1. Introduction
Equine herpesvirus 1 (EHV-1) is a member of the
Varicellovirus genus, in the Alphaherpesvirus sub-family [
1]. EHV-1 is known to infect horses as principal hosts but some cases have been reported in other equids and non-equid mammals [
2,
3,
4,
5]. Virus transmission occurs through direct contact between horses, infectious aerosols, fomites, and/or indirectly by humans. More recently, transmission of herpesvirus after survival in water was reported. Experimentally, EHV-1 was shown to be stable and infectious in water for over a week under different conditions of pH, salinity, temperature, and turbidity, and up to three weeks under some of these conditions [
6]. EHV-1 infection may induce several clinical forms of disease including respiratory infection (also called rhinopneumonitis) associated with pyrexia, cough, and respiratory distress, abortion in pregnant mares, stillbirth and neonatal death, and neurological disorders with symptoms ranging from mild ataxia to complete paralysis of the animal (EHV-1 inducing neurological disease is usually referred as Equine Herpesvirus Encephalomyelitis (EHM)) [
7]. However, numerous factors may affect the nature and the extent of clinical signs of disease, such as age, sex, physical condition, prior infection history, and/or the nature of the EHV-1 strain [
8]. After infection, the trigeminal ganglia and leukocytes have been identified as the sites of latency for EHV-1 [
9,
10]. The virus can be reactivated after environmental stimuli (e.g., stress) or therapeutic treatment (e.g., dexamethasone) and replicate in mucous tissues with subsequent dissemination to other hosts [
11]. EHV-1 latency mechanisms are poorly understood, although gene regulation has been shown to play a major role in the process [
12].
The linear double-stranded genome of EHV-1 is 150 kb long and consists of a unique long (U
L) and unique short region (U
S). Each of these regions is surrounded by small inverted sequences (Terminal Repeat Long (TRL)/Internal Repeat Long (IRL) and Terminal Repeat Short (TRS)/Internal repeat Short (IRS), respectively). The genome contains 80 open reading frames (ORFs), some of which show more genetic variability than others and contain sufficient variation for phylogenetic analysis [
