Persistent Infection with Herpes Simplex Virus 1 and Alzheimer’s Disease—A Call to Study How Variability in Both Virus and Host may Impact Disease



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Definitions of Latent, Persistent, and Chronic Viral Infection
2.1. Definitions
2.2. HSV-1 Causes a Persistent, Latent Infection in Neurons
3. Neuroinflammation, β Amyloid Plaques, and Neurodegeneration—Connections with Cognitive Decline and Alzheimer’s Disease
3.1. Expression of Immune Proteins in the Brain—Contributions of Neuroinflammation to Synaptic Dysfunction
3.2. Pathological Hallmarks of AD
4. Possible Connections between Persistent HSV-1 Infection and AD
4.1. Persistent HSV-1 Infection and Neuroinflammation
4.2. Implications for HSV-1 Infection in the Development of AD Pathology
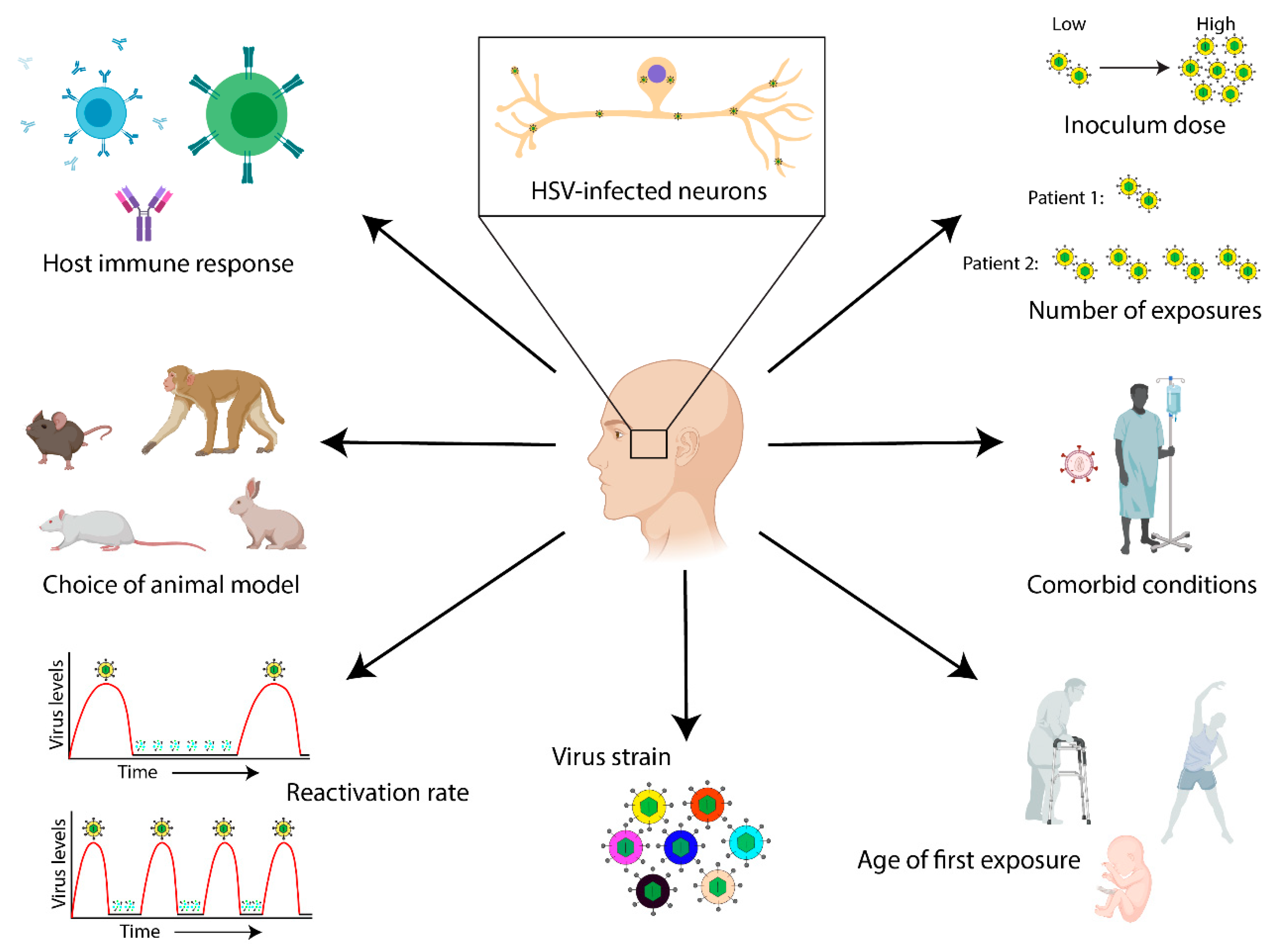
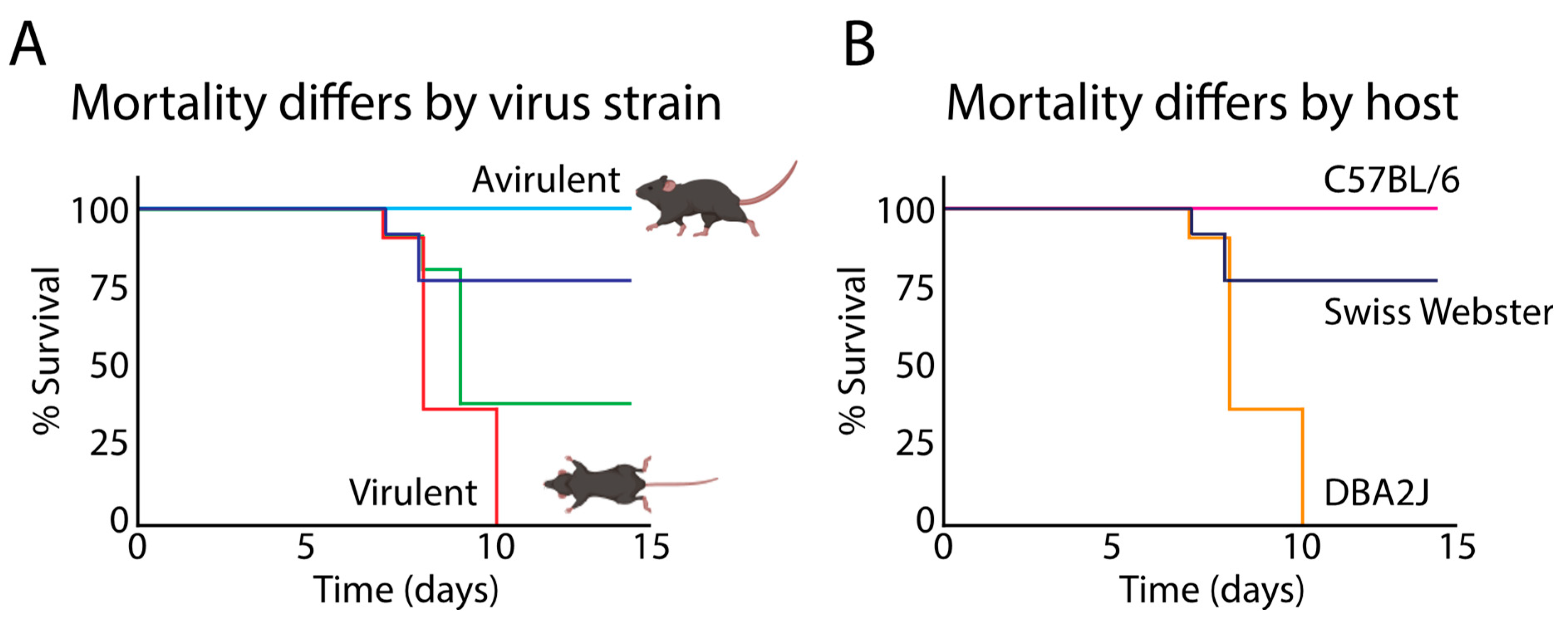
5. Strain-Specific Differences in HSV-1 Neurovirulence and the Potential Impact on AD Development
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- World Population Prospects—Population Division—United Nations. Available online: https://population.un.org/wpp/ (accessed on 19 October 2019).
