, 122–131. [Google Scholar] [CrossRef]
- Hagenbüchle, O.; Santer, M.; Steitz, J.A.; Mans, R.J. Conservation of the primary structure at the 3′ end of 18S rRNA from eucaryotic cells. Cell 1978, 13, 551–563. [Google Scholar] [CrossRef]
- Taniguchi, T.; Palmieri, M.; Weissmann, C. QB DNA-containing hybrid plasmids giving rise to QB phage formation in the bacterial host. Nature 1978, 274, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Both, G.W.; Bellamy, A.R.; Street, J.E.; Siegman, L.J. A general strategy for cloning double-stranded RNA: Nucleotide sequence of the Simian-11 rotavirus gene 8. Nucleic Acids Res. 1982, 10, 7075–7088. [Google Scholar] [CrossRef] [PubMed]
- Ghibelli, L.; Usala, S.J.; Mukhopadhyay, R.; Haselkorn, R. Polyadenylation and reverse transcription of bacteriophage φ6 double-stranded RNA. Virology 1982, 120, 318–328. [Google Scholar] [CrossRef]
- Grybchuk, D.; Akopyants, N.S.; Kostygov, A.Y.; Konovalovas, A.; Lye, L.-F.; Dobson, D.E.; Zangger, H.; Fasel, N.; Butenko, A.; Frolov, A.O.; et al. Viral discovery and diversity in trypanosomatid protozoa with a focus on relatives of the human parasite Leishmania. Proc. Natl. Acad. Sci. USA 2018, 115, E506–E515. [Google Scholar] [CrossRef]
- Imai, M.; Richardson, M.A.; Ikegami, N.; Shatkin, A.J.; Furuichi, Y. Molecular cloning of double-stranded RNA virus genomes. Proc. Natl. Acad. Sci. USA 1983, 80, 373–377. [Google Scholar] [CrossRef]
- Potgieter, A.C.; Page, N.A.; Liebenberg, J.; Wright, I.M.; Landt, O.; van Dijk, A.A. Improved strategies for sequence-independent amplification and sequencing of viral double-stranded RNA genomes. J. Gen. Virol. 2009, 90, 1423–1432. [Google Scholar] [CrossRef] [Green Version]
- Bobek, L.A.; Bruenn, J.A.; Field, L.J.; Gross, K.W. Cloning of cDNA to a yeast viral double-stranded RNA and comparison of three viral RNAs. Gene 1982, 19, 225–230. [Google Scholar] [CrossRef]
- Roossinck, M.J.; Saha, P.; Wiley, G.B.; Quan, J.; White, J.D.; Lai, H.; Chavarría, F.; Shen, G.; Roe, B.A. Ecogenomics: Using massively parallel pyrosequencing to understand virus ecology. Mol. Ecol. 2010, 19, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Decker, C.J.; Parker, R. Analysis of double-stranded RNA from microbial communities identifies double-stranded RNA virus-like elements. Cell Rep. 2014, 7, 898–906. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, B.; Freeborough, M.-J.; Maree, H.J.; Celton, J.-M.; Rees, D.J.G.; Burger, J.T. Deep sequencing analysis of viruses infecting grapevines: Virome of a vineyard. Virology 2010, 400, 157–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackevicien, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef] [PubMed]
- Baym, M.; Kryazhimskiy, S.; Lieberman, T.D.; Chung, H.; Desai, M.M.; Kishony, R. Inexpensive multiplexed library preparation for megabase-sized genomes. PLoS ONE 2015, 10, e0128036. [Google Scholar] [CrossRef]
- Okada, R.; Kiyota, E.; Moriyama, H.; Fukuhara, T.; Natsuaki, T. A simple and rapid method to purify viral dsRNA from plant and fungal tissue. J. Gen. Plant Pathol. 2015, 81, 103–107. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Alberti, S.; Gitler, A.D.; Lindquist, S. A suite of Gateway cloning vectors for high-throughput genetic analysis in Saccharomyces cerevisiae. Yeast 2007, 24, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Rowley, P.A.; Ho, B.; Bushong, S.; Johnson, A.; Sawyer, S.L. XRN1 Is a Species-Specific Virus Restriction Factor in Yeasts. PLoS Pathog. 2016, 12, e1005890. [Google Scholar] [CrossRef]