13]. Previous studies performed ORF and/or whole genome sequencing to study genetic polymorphism [
14,
15]. Whole genome comparison of two characterised strains, Ab4 [
16] and V592 [
17], identified a polymorphic region within ORF68 (herpes simplex virus type 1 US2 homologue), which was used to examine the genetic heterogeneity of EHV-1 isolates in several different countries [
15,
18]. Furthermore, a single point mutation of adenine to guanine at nucleotide position 2254 within ORF30 (DNA polymerase catalytic subunit) is often associated with EHV-1 different forms of disease [
19,
20,
21,
22,
23,
24]. Although it is not exclusive, A
2254 (non-neuropathogenic) and G
2254 (neuropathogenic) strains were more frequently associated with abortion and neurological disease, respectively [
19,
25]. Additionally, genome comparison of Ab4 and V592 identified non-synonymous substitutions that could be used for multi-locus sequence typing (MLST) of EHV-1 strains [
19,
26].
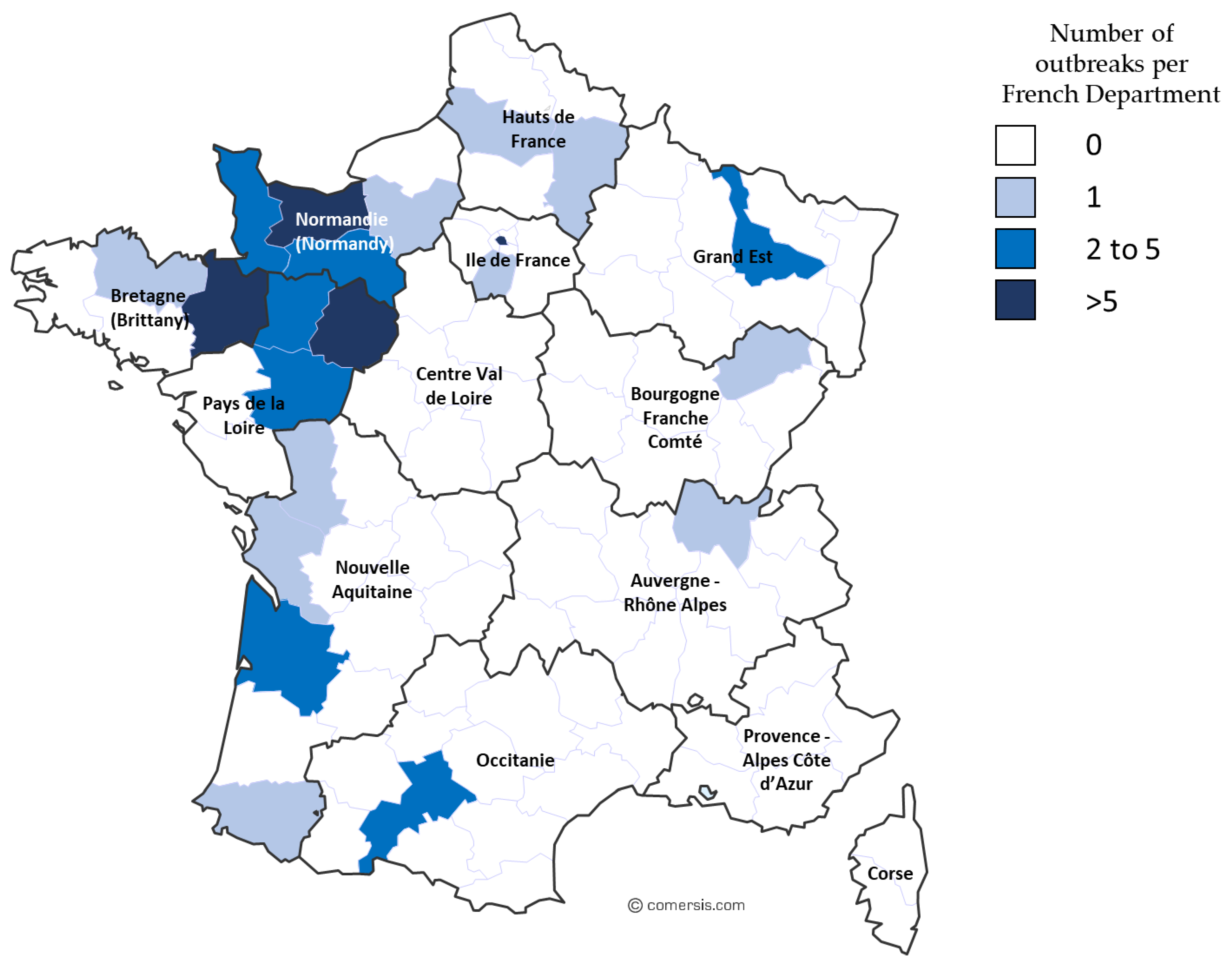
Due to its impact on animal welfare and performance, EHV-1 represents a major threat to the equine industry and a great interest and challenge for the equine veterinary and scientific community. This report aims to illustrate how challenging EHV-1 infection could be in the field through the description of a multi-syndromic outbreak that lasted over 9 months in 2009 and 2010. This outbreak involved 28 animals and different forms of disease were observed, including two EHM cases and four abortions. Both neuropathogenic and non-neuropathogenic strains were isolated from this major outbreak and were investigated alongside samples collected from 2012 to 2018. Different molecular analytic methods were applied to specimens from these outbreaks (2009, and from 2012 to 2018), including the ORF30 substitution A2254G typing, complete ORF30 sequencing, and Multi Locus Sequence Typing (MLST). These samples included EHV-1 strains isolated from the 2018 EHV-1 epizootic, which is considered to be the most important recorded in France for the last 30 years, with at least 56 outbreaks reported and leading to the cancellation of numerous equestrian events.