- Elobeid, A.; Libard, S.; Leino, M.; Popova, S.N.; Alafuzoff, I. Altered Proteins in the Aging Brain. J. Neuropathol. Exp. Neurol. 2016, 75, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrak, R.E.; Griffin, S.T.; Graham, D.I. Aging-associated changes in human brain. J. Neuropathol. Exp. Neurol. 1997, 56, 1269–1275. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafè, M.; Valensin, S.; Olivieri, F.; Luca, M.D.; Ottaviani, E.; Benedictis, G.D. Inflamm-aging: An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Carson, M.J.; El Khoury, J.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef]
- Lucin, K.M.; Wyss-Coray, T. Immune activation in brain aging and neurodegeneration: Too much or too little? Neuron 2009, 64, 110–122. [Google Scholar] [CrossRef]
- Nichols, E.; Szoeke, C.E.I.; Vollset, S.E.; Abbasi, N.; Abd-Allah, F.; Abdela, J.; Aichour, M.T.E.; Akinyemi, R.O.; Alahdab, F.; Asgedom, S.W.; et al. Global, regional, and national burden of Alzheimer’s disease and other dementias, 1990–2016: A systematic analysis for the Global Burden of Disease Study Lancet Neurol. 2019, 18, 88–106. Lancet Neurol. 2019, 18, 88–106. [Google Scholar] [CrossRef]
- Pinto, T.; Lanctôt, K.L.; Herrmann, N. Revisiting the cholinergic hypothesis of behavioral and psychological symptoms in dementia of the Alzheimer’s type. Ageing Res. Rev. 2011, 10, 404–412. [Google Scholar] [CrossRef]
- Ray, W.J.; Ashall, F.; Goate DPhil, A.M. Molecular pathogenesis of sporadic and familial forms of Alzheimer’s disease. Mol. Med. Today 1998, 4, 151–157. [Google Scholar] [CrossRef]
- Menéndez, M. Pathological and clinical heterogeneity of presenilin 1 gene mutations. J. Alzheimers Dis. 2004, 6, 475–482. [Google Scholar] [CrossRef]
- Gómez-Isla, T.; Growdon, W.B.; McNamara, M.J.; Nochlin, D.; Bird, T.D.; Arango, J.C.; Lopera, F.; Kosik, K.S.; Lantos, P.L.; Cairns, N.J.; et al. The impact of different presenilin 1 and presenilin 2 mutations on amyloid deposition, neurofibrillary changes and neuronal loss in the familial Alzheimer’s disease brainEvidence for other phenotype-modifying factors. Brain 1999, 122, 1709–1719. [Google Scholar] [CrossRef]
- Cruts, M.; van Duijn, C.M.; Backhovens, H.; Van den Broeck, M.; Wehnert, A.; Serneels, S.; Sherrington, R.; Hutton, M.; Hardy, J.; St George-Hyslop, P.H.; et al. Estimation of the genetic contribution of presenilin-1 and -2 mutations in a population-based study of presenile Alzheimer disease. Hum. Mol. Genet. 1998, 7, 43–51. [Google Scholar] [CrossRef] [PubMed]
- Lippa, C.F.; Saunders, A.M.; Smith, T.W.; Swearer, J.M.; Drachman, D.A.; Ghetti, B.; Nee, L.; Pulaski-Salo, D.; Dickson, D.; Robitaille, Y.; et al. Familial and sporadic Alzheimer’s disease. Neurology 1996, 46, 406. [Google Scholar] [CrossRef] [PubMed]
- Goate, A. Segregation of a missense mutation in the amyloid beta-protein precursor gene with familial Alzheimer’s disease. J. Alzheimers Dis. 2006, 9, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Hellström-Lindahl, E.; Viitanen, M.; Marutle, A. Comparison of Aβ levels in the brain of familial and sporadic Alzheimer’s disease. Neurochem. Int. 2009, 55, 243–252. [Google Scholar] [CrossRef]
- Rogaev, E.I.; Sherrington, R.; Rogaeva, E.A.; Levesque, G.; Ikeda, M.; Liang, Y.; Chi, H.; Lin, C.; Holman, K.; Tsuda, T. Familial Alzheimer’s disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer’s disease type 3 gene. Nature 1995, 376, 775–778. [Google Scholar] [CrossRef]
- Huang, H.-C.; Jiang, Z.-F. Accumulated amyloid-beta peptide and hyperphosphorylated tau protein: Relationship and links in Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 15–27. [Google Scholar] [CrossRef]
- Wyss-Coray, T.; Rogers, J. Inflammation in Alzheimer Disease—A Brief Review of the Basic Science and Clinical Literature. Cold Spring Harb. Perspect Med. 2012, 2, a006346. [Google Scholar] [CrossRef]
- De Chiara, G.; Marcocci, M.E.; Sgarbanti, R.; Civitelli, L.; Ripoli, C.; Piacentini, R.; Garaci, E.; Grassi, C.; Palamara, A.T. Infectious Agents and Neurodegeneration. Mol. Neurobiol. 2012, 46, 614–638. [Google Scholar] [CrossRef] [Green Version]
- Dehhaghi, M.; Kazemi Shariat Panahi, H.; Guillemin, G.J. Microorganisms’ Footprint in Neurodegenerative Diseases. Front. Cell Neurosci. 2018, 12, 466. [Google Scholar] [CrossRef]
- Gosztyla, M.L.; Brothers, H.M.; Robinson, S.R. Alzheimer’s Amyloid-β is an Antimicrobial Peptide: A Review of the Evidence. J. Alzheimer’s Dis. 2018, 62, 1495–1506. [Google Scholar] [CrossRef]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2018, 14, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Sochocka, M.; Zwolińska, K.; Leszek, J. The Infectious Etiology of Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 996–1009. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Piacentini, R.; Fabiani, M.; Mastrodonato, A.; Marcocci, M.E.; Limongi, D.; Napoletani, G.; Protto, V.; Coluccio, P.; Celestino, I.; et al. Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog. 2019, 15, e1007617. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, F.B.; Boronow, J.J.; Stallings, C.; Origoni, A.E.; Ruslanova, I.; Yolken, R.H. Association of serum antibodies to herpes simplex virus 1 with cognitive deficits in individuals with schizophrenia. Arch. Gen. Psychiatry 2003, 60, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Harris, S.A.; Harris, E.A. Molecular Mechanisms for Herpes Simplex Virus Type 1 Pathogenesis in Alzheimer’s Disease. Front. Aging Neurosci. 2018, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Prasad, K.M.; Watson, A.M.M.; Dickerson, F.B.; Yolken, R.H.; Nimgaonkar, V.L. Exposure to herpes simplex virus type 1 and cognitive impairments in individuals with schizophrenia. Schizophr. Bull. 2012, 38, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.E.; Vickerman, P.; Gottlieb, S.L.; Newman, L.M. Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS ONE 2015, 10, e0140765. [Google Scholar] [CrossRef]