- Chang, S.-L.; Leu, J.-Y.; Chang, T.-H. A Population Study of Killer Viruses Reveals Different Evolutionary Histories of Two Closely Related Saccharomyces sensu stricto Yeasts. Mol. Ecol. 2015, 24, 4312–4322. [Google Scholar] [CrossRef]
- Rodríguez-Cousiño, N.; Gómez, P.; Esteban, R. Variation and Distribution of L-A Helper Totiviruses in Saccharomyces sensu stricto Yeasts Producing Different Killer Toxins. Toxins 2017, 9, 313. [Google Scholar] [CrossRef] [PubMed]
- Rowley, P.A. The frenemies within: Viruses, retrotransposons and plasmids that naturally infect Saccharomyces yeasts. Yeast 2017, 49, 111–292. [Google Scholar] [CrossRef] [PubMed]
- Russell, P.J.; Bennett, A.M.; Love, Z.; Baggott, D.M. Cloning, sequencing and expression of a full-length cDNA copy of the M1 double-stranded RNA virus from the yeast, Saccharomyces cerevisiae. Yeast 1997, 13, 829–836. [Google Scholar] [CrossRef]
- Shapouri, M.R.; Kane, M.; Letarte, M.; Bergeron, J.; Arella, M.; Silim, A. Cloning, sequencing and expression of the S1 gene of avian reovirus. J. Gen. Virol. 1995, 76 Pt 6, 1515–1520. [Google Scholar] [CrossRef] [Green Version]
- Rodríguez-Cousiño, N.; Esteban, R. Relationships and Evolution of Double-Stranded RNA Totiviruses of Yeasts Inferred from Analysis of L-A-2 and L-BC Variants in Wine Yeast Strain Populations. Appl. Environ. Microbiol. 2017, 83, e02991-16. [Google Scholar] [CrossRef]
- Bruenn, J.A. A structural and primary sequence comparison of the viral RNA-dependent RNA polymerases. Nucleic Acids Res. 2003, 31, 1821–1829. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef]
- Lau, M.M.; Neufeld, E.F. A frameshift mutation in a patient with Tay-Sachs disease causes premature termination and defective intracellular transport of the alpha-subunit of beta-hexosaminidase. J. Biol. Chem. 1989, 264, 21376–21380. [Google Scholar] [PubMed]
- White, M.B.; Amos, J.; Hsu, J.; Gerrard, B.; Finn, P.; Dean, M. A Frame-Shift Mutation in the Cystic-Fibrosis Gene. Nature 1990, 344, 665–667. [Google Scholar] [CrossRef] [PubMed]
- Raes, J.; Van de Peer, Y. Functional divergence of proteins through frameshift mutations. Trends Genet. 2005, 21, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Bussey, H. Mutational analysis of the functional domains of yeast K1 killer toxin. Mol. Cell. Biol. 1991, 11, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Rosini, G. The Occurrence of Killer Characters in Yeasts. Can. J. Microbiol. 1983, 29, 1462–1464. [Google Scholar] [CrossRef] [PubMed]
- Young, T.W.; Yagiu, M. A comparison of the killer character in different yeasts and its classification. Antonie Van Leeuwenhoek 1978, 44, 59–77. [Google Scholar] [CrossRef]
- Kandel, J.S.; Stern, T.A. Killer Phenomenon in Pathogenic Yeast. Antimicrob. Agents Chemother. 1979, 15, 568–571. [Google Scholar] [CrossRef] [Green Version]
- Philliskirk, G.; Young, T.W. The occurrence of killer character in yeasts of various genera. Antonie Van Leeuwenhoek 1975, 41, 147–151. [Google Scholar] [CrossRef]
- Starmer, W.T.; Ganter, P.F.; Aberdeen, V. Geographic distribution and genetics of killer phenotypes for the yeast Pichia kluyveri across the United States. Appl. Environ. Microbiol. 1992, 58, 990–997. [Google Scholar]