4. Discussion
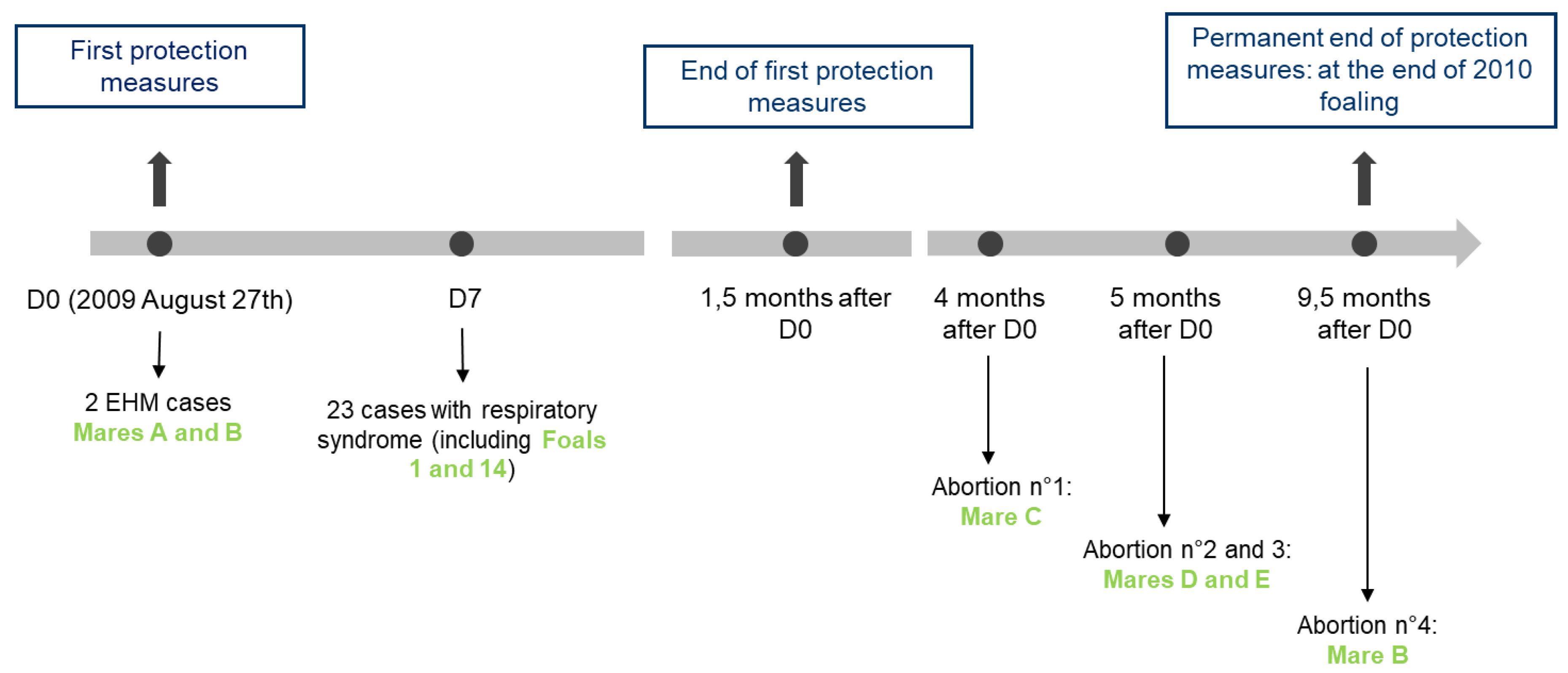
The 2009 EHV1 outbreak is regretfully a good example of the impact that an EHV-1 infection can have in stud farms and illustrates the diversity of diseases that could be observed and faced by veterinarians. The three clinical forms of disease induced by EHV-1 (respiratory, neurological disorder and abortion) were observed over a nine-and-a-half-month period on a thoroughbred farm (169 horses), which raises questions about the source of EHV-1 infection and its transmission during this outbreak. The significance of this 2009 EHV-1 outbreak was also the observation of the three clinical forms of disease in the same premise over a long period of time. This phenomenon is rarely described in literature [
39], and no other outbreak involving all three forms of disease was reported in France since 2009. The number of horses on this premise and the breeding activities may be factors that have contributed to increase the number of cases, which could have been higher in the absence of vaccination. The identification of two different strains, which may be the result from frequent horse movements in France and abroad, might also have influenced the diseases observed during this unusual outbreak. EHV-1 outbreaks are frequent but are usually limited to the report of one or two forms of the disease for the same outbreak (RESPE, personal communication). However, epidemiological links between outbreaks separated in time are not always available or identified. For example, the 2009 outbreak described here lasted nine and a half months with at least two and a half months between the last respiratory infection and the first abortion. Occurrence of multiple forms of disease may be more frequent than currently imagined.
The three clinical forms of disease were reported on a regular basis from 2009 to 2018 on the different outbreaks of this study. In 2018, a large number of cases were reported during a short period (March 2018 to May 2018), with exceptional sanitary measures needed to control contaminations between horses. This EHV-1 crisis is likely to be associated with the fact that a vaccine shortage occurred in 2016, implying a lower vaccination rate.