- Corey, L.; Spear, P.G. Infections with Herpes Simplex Viruses (Second of Two Parts). N. Engl. J. Med. 1986, 314, 749–757. [Google Scholar] [CrossRef]
- Looker, K.J.; Magaret, A.S.; May, M.T.; Turner, K.M.; Vickerman, P.; Newman, L.M.; Gottlieb, S.L. First estimates of the global and regional incidence of neonatal herpes infection. Lancet Glob. Health 2017, 5, e300–e309. [Google Scholar] [CrossRef] [Green Version]
- Roizman, B.; Knipe, D.M.; Whitley, R. Herpes Simplex Viruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1823–1897. [Google Scholar]
- Deshmane, S.L.; Fraser, N.W. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 1989, 63, 943–947. [Google Scholar] [Green Version]
- Nicoll, M.P.; Proença, J.T.; Efstathiou, S. The molecular basis of herpes simplex virus latency. FEMS Microbiol. Rev. 2012, 36, 684–705. [Google Scholar] [CrossRef] [PubMed]
- De Chiara, G.; Marcocci, M.E.; Civitelli, L.; Argnani, R.; Piacentini, R.; Ripoli, C.; Manservigi, R.; Grassi, C.; Garaci, E.; Palamara, A.T. APP Processing Induced by Herpes Simplex Virus Type 1 (HSV-1) Yields Several APP Fragments in Human and Rat Neuronal Cells. PLoS ONE 2010, 5, e13989. [Google Scholar] [CrossRef] [PubMed]
- Lövheim, H.; Gilthorpe, J.; Johansson, A.; Eriksson, S.; Hallmans, G.; Elgh, F. Herpes simplex infection and the risk of Alzheimer’s disease: A nested case-control study. Alzheimers Dement. 2015, 11, 587–592. [Google Scholar] [CrossRef] [PubMed]
- Lövheim, H.; Gilthorpe, J.; Adolfsson, R.; Nilsson, L.-G.; Elgh, F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 593–599. [Google Scholar] [CrossRef]
- Piacentini, R.; De Chiara, G.; Li Puma, D.D.; Ripoli, C.; Marcocci, M.E.; Garaci, E.; Palamara, A.T.; Grassi, C. HSV-1 and Alzheimer’s disease: More than a hypothesis. Front. Pharmacol. 2014, 5, 1–9. [Google Scholar] [CrossRef]
- Piacentini, R.; Puma, D.D.L.; Ripoli, C.; Marcocci, M.E.; Chiara, G.D.; Garaci, E.; Palamara, A.T.; Grassi, C. Herpes Simplex Virus type-1 infection induces synaptic dysfunction in cultured cortical neurons via GSK-3 activation and intraneuronal amyloid-β protein accumulation. Sci. Rep. 2015, 5, 15444. [Google Scholar] [CrossRef]
- Readhead, B.; Haure-Mirande, J.-V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018, 99, 64–82.e7. [Google Scholar] [CrossRef]
- Bu, X.L.; Yao, X.Q.; Jiao, S.S.; Zeng, F.; Liu, Y.H.; Xiang, Y.; Liang, C.R.; Wang, Q.H.; Cao, H.Y.; Yi, X.; et al. A Study on the Association Between Infectious Burden and Alzheimer’s Disease. Eur. J. Neurol. 2015, 22, 1519–1525. [Google Scholar] [CrossRef]
- Tzeng, N.-S.; Chung, C.-H.; Lin, F.-H.; Chiang, C.-P.; Yeh, C.-B.; Huang, S.-Y.; Lu, R.-B.; Chang, H.-A.; Kao, Y.-C.; Yeh, H.-W.; et al. Anti-herpetic Medications and Reduced Risk of Dementia in Patients with Herpes Simplex Virus Infections—A Nationwide, Population-Based Cohort Study in Taiwan. Neurotherapeutics 2018, 15, 417–429. [Google Scholar] [CrossRef]
- Bowen, C.D.; Renner, D.W.; Shreve, J.T.; Tafuri, Y.; Payne, K.M.; Dix, R.D.; Kinchington, P.R.; Gatherer, D.; Szpara, M.L. Viral forensic genomics reveals the relatedness of classic herpes simplex virus strains KOS, KOS63, and KOS. Virology 2016, 492, 179–186. [Google Scholar] [CrossRef]
- Kolb, A.W.; Adams, M.; Cabot, E.L.; Craven, M.; Brandt, C.R. Multiplex sequencing of seven ocular herpes simplex virus type-1 genomes: Phylogeny, sequence variability, and SNP distribution. Investig. Ophthalmol. Vis. Sci. 2011, 52, 9061–9073. [Google Scholar] [CrossRef] [PubMed]
- Parsons, L.R.; Tafuri, Y.R.; Shreve, J.T.; Bowen, C.D.; Shipley, M.M.; Enquist, L.W.; Szpara, M.L. Rapid genome assembly and comparison decode intrastrain variation in human alphaherpesviruses. mBio 2015, 6, e02213-14. [Google Scholar] [CrossRef] [PubMed]
- Pfaff, F.; Groth, M.; Sauerbrei, A.; Zell, R. Genotyping of herpes simplex virus type 1 by whole-genome sequencing. J. Gen. Virol. 2016, 97, 2732–2741. [Google Scholar] [CrossRef] [PubMed]
- Sijmons, S.; Thys, K.; Mbong Ngwese, M.; Van Damme, E.; Dvorak, J.; Van Loock, M.; Li, G.; Tachezy, R.; Busson, L.; Aerssens, J.; et al. High-Throughput Analysis of Human Cytomegalovirus Genome Diversity Highlights the Widespread Occurrence of Gene-Disrupting Mutations and Pervasive Recombination. J. Virol. 2015, 89, 7673–7695. [Google Scholar] [CrossRef] [Green Version]
- Szpara, M.L.; Gatherer, D.; Ochoa, A.; Greenbaum, B.; Dolan, A.; Bowden, R.J.; Enquist, L.W.; Legendre, M.; Davison, A.J. Evolution and diversity in human herpes simplex virus genomes. J. Virol. 2014, 88, 1209–1227. [Google Scholar] [CrossRef]
- Szpara, M.L.; Parsons, L.; Enquist, L.W. Sequence variability in clinical and laboratory isolates of herpes simplex virus 1 reveals new mutations. J. Virol. 2010, 84, 5303–5313. [Google Scholar] [CrossRef]
- Dix, R.D.; McKendall, R.R.; Baringer, J.R. Comparative neurovirulence of herpes simplex virus type 1 strains after peripheral or intracerebral inoculation of BALB/c mice. Infect. Immun. 1983, 40, 103–112. [Google Scholar]
- Wang, H.; Davido, D.J.; Morrison, L.A. HSV-1 strain McKrae is more neuroinvasive than HSV-1 KOS after corneal or vaginal inoculation in mice. Virus Res. 2013, 173, 436–440. [Google Scholar] [CrossRef] [Green Version]
- Kriesel, J.D.; Jones, B.B.; Matsunami, N.; Patel, M.K.; St. Pierre, C.A.; Kurt-Jones, E.A.; Finberg, R.W.; Leppert, M.; Hobbs, M.R. C21orf91 Genotypes Correlate with Herpes Simplex Labialis (Cold Sore) Frequency: Description of a Cold Sore Susceptibility Gene. J. Infect. Dis. 2011, 204, 1654–1662. [Google Scholar] [CrossRef]
- Thompson, R.L.; Williams, R.W.; Kotb, M.; Sawtell, N.M. A Forward Phenotypically Driven Unbiased Genetic Analysis of Host Genes That Moderate Herpes Simplex Virus Virulence and Stromal Keratitis in Mice. PLoS ONE 2014, 9, e92342. [Google Scholar] [CrossRef]