- Stumm, C.; Hermans, J.; Middelbeek, E.J.; Croes, A.F.; de Vries, G. Killer-Sensitive Relationships in Yeasts From Natural Habitats. Antonie Van Leeuwenhoek 1977, 43, 125–128. [Google Scholar] [CrossRef]
- Pieczynska, M.D.; de Visser, J.A.G.M.; Korona, R. Incidence of symbiotic dsRNA “killer” viruses in wild and domesticated yeast. FEMS Yeast Res. 2013, 13, 856–859. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Criteria | 25 Units Poly(A) Polymerase | 0.02 Units Poly(A) Polymerase | Fold Increase |
|---|---|---|---|
| Number of reads | 22,521 | 471,742 | 21 |
| Homopolymeric reads | 68% | 0.6% | 0.01 |
| De novo assembled contigs > 100nt | 21 | 98 | 5 |
| Length of largest contig | 925 | 5022 | 5 |
| Mean coverage of viral contigs | 1 | 610 | 610 |
| dsRNA Element | Start Position (bp) | Polymorphism Type | Mutation | Coding Change | Coverage | Freq. (%) |
|---|---|---|---|---|---|---|
| ScV-L-A1 | 89 | SNP (transition) | U to C | Non-synonymous | 1002 | 5.8 |
| ScV-L-A1 | 269 | Indel | Insertion U | Gag truncation | 1332 | 5.4 |
| ScV-L-A1 | 407 | SNP (transition) | C to U | Synonymous | 848 | 31.8 |
| ScV-L-A1 | 1526 | SNP (transition) | U to C | Synonymous | 1297 | 7.9 |
| ScV-L-A1 | 2362 | Indel | Insertion A | +1 frameshift | 583 | 20.8 |
| ScV-L-A1 | 2417 | Indel | Deletion U | -1 frameshift | 618 | 20.9 |
| ScV-L-A1 | 2860 | SNP (transition) | G to A | Synonymous | 43,300 | 68.4 |
| ScV-L-A1 | 4557 | Indel | Insertion A | n/a | 140 | 7.1 |
| ScV-L-BC | 10 | SNP (transition) | G to A | n/a | 245 | 81.6 |
| ScV-L-BC | 10 | SNP (transversion) | G to U | n/a | 245 | 8.6 |
| ScV-L-BC | 455 | SNP (transition) | G to A | Synonymous | 1882 | 8.3 |
| ScV-L-BC | 884 | SNP (transition) | A to G | Synonymous | 13,398 | 8.9 |
| ScV-L-BC | 1551 | Indel | Insertion U | Gag truncation | 207 | 8.9 |
| ScV-L-BC | 1569 | Indel | Deletion U | Gag truncation | 203 | 13.3 |
| ScV-L-BC | 3424 | Indel | Insertion U | Pol truncation | 8674 | 5.4 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crabtree, A.M.; Kizer, E.A.; Hunter, S.S.; Van Leuven, J.T.; New, D.D.; Fagnan, M.W.; Rowley, P.A. A Rapid Method for Sequencing Double-Stranded RNAs Purified from Yeasts and the Identification of a Potent K1 Killer Toxin Isolated from Saccharomyces cerevisiae. Viruses 2019, 11, 70. https://doi.org/10.3390/v11010070
Crabtree AM, Kizer EA, Hunter SS, Van Leuven JT, New DD, Fagnan MW, Rowley PA. A Rapid Method for Sequencing Double-Stranded RNAs Purified from Yeasts and the Identification of a Potent K1 Killer Toxin Isolated from Saccharomyces cerevisiae. Viruses. 2019; 11(1):70. https://doi.org/10.3390/v11010070
Chicago/Turabian StyleCrabtree, Angela M., Emily A. Kizer, Samuel S. Hunter, James T. Van Leuven, Daniel D. New, Matthew W. Fagnan, and Paul A. Rowley. 2019. "A Rapid Method for Sequencing Double-Stranded RNAs Purified from Yeasts and the Identification of a Potent K1 Killer Toxin Isolated from Saccharomyces cerevisiae" Viruses 11, no. 1: 70. https://doi.org/10.3390/v11010070
APA StyleCrabtree, A. M., Kizer, E. A., Hunter, S. S., Van Leuven, J. T., New, D. D., Fagnan, M. W., & Rowley, P. A. (2019). A Rapid Method for Sequencing Double-Stranded RNAs Purified from Yeasts and the Identification of a Potent K1 Killer Toxin Isolated from Saccharomyces cerevisiae. Viruses, 11(1), 70. https://doi.org/10.3390/v11010070