At the time of the 2009 outbreak, only a few tools were available to conduct EHV-1 molecular investigation. The PCR designed by Diallo et al. (2006, [
28]) was used as an EHV-1 detection and quantification test. Viral loads of samples were quantified, and sanitary measures were lifted when undetected. Results obtained at the time indicated that virus titers in total blood samples were lower than in swab extracts, but these two compartments are not always correlated to each other. Monitoring viral loads provided an overview of the virus excretion and risk of transmission. When none of the group 1 foals tested positive after a seven-week isolation period, the day to day management of the stud farm returned to normal. EHV-1 strains were typed (ORF30 SNP A2254G) and both types were identified (neuropathogenic and non-neuropathogenic).
Although those tools gave useful indicative information concerning the different strains isolated from this outbreak, they proved to be limited to establish potential relationship between cases and virus strains. Other tools have been developed (Nugent et al. 2006, Bryant et al. 2018 and Garvey et al. 2019 [
14,
19,
26]) that have motivated our retrospective and molecular analysis of French and Belgian EHV-1 strains isolated from 2009 outbreak to the major 2018 EHV-1 epizootic. Three different molecular analysis were performed: the ORF30 A2254G typing, the complete ORF30 sequencing and the MLST. In 2006, Nugent et al. [
19] reported a significant association between the A2254G SNP in the DNA polymerase gene (ORF30), neuropathogenicity. This single point substitution (A2254G) involves an amino acid change (N752D). N752 (A