- Kriesel, J.D.; Bhatia, A.; Thomas, A. Cold sore susceptibility gene-1 genotypes affect the expression of herpes labialis in unrelated human subjects. Hum. Genome Var. 2014, 1, 14024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, U.; Renner, D.W.; Thompson, R.L.; Szpara, M.L.; Sawtell, N.M. Inferred father-to-son transmission of herpes simplex virus results in near-perfect preservation of viral genome identity and in vivo phenotypes. Sci. Rep. 2017, 7, 13666. [Google Scholar] [CrossRef] [PubMed]
- Minaya, M.A.; Jensen, T.L.; Goll, J.B.; Korom, M.; Datla, S.H.; Belshe, R.B.; Morrison, L.A. Molecular evolution of herpes simplex virus 2 complete genomes: Comparison between primary and recurrent infections. J. Virol. 2017, 91, e00942-17. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.; Magaret, A.; Roychoudhury, P.; Greninger, A.L.; Reeves, D.; Schiffer, J.; Jerome, K.R.; Sather, C.; Diem, K.; Lingappa, J.R.; et al. Dual-strain genital herpes simplex virus type 2 (HSV-2) infection in the US, Peru, and 8 countries in sub-Saharan Africa: A nested cross-sectional viral genotyping study. PLoS Med. 2017, 14, e1002475. [Google Scholar] [CrossRef] [PubMed]
- Kleinstein, S.E.; Shea, P.R.; Allen, A.S.; Koelle, D.M.; Wald, A.; Goldstein, D.B. Genome-wide association study (GWAS) of human host factors influencing viral severity of herpes simplex virus type 2 (HSV-2). Genes Immun. 2019, 20, 112. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, L.N.; Bowen, C.D.; Renner, D.W.; Pandey, U.; Della Fera, A.N.; Kimberlin, D.W.; Prichard, M.N.; Whitley, R.J.; Weitzman, M.D.; Szpara, M.L. Genotypic and Phenotypic Diversity of Herpes Simplex Virus 2 within the Infected Neonatal Population. mSphere 2019, 4, e00590-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramchandani, M.S.; Jing, L.; Russell, R.M.; Tran, T.; Laing, K.J.; Magaret, A.S.; Selke, S.; Cheng, A.; Huang, M.-L.; Xie, H.; et al. Viral Genetics Modulate Orolabial Herpes Simplex Virus Type 1 Shedding in Humans. J. Infect. Dis. 2019, 219, 1058–1066. [Google Scholar] [CrossRef]
- Boldogh, I.; Albrecht, T.; Porter, D.D. Persistent Viral Infections. In Medical Microbiology; Baron, S., Ed.; University of Texas Medical Branch at Galveston: Galveston, TX, USA, 1996; ISBN 978-0-96-311721-2. [Google Scholar]
- Alawieh, A.; Elvington, A.; Tomlinson, S. Complement in the Homeostatic and Ischemic Brain. Front. Immunol. 2015, 6, 417. [Google Scholar] [CrossRef]
- Boulanger, L.M. Immune Proteins in Brain Development and Synaptic Plasticity. Neuron 2009, 64, 93–109. [Google Scholar] [CrossRef] [Green Version]
- Hammad, A.; Westacott, L.; Zaben, M. The role of the complement system in traumatic brain injury: A review. J. Neuroinflamm. 2018, 15, 24. [Google Scholar] [CrossRef]
- McAllister, A.K. Major histocompatibility complex I in brain development and schizophrenia. Biol. Psychiatry 2014, 75, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Needleman, L.A.; McAllister, A.K. The Major Histocompatibility Complex and Autism Spectrum Disorder. Dev. Neurobiol. 2012, 72, 1288–1301. [Google Scholar] [CrossRef] [PubMed]
- Shatz, C.J. MHC class I: An unexpected role in neuronal plasticity. Neuron 2009, 64, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Stephan, A.H.; Barres, B.A.; Stevens, B. The Complement System: An Unexpected Role in Synaptic Pruning During Development and Disease. Annu. Rev. Neurosci. 2012, 35, 369–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef]
- Wilson, A.C.; Mohr, I. A cultured affair: HSV latency and reactivation neurons. Trends Microbiol. 2012, 20, 604–611. [Google Scholar] [CrossRef]
- Mehta, A.; Maggioncalda, J.; Bagasra, O.; Thikkavarapu, S.; Saikumari, P.; Valyi-Nagy, T.; Fraser, N.W.; Block, T.M. In situ DNA PCR and RNA hybridization detection of herpes simplex virus sequences in trigeminal gangliaof latently infected mice. Virology 1995, 206, 633–640. [Google Scholar] [CrossRef] [Green Version]
- Maggioncalda, J.; Mehta, A.; Su, Y.H.; Fraser, N.W.; Block, T.M. Correlation between Herpes Simplex Virus Type 1 Rate of Reactivation from Latent Infection and the Number of Infected Neurons in Trigeminal Ganglia. Virology 1996, 225, 72–81. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.; Lau, T.Y.; Morales, M.; Mont, E.K.; Straus, S.E. Laser-capture microdissection: Refining estimates of the quantity and distribution of latent herpes simplex virus 1 and varicella-zoster virus DNA in human trigeminal Ganglia at the single-cell level. J. Virol. 2005, 79, 14079–14087. [Google Scholar] [CrossRef]
- Stevens, J.G.; Wagner, E.K.; Devi-Rao, G.B.; Cook, M.L.; Feldman, L.T. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 1987, 235, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Wagner, E.K.; Bloom, D.C. Experimental investigation of herpes simplex virus latency. Clin. Microbiol. Rev. 1997, 10, 419–443. [Google Scholar] [CrossRef] [PubMed]
- Zabolotny, J.M.; Krummenacher, C.; Fraser, N.W. The herpes simplex virus type 1 2.0-kilobase latency-associated transcript is a stable intron which branches at a guanosine. J. Virol. 1997, 71, 4199–4208. [Google Scholar] [PubMed]
- Chen, S.H.; Kramer, M.F.; Schaffer, P.A.; Coen, D.M. A viral function represses accumulation of transcripts from productive-cycle genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1997, 71, 5878–5884. [Google Scholar] [Green Version]
- Mador, N.; Goldenberg, D.; Cohen, O.; Panet, A.; Steiner, I. Herpes Simplex Virus Type 1 Latency-Associated Transcripts Suppress Viral Replication and Reduce Immediate-Early Gene mRNA Levels in a Neuronal Cell Line. J. Virol. 1998, 72, 5067–5075. [Google Scholar] [Green Version]
- Nicoll, M.P.; Hann, W.; Shivkumar, M.; Harman, L.E.R.; Connor, V.; Coleman, H.M.; Proença, J.T.; Efstathiou, S. The HSV-1 Latency-Associated Transcript Functions to Repress Latent Phase Lytic Gene Expression and Suppress Virus Reactivation from Latently Infected Neurons. PLoS Pathog. 2016, 12, e1005539. [Google Scholar] [CrossRef]