2254) strains were strongly associated to non-neuropathogenic infection cases, while D752 (G
2254) strains were strongly associated to neuropathogenic cases [
19]. A2254G typing was subsequently used on a regular basis for strain discrimination [
40,
41]. In our study, 137 strains collected from 2009 to 2018 were typed, and A
2254 strains were more significantly associated with abortion cases than to neurological cases (
p = 0.0002), as recently described by Lechmann et al. (2019) [
42]. However, no significant correlation between G
2254 strains and the neurological form of disease was measured. This observation is in agreement with some recent studies showing that A2254G mutation is not exclusively associated to EHM but could be part of a more complex mechanism affecting strains virulence [
41]. Both neuropathogenic and non-neuropathogenic strains could be typed among 2018 strains, suggesting that more than one strain was circulating during the crisis. Strain A2254G typing is also interesting as it was reported that the point mutation could change sensitivity to some drugs targeting DNA polymerase activity as it was demonstrated in a study showing that a N752 variant was more sensitive to aphidicolin than the D752 variants. Aphidicolin inhibits some dNTPs binding to a family of DNA polymerase which include herpesvirus DNA polymerase [
17,
19].
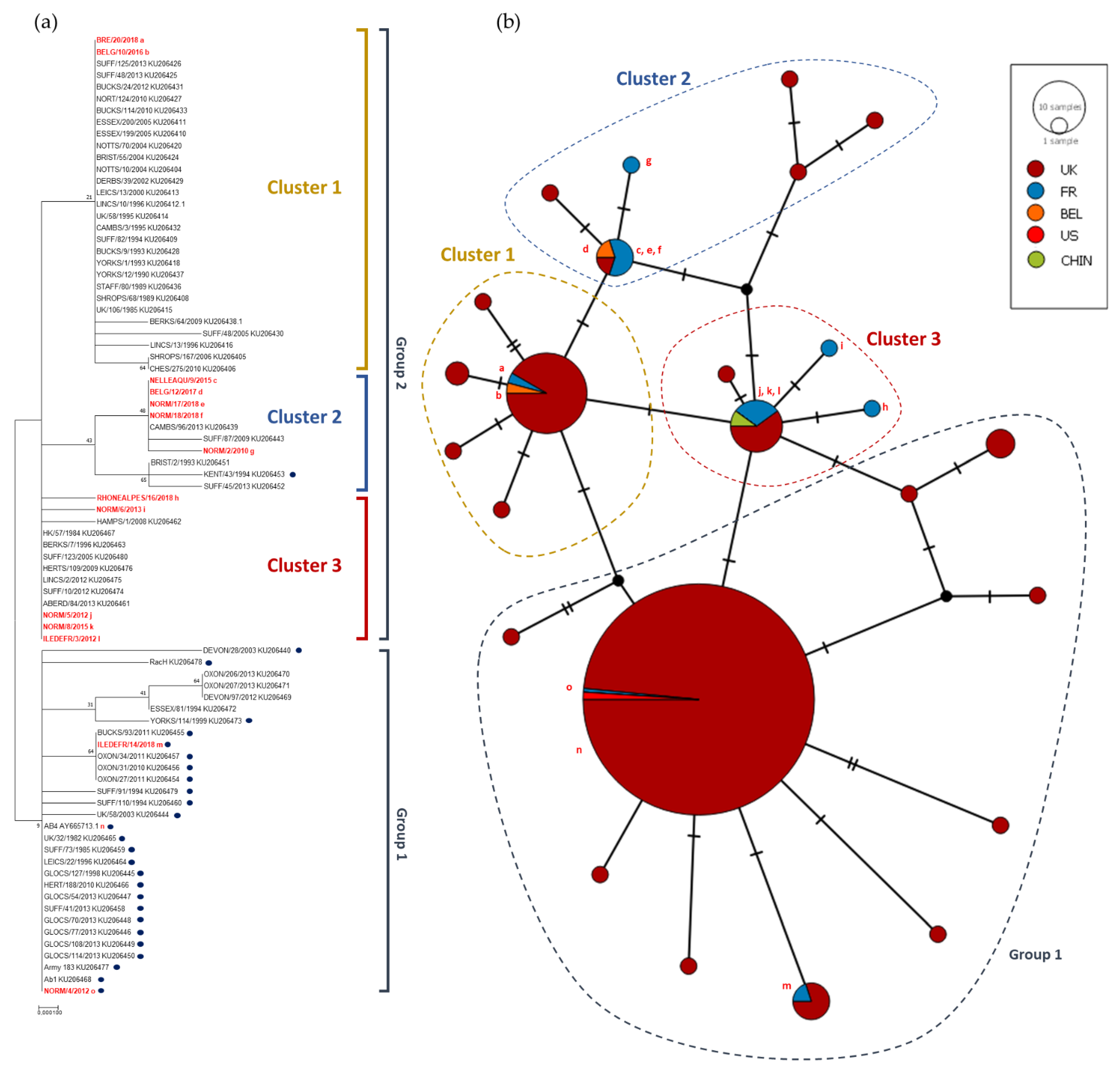
The A2254G typing was completed with ORF30 sequencing on 14 French and Belgian strains isolated between 2012 and 2018 in order to compare sequences and to perform a phylogenetic analysis. Several synonymous and non-synonymous substitutions were identified. Equine herpesvirus DNA polymerase subunit structures and mechanisms are not well known. Despite a low homology between EHV-1 and Human Simplex Virus (HSV) polymerase amino acid sequence (54%), the latter has been described as closest to α polymerase structure [