- Thompson, R.L.; Sawtell, N.M. The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J. Virol. 1997, 71, 5432–5440. [Google Scholar] [Green Version]
- Thompson, R.L.; Sawtell, N.M. The herpes simplex virus type 1 latency associated transcript locus is required for the maintenance of reactivation competent latent infections. J. Neurovirol. 2011, 17, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Thompson, R.L.; Sawtell, N.M. Herpes simplex virus type 1 latency-associated transcript gene promotes neuronal survival. J. Virol. 2001, 75, 6660–6675. [Google Scholar] [CrossRef]
- Perng, G.C.; Dunkel, E.C.; Geary, P.A.; Slanina, S.M.; Ghiasi, H.; Kaiwar, R.; Nesburn, A.B.; Wechsler, S.L. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J. Virol. 1994, 68, 8045–8055. [Google Scholar] [Green Version]
- Shen, W.; e Silva, M.S.; Jaber, T.; Vitvitskaia, O.; Li, S.; Henderson, G.; Jones, C. Two Small RNAs Encoded within the First 1.5 Kilobases of the Herpes Simplex Virus Type 1 Latency-Associated Transcript Can Inhibit Productive Infection and Cooperate to Inhibit Apoptosis. J. Virol. 2009, 83, 9131–9139. [Google Scholar] [CrossRef] [PubMed]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Green, M.T.; Courtney, R.J.; Dunkel, E.C. Detection of an immediate early herpes simplex virus type 1 polypeptide in trigeminal ganglia from latently infected animals. Infect. Immun. 1981, 34, 987–992. [Google Scholar] [PubMed]
- Kramer, M.F.; Coen, D.M. Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J. Virol. 1995, 69, 1389–1399. [Google Scholar] [PubMed]
- Feldman, L.T.; Ellison, A.R.; Voytek, C.C.; Yang, L.; Krause, P.; Margolis, T.P. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 978–983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khanna, K.M.; Bonneau, R.H.; Kinchington, P.R.; Hendricks, R.L. Herpes Simplex Virus-Specific Memory CD8^ T Cells Are Selectively Activated and Retained in Latently Infected Sensory Ganglia. Immunity 2003, 18, 593–603. [Google Scholar] [CrossRef]
- Cliffe, A.R.; Wilson, A.C. Restarting Lytic Gene Transcription at the Onset of Herpes Simplex Virus Reactivation. J. Virol. 2017, 91, e01419-16. [Google Scholar] [CrossRef]
- Linderman, J.A.; Kobayashi, M.; Rayannavar, V.; Fak, J.J.; Darnell, R.B.; Chao, M.V.; Wilson, A.C.; Mohr, I. Immune Escape via a Transient Gene Expression Program Enables Productive Replication of a Latent Pathogen. Cell Rep. 2017, 18, 1312–1323. [Google Scholar] [CrossRef]
- Kang, M.-S.; Kieff, E. Epstein–Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef]
- Longnecker, R.M.; Kieff, E.; Cohen, J.I. Epstein-barr virus. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P., Eds.; Wolters Kluwer: Alphen aan den Rijn, The Netherlands, 2013. [Google Scholar]
- Khanna, K.M.; Lepisto, A.J.; Hendricks, R.L. Immunity to latent viral infection: Many skirmishes but few fatalities. Trends Immunol. 2004, 25, 230–234. [Google Scholar] [CrossRef]
- Medzhitov, R. Origin and physiological roles of inflammation. Nat. Lond. 2008, 454, 428–435. [Google Scholar] [CrossRef] [PubMed]
- Warren, K.G.; Wroblewska, Z.; Okabe, H.; Brown, S.M.; Gilden, D.H.; Koprowski, H.; Rorke, L.B.; Subak-Sharpe, J.; Yonezawa, T. Virology and histopathology of the trigeminal ganglia of Americans and Japanese. Can. J. Neurol. Sci. 1978, 5, 425–430. [Google Scholar] [CrossRef]
- Joly, E.; Mucke, L.; Oldstone, M.B. Viral persistence in neurons explained by lack of major histocompatibility class I expression. Science 1991, 253, 1283–1285. [Google Scholar] [CrossRef] [PubMed]
- Neuwelt, E.A.; Clark, W.K. Unique aspects of central nervous system immunology. Neurosurgery 1978, 3, 419–430. [Google Scholar]
- Shirai, Y. On the transplantation of the rat sarcoma in adult heterogeneous animals. Jpn. Med. World 1921, 1, 14–15. [Google Scholar]
- Murphy, J.B.; Sturm, E. Conditions determining the transplantability of tissues in the brain. J. Exp. Med. 1923, 38, 183–197. [Google Scholar] [CrossRef]
- Cebrián, C.; Loike, J.D.; Sulzer, D. Neuronal MHC-I expression and its implications in synaptic function, axonal regeneration and Parkinson’s and other brain diseases. Front. Neuroanat. 2014, 8, 114. [Google Scholar] [CrossRef]
- Mangold, C.A.; Masser, D.R.; Stanford, D.R.; Bixler, G.V.; Pisupati, A.; Giles, C.B.; Wren, J.D.; Ford, M.M.; Sonntag, W.E.; Freeman, W.M. CNS-wide Sexually Dimorphic Induction of the Major Histocompatibility Complex 1 Pathway with Aging. J. Gerontol. A Biol. Sci. Med. Sci. 2017, 72, 16–29. [Google Scholar] [CrossRef]
- Mangold, C.A.; Wronowski, B.; Du, M.; Masser, D.R.; Hadad, N.; Bixler, G.V.; Brucklacher, R.M.; Ford, M.M.; Sonntag, W.E.; Freeman, W.M. Sexually divergent induction of microglial-associated neuroinflammation with hippocampal aging. J. Neuroinflamm. 2017, 14, 141. [Google Scholar] [CrossRef]
- Starkey, H.D.V.; Van Kirk, C.A.; Bixler, G.V.; Imperio, C.G.; Kale, V.P.; Serfass, J.M.; Farley, J.A.; Yan, H.; Warrington, J.P.; Han, S.; et al. Neuroglial Expression of the MHCI Pathway and PirB Receptor Is Upregulated in the Hippocampus with Advanced Aging. J. Mol. Neurosci. 2012, 48, 111–126. [Google Scholar] [CrossRef] [Green Version]
- Long, L.-H.; Liu, R.-L.; Wang, F.; Liu, J.; Hu, Z.-L.; Xie, N.; Jin, Y.; Fu, H.; Chen, J.-G. Age-related synaptic changes in the CA1 stratum radiatum and spatial learning impairment in rats. Clin. Exp. Pharm. Physiol. 2009, 36, 675–681. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Linville, M.C.; Tucker, E.W.; Sonntag, W.E.; Brunso-Bechtold, J.K. Differential effects of aging and insulin-like growth factor-1 on synapses in CA1 of rat hippocampus. Cereb. Cortex 2005, 15, 571–577. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Fu, A.K.Y.; Ip, N.Y. Synaptic dysfunction in Alzheimer’s disease: Mechanisms and therapeutic strategies. Pharmacol. Ther. 2019, 195, 186–198. [Google Scholar] [CrossRef] [PubMed]
- Lassmann, H.; Weiler, R.; Fischer, P.; Bancher, C.; Jellinger, K.; Floor, E.; Danielczyk, W.; Seitelberger, F.; Winkler, H. Synaptic pathology in Alzheimer’s disease: Immunological data for markers of synaptic and large dense-core vesicles. Neuroscience 1992, 46, 1–8. [Google Scholar] [CrossRef]
- Masliah, E.; Mallory, M.; Alford, M.; DeTeresa, R.; Hansen, L.A.; McKeel, D.W.; Morris, J.C. Altered expression of synaptic proteins occurs early during progression of Alzheimer’s disease. Neurology 2001, 56, 127–129. [Google Scholar] [CrossRef] [PubMed]
- Rajmohan, R.; Reddy, P.H. Amyloid-Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimers Dis. 2017, 57, 975–999. [Google Scholar] [CrossRef] [PubMed]
- Reddy, P.H.; Mani, G.; Park, B.S.; Jacques, J.; Murdoch, G.; Whetsell, W.; Kaye, J.; Manczak, M. Differential loss of synaptic proteins in Alzheimer’s disease: Implications for synaptic dysfunction. J. Alzheimers Dis. 2005, 7, 103–117, discussion 173–180. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer Disease: Mechanistic Understanding Predicts Novel Therapies. Ann. Intern. Med. Phila. 2004, 140, 627–638. [Google Scholar] [CrossRef]
- Cárdenas-Aguayo, M.D.C.; Silva-Lucero, M.D.C.; Cortes-Ortiz, M.; Jiménez-Ramos, B.; Gómez-Virgilio, L.; Ramírez-Rodríguez, G.; Vera-Arroyo, E.; Fiorentino-Pérez, R.; García, U.; Luna-Muñoz, J.; et al. Physiological Role of Amyloid Beta in Neural Cells: The Cellular Trophic Activity. Neurochemistry 2014, 257–281. [Google Scholar] [CrossRef]
- Bouras, C.; Hof, P.R.; Giannakopoulos, P.; Michel, J.P.; Morrison, J.H. Regional distribution of neurofibrillary tangles and senile plaques in the cerebral cortex of elderly patients: A quantitative evaluation of a one-year autopsy population from a geriatric hospital. Cereb. Cortex 1994, 4, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.J.; Pike, C.J.; Shankle, R.; Cotman, C.W. Beta-amyloid deposition and other measures of neuropathology predict cognitive status in Alzheimer’s disease. Neurobiol. Aging 1996, 17, 921–933. [Google Scholar] [CrossRef]
- Nelson, P.T.; Alafuzoff, I.; Bigio, E.H.; Bouras, C.; Braak, H.; Cairns, N.J.; Castellani, R.J.; Crain, B.J.; Davies, P.; Del Tredici, K.; et al. Correlation of Alzheimer Disease Neuropathologic Changes with Cognitive Status: A Review of the Literature. J. Neuropathol. Exp. Neurol 2012, 71, 362–381. [Google Scholar] [CrossRef] [PubMed]
- Näslund, J.; Haroutunian, V.; Mohs, R.; Davis, K.L.; Davies, P.; Greengard, P.; Buxbaum, J.D. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA 2000, 283, 1571–1577. [Google Scholar] [CrossRef]
- Blessed, G.; Tomlinson, B.E.; Roth, M. The Association Between Quantitative Measures of Dementia and of Senile Change in the Cerebral Grey Matter of Elderly Subjects. Br. J. Psychiatry 1968, 114, 797–811. [Google Scholar] [CrossRef]
- Moya, K.L.; Benowitz, L.I.; Schneider, G.E.; Allinquant, B. The amyloid precursor protein is developmentally regulated and correlated with synaptogenesis. Dev. Biol. 1994, 161, 597–603. [Google Scholar] [CrossRef]
- Mileusnic, R.; Lancashire, C.L.; Johnston, A.N.; Rose, S.P. APP is required during an early phase of memory formation. Eur. J. Neurosci. 2000, 12, 4487–4495. [Google Scholar]
- Kolarova, M.; García-Sierra, F.; Bartos, A.; Ricny, J.; Ripova, D. Structure and Pathology of Tau Protein in Alzheimer Disease. Int. J. Alzheimer’s Dis. 2012, 2012, 731526. [Google Scholar] [CrossRef]
- Braak, E.; Braak, H.; Mandelkow, E.M. A sequence of cytoskeleton changes related to the formation of neurofibrillary tangles and neuropil threads. Acta Neuropathol. 1994, 87, 554–567. [Google Scholar] [CrossRef]
- Mandelkow, E.-M.; Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E. Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol. Aging 2003, 24, 1079–1085. [Google Scholar] [CrossRef]
- Buée, L.; Bussière, T.; Buée-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders11These authors contributed equally to this work. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Lovestone, S.; Reynolds, C.H. The phosphorylation of tau: A critical stage in neurodevelopment and neurodegenerative processes. Neuroscience 1997, 78, 309–324. [Google Scholar] [PubMed]
- Vermersch, P.; David, J.P.; Frigard, B.; Fallet-Bianco, C.; Wattez, A.; Petit, H.; Delacourte, A. Cortical mapping of Alzheimer pathology in brains of aged non-demented subjects. Prog. Neuropsychopharmacol. Biol. Psychiatry 1995, 19, 1035–1047. [Google Scholar] [CrossRef]
- Vermersch, P.; Frigard, B.; David, J.P.; Fallet-Bianco, C.; Delacourte, A. Presence of abnormally phosphorylated Tau proteins in the entorhinal cortex of aged non-demented subjects. Neurosci. Lett. 1992, 144, 143–146. [Google Scholar] [CrossRef]
- Castellani, R.J.; Lee, H.-G.; Zhu, X.; Perry, G.; Smith, M.A. Alzheimer disease pathology as a host response. J. Neuropathol. Exp. Neurol. 2008, 67, 523–531. [Google Scholar] [CrossRef]
- Benditt, E.P.; Barrett, T.; McDougall, J.K. Viruses in the etiology of atherosclerosis. Proc. Natl. Acad. Sci. USA 1983, 80, 6386–6389. [Google Scholar] [CrossRef]
- Gyorkey, F.; Melnick, J.L.; Guinn, G.A.; Gyorkey, P.; DeBakey, M.E. Herpesviridae in the endothelial and smooth muscle cells of the proximal aorta in arteriosclerotic patients. Exp. Mol. Pathol. 1984, 40, 328–339. [Google Scholar] [CrossRef]
- Yamashiroya, H.M.; Ghosh, L.; Yang, R.; Robertson, A.L., Jr. Herpesviridae in the coronary arteries and aorta of young trauma victims. Am. J. Pathol. 1988, 130, 71. [Google Scholar]
- Linnavuori, K.H. History of recurrent mucocutaneous herpes correlates with relatively low interferon production by herpes simplex virus-exposed cultured monocytes. J. Med. Virol. 1988, 25, 61–68. [Google Scholar] [CrossRef]
- Roivainen, M.; Viik-Kajander, M.; Palosuo, T.; Toivanen, P.; Leinonen, M.; Saikku, P.; Tenkanen, L.; Manninen, V.; Hovi, T.; Mänttäri, M. Infections, inflammation, and the risk of coronary heart disease. Circulation 2000, 101, 252–257. [Google Scholar] [CrossRef]