43]. On this basis, SNP found in EHV-1 strains could be attributed to structure domains identified in HSV polymerase [
43]. Amino acid substitutions could be localised in the pre-NH2 terminal domain (R59G), in the 3′-5′ exonuclease domain (S419L and R429K), in the palm domain (A694V and D752N), in the thumb domain (E990K). Although some of the domain activities have been studied for human herpesviruses [
43,
44], it is hard to predict the impact of the substitutions observed in EHV-1 strains on the protein activity. A strong homology (99.86% to 100%) was measured among those strains with no obvious evolutionary tendency. Results were in agreement with those published by Bryant et al. in 2018 [
14]. According to ORF30 sequencing, EHV-1 evolution is not linked to sampling location or year of collection, with the exception of the strains NORM/17/2018 and NORM/18/2018 that were isolated from the same stud farm, a few weeks apart, and have similar ORF30 sequences. All G
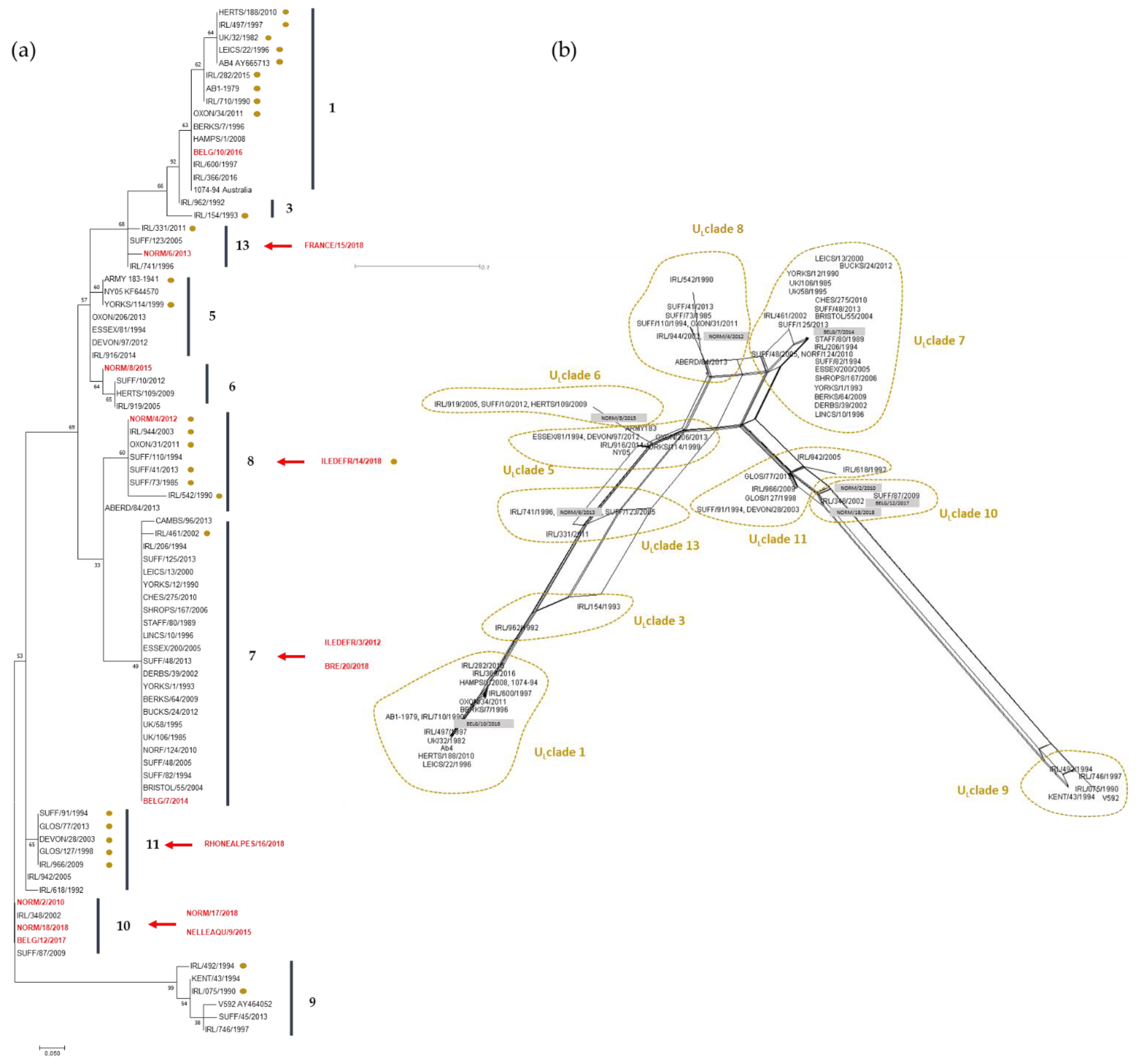
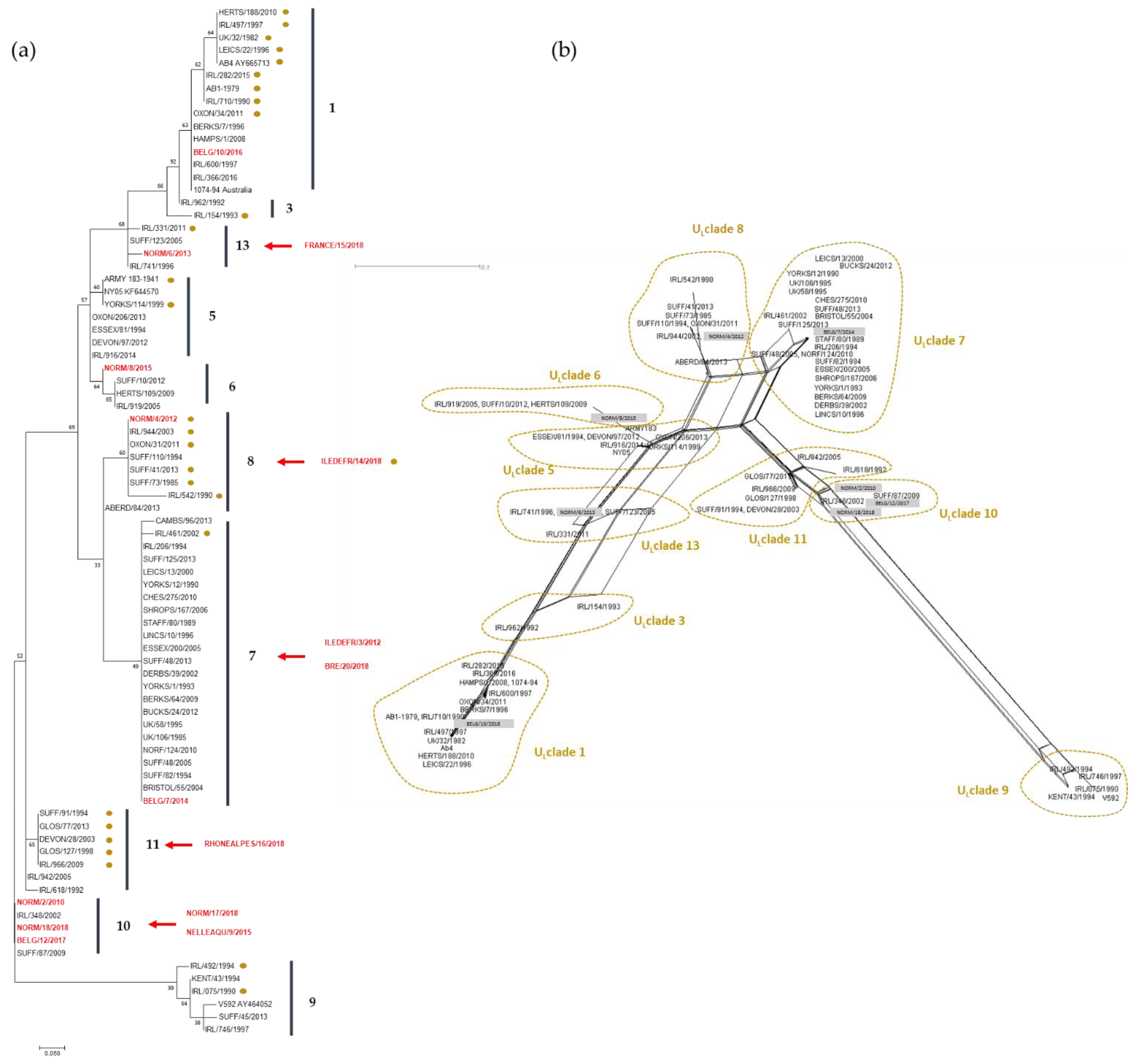
2254 strains were grouped in the same cluster, suggesting that ORF30 sequencing does not provide further information when compared with the A2254G typing. However, three clusters were identified for A2254 strains, primarily differentiated by one SNP (e.g A2968G). The significance of these different clusters is unknown. MLST analysis provides a more global view