- Siscovick, D.S.; Schwartz, S.M.; Corey, L.; Grayston, J.T.; Ashley, R.; Wang, S.P.; Psaty, B.M.; Tracy, R.P.; Kuller, L.H.; Kronmal, R.A. Chlamydia pneumoniae, herpes simplex virus type 1, and cytomegalovirus and incident myocardial infarction and coronary heart disease death in older adults: The Cardiovascular Health Study. Circulation 2000, 102, 2335–2340. [Google Scholar] [CrossRef] [PubMed]
- Vankirk, C.A. Characterization and Regulation of the Major Histocompatibility Complex Class I in the Cns: Functional Implications for Brain Aging and Sexually Dimorphic Differences in Neuroinflammation. Ph.D. Thesis, Pennsylvania State University, Centre County, PA, USA, 2013. [Google Scholar]
- Popescu, B.O.; Toescu, E.C.; Popescu, L.M.; Bajenaru, O.; Muresanu, D.F.; Schultzberg, M.; Bogdanovic, N. Blood-brain barrier alterations in ageing and dementia. J. Neurol. Sci. 2009, 283, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Weiskopf, D.; Weinberger, B.; Grubeck-Loebenstein, B. The aging of the immune system. Transpl. Int. 2009, 22, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Stowe, R.P.; Kozlova, E.V.; Yetman, D.L.; Walling, D.M.; Goodwin, J.S.; Glaser, R. Chronic herpesvirus reactivation occurs in aging. Exp. Gerontol. 2007, 42, 563–570. [Google Scholar] [CrossRef] [Green Version]
- Ershler, W.B. Interleukin-6: A cytokine for gerontologists. J. Am. Geriatr. Soc. 1993, 41, 176–181. [Google Scholar] [CrossRef]
- Saldanha, J.; Sutton, R.N.; Gannicliffe, A.; Farragher, B.; Itzhaki, R.F. Detection of HSV1 DNA by in situ hybridisation in human brain after immunosuppression. J. Neurol. Neurosurg. Psychiatry 1986, 49, 613–619. [Google Scholar] [CrossRef]
- Cheon, M.S.; Bajo, M.; Gulesserian, T.; Cairns, N.; Lubec, G. Evidence for the relation of herpes simplex virus type 1 to Down syndrome and Alzheimer’s disease. Electrophoresis 2001, 22, 445–448. [Google Scholar] [CrossRef]
- Itzhaki, R.F.; Lin, W.R.; Shang, D.; Wilcock, G.K.; Faragher, B.; Jamieson, G.A. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 1997, 349, 241–244. [Google Scholar] [CrossRef]
- Jamieson, G.A.; Maitland, N.J.; Wilcock, G.K.; Craske, J.; Itzhaki, R.F. Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J. Med. Virol. 1991, 33, 224–227. [Google Scholar] [CrossRef]
- Jamieson, G.A.; Maitland, N.J.; Wilcock, G.K.; Yates, C.M.; Itzhaki, R.F. Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J. Pathol. 1992, 167, 365–368. [Google Scholar] [CrossRef]
- Jamieson, G.A.; Maitland, N.J.; Craske, J.; Wilcock, G.K.; Itzhaki, R.F. Detection of herpes simplex virus type 1 DNA sequences in normal and Alzheimer’s disease brain using polymerase chain reaction. Biochem. Soc. Trans. 1991, 19, 122S. [Google Scholar] [CrossRef] [PubMed]
- Wozniak, M.A.; Shipley, S.J.; Combrinck, M.; Wilcock, G.K.; Itzhaki, R.F. Productive herpes simplex virus in brain of elderly normal subjects and Alzheimer’s disease patients. J. Med. Virol. 2005, 75, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.J. Limbic predilection in Alzheimer dementia: Is reactivated herpesvirus involved? Can. J. Neurol. Sci. 1982, 9, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Nicoll, J.A.; Love, S.; Kinrade, E. Distribution of herpes simplex virus DNA in the brains of human long-term survivors of encephalitis. Neurosci. Lett. 1993, 157, 215–218. [Google Scholar] [CrossRef]
- Wozniak, M.; Mee, A.; Itzhaki, R. Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J. Pathol. 2009, 217, 131–138. [Google Scholar] [CrossRef]
- Hemling, N.; Röyttä, M.; Rinne, J.; Pöllänen, P.; Broberg, E.; Tapio, V.; Vahlberg, T.; Hukkanen, V. Herpesviruses in brains in Alzheimer’s and Parkinson’s diseases. Ann. Neurol. 2003, 54, 267–271. [Google Scholar] [CrossRef]
- Marques, A.R.; Straus, S.E.; Fahle, G.; Weir, S.; Csako, G.; Fischer, S.H. Lack of association between HSV-1 DNA in the brain, Alzheimer’s disease and apolipoprotein Ejournal. Neurovirology 2001, 7, 82–83. [Google Scholar]
- Wozniak, M.A.; Itzhaki, R.F.; Shipley, S.J.; Dobson, C.B. Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci. Lett. 2007, 429, 95–100. [Google Scholar] [CrossRef]
- Li Puma, D.D.; Piacentini, R.; Leone, L.; Gironi, K.; Marcocci, M.E.; Chiara, G.D.; Palamara, A.T.; Grassi, C. Herpes Simplex Virus Type-1 Infection Impairs Adult Hippocampal Neurogenesis via Amyloid-β Protein Accumulation: Herpes Simplex Virus Impairs Adult Neurogenesis. Stem Cells 2019. [Google Scholar] [CrossRef]
- Alvarez, G.; Aldudo, J.; Alonso, M.; Santana, S.; Valdivieso, F. Herpes simplex virus type 1 induces nuclear accumulation of hyperphosphorylated tau in neuronal cells. J. Neurosci. Res. 2012, 90, 1020–1029. [Google Scholar] [CrossRef]
- Wozniak, M.A.; Frost, A.L.; Itzhaki, R.F. Alzheimer’s disease-specific tau phosphorylation is induced by herpes simplex virus type 1. J. Alzheimers Dis. 2009, 16, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, A.; Solis, L.; Salvadores, N.; Cortés, M.; Lerchundi, R.; Otth, C. Neuronal cytoskeletal dynamic modification and neurodegeneration induced by infection with herpes simplex virus type 1. J. Alzheimers Dis. 2008, 14, 259–269. [Google Scholar] [CrossRef] [PubMed]
- Martin, C.; Aguila, B.; Araya, P.; Vio, K.; Valdivia, S.; Zambrano, A.; Concha, M.I.; Otth, C. Inflammatory and neurodegeneration markers during asymptomatic HSV-1 reactivation. J. Alzheimers Dis. 2014, 39, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Shimeld, C.; Whiteland, J.L.; Nicholls, S.M.; Grinfeld, E.; Easty, D.L.; Gao, H.; Hill, T.J. Immune cell infiltration and persistence in the mouse trigeminal ganglion after infection of the cornea with herpes simplex virus type 1. J. Neuroimmunol. 1995, 61, 7–16. [Google Scholar] [CrossRef]