concerning EHV-1 strains evolution as it takes into account 37 loci in 26 different ORFs. As observed with ORF30 analysis, there is no obvious correlation between year and location of collection, with the exception of NORM/17/2018 and NORM/18/2018, both located in clade 10, which support a co-circulation of EHV-1 strains from different clades as already described by Bryant et al. and Garvey et al. [
14,
26]. It is interesting to note that four French strains and one Belgium strain are localised in clade 10. The U
L Clade 12 described by Bryant et al. (2018) [
14], which contains a strain from the UK, is not shown here. U
L Clades 2 and 4 were not represented by any of the European strains analysed in this study. It is important to note that the MLST method cannot distinguish U
L Clade 2 and 12 from MLST Clade 1 and 10, respectively [
26]. As all the strains compared in this study are from Europe, a broader strain selection would be needed to identify a potential geographical effect on clade differentiation. Finally, two abortion strains isolated from the same premise in a one-month interval after two mare abortions had the exact same MLST profile suggesting that the same strain infected the mares. Obviously, EHV-1 strain surveillance is complicated by the fact that EHV-1 can also establish latency in different sites, implying no viral replication as the viral genome maintains an episomal form blocking transcription and translation of its genes (limited transcription with LATs) [
9,
11]. This implies that strains circulation and outbreaks are potentially dependent on latency and re-activation.


 and
and 




{kind=link}
{kind=link}
{kind=link}
{kind=link}