- Shimeld, C.; Whiteland, J.L.; Williams, N.A.; Easty, D.L.; Hill, T.J. Cytokine production in the nervous system of mice during acute and latent infection with herpes simplex virus type 1. J. Gen. Virol. 1997, 78 Pt 12, 3317–3325. [Google Scholar] [CrossRef]
- Szpara, M.L.; Kobiler, O.; Enquist, L.W. A common neuronal response to alphaherpesvirus infection. J. Neuroimmune Pharmacol. 2010, 5, 418–427. [Google Scholar] [CrossRef]
- Bowen, C.D.; Paavilainen, H.; Renner, D.W.; Palomäki, J.; Lehtinen, J.; Vuorinen, T.; Norberg, P.; Hukkanen, V.; Szpara, M. HSV-1 strains circulating in Finland demonstrate uncoupling of geographic and phenotypic variation. J. Virol. 2018, 93, e01824-18. [Google Scholar] [CrossRef]
- Bower, J.R.; Mao, H.; Durishin, C.; Rozenbom, E.; Detwiler, M.; Rempinski, D.; Karban, T.L.; Rosenthal, K.S. Intrastrain variants of herpes simplex virus type 1 isolated from a neonate with fatal disseminated infection differ in the ICP34.5 gene, glycoprotein processing, and neuroinvasiveness. J. Virol. 1999, 73, 3843–3853. [Google Scholar]
- Mao, H.; Rosenthal, K.S. Strain-Dependent Structural Variants of Herpes Simplex Virus Type 1 ICP34.5 Determine Viral Plaque Size, Efficiency of Glycoprotein Processing, and Viral Release and Neuroinvasive Disease Potential. J. Virol. 2003, 77, 3409–3417. [Google Scholar] [CrossRef] [Green Version]
- Velandia, M.L.; Castellanos, J.E. Flavivirus Neurotropism, Neuroinvasion, Neurovirulence and Neurosusceptibility: Clues to Understanding Flavivirus- and Dengue-Induced Encephalitis. Viral Genomes Mol. Struct. Divers. Gene Expr. Mech. Host Virus Interact. 2012. [Google Scholar] [CrossRef] [Green Version]
- Stroop, W.G.; Schaefer, D.C. Severity of experimentally reactivated herpetic eye disease is related to the neurovirulence of the latent virus. Invest. Ophthalmol. Vis. Sci. 1987, 28, 229–237. [Google Scholar] [PubMed]
- Kolb, A.W.; Lee, K.; Larsen, I.; Craven, M.; Brandt, C.R. Quantitative Trait Locus Based Virulence Determinant Mapping of the HSV-1 Genome in Murine Ocular Infection: Genes Involved in Viral Regulatory and Innate Immune Networks Contribute to Virulence. PLoS Pathog. 2016, 12, e1005499. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.M.; Rayfield, M.A.; Haruta, Y. Strain specificity of spontaneous and adrenergically induced HSV-1 ocular reactivation in latently infected rabbits. Curr. Eye Res. 1987, 6, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, S.; Naderi, M.; Chouljenko, V.N.; Walker, J.D.; Kousoulas, K.G. Amino acid differences in glycoproteins B (gB), C (gC), H (gH) and L (gL) are associated with enhanced herpes simplex virus type-1 (McKrae) entry via the paired immunoglobulin-like type-2 receptor α. Virol. J. 2012, 9, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Watson, G.; Xu, W.; Reed, A.; Babra, B.; Putman, T.; Wick, E.; Wechsler, S.L.; Rohrmann, G.F.; Jin, L. Sequence and comparative analysis of the genome of HSV-1 strain McKrae. Virology 2012, 433, 528–537. [Google Scholar] [CrossRef] [Green Version]
- Perng, G.-C.; Mott, K.R.; Osorio, N.; Yukht, A.; Salina, S.; Nguyen, Q.-H.; Nesburn, A.B.; Wechsler, S.L. Herpes simplex virus type 1 mutants containing the KOS strain ICP34.5 gene in place of the McKrae ICP34.5 gene have McKrae-like spontaneous reactivation but non-McKrae-like virulence. J. Gen. Virol. 2002, 83, 2933–2942. [Google Scholar] [CrossRef] [Green Version]
- Bolovan, C.A.; Sawtell, N.M.; Thompson, R.L. ICP34.5 mutants of herpes simplex virus type 1 strain 17syn+ are attenuated for neurovirulence in mice and for replication in confluent primary mouse embryo cell cultures. J. Virol. 1994, 68, 48–55. [Google Scholar] [Green Version]
- Li, Y.; Zhang, C.; Chen, X.; Yu, J.; Wang, Y.; Yang, Y.; Du, M.; Jin, H.; Ma, Y.; He, B.; et al. ICP34.5 Protein of Herpes Simplex Virus Facilitates the Initiation of Protein Translation by Bridging Eukaryotic Initiation Factor 2α (eIF2α) and Protein Phosphatase. J. Biol. Chem. 2011, 286, 24785–24792. [Google Scholar] [CrossRef]
- Orvedahl, A.; Alexander, D.; Tallóczy, Z.; Sun, Q.; Wei, Y.; Zhang, W.; Burns, D.; Leib, D.A.; Levine, B. HSV-1 ICP34.5 Confers Neurovirulence by Targeting the Beclin 1 Autophagy Protein. Cell Host Microbe 2007, 1, 23–35. [Google Scholar] [CrossRef] [Green Version]
- Perng, G.C.; Thompson, R.L.; Sawtell, N.M.; Taylor, W.E.; Slanina, S.M.; Ghiasi, H.; Kaiwar, R.; Nesburn, A.B.; Wechsler, S.L. An avirulent ICP34.5 deletion mutant of herpes simplex virus type 1 is capable of in vivo spontaneous reactivation. J. Virol. 1995, 69, 3033–3041. [Google Scholar] [Green Version]
- Mao, H.; Rosenthal, K.S. An N-terminal Arginine-rich Cluster and a Proline-Alanine-Threonine Repeat Region Determine the Cellular Localization of the Herpes Simplex Virus Type 1 ICP34.5 Protein and Its Ligand, Protein Phosphatase. J. Biol. Chem. 2002, 277, 11423–11431. [Google Scholar] [CrossRef] [PubMed]
- Eimer, W.A.; Vijaya Kumar, D.K.; Navalpur Shanmugam, N.K.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; György, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated β-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 99, 56–63.e3. [Google Scholar] [CrossRef] [PubMed]
- Ezzat, K.; Pernemalm, M.; Pålsson, S.; Roberts, T.C.; Järver, P.; Dondalska, A.; Bestas, B.; Sobkowiak, M.J.; Levänen, B.; Sköld, M.; et al. The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat. Commun. 2019, 10, 2331. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.-B.; Ferland, P.; Webster, P.; Bearer, E.L. Herpes Simplex Virus Dances with Amyloid Precursor Protein while Exiting the Cell. PLoS ONE 2011, 6, e17966. [Google Scholar] [CrossRef] [PubMed]
- Santana, S.; Recuero, M.; Bullido, M.J.; Valdivieso, F.; Aldudo, J. Herpes simplex virus type I induces the accumulation of intracellular β-amyloid in autophagic compartments and the inhibition of the non-amyloidogenic pathway in human neuroblastoma cells. Neurobiol. Aging 2012, 33, 430.e19–430.e33. [Google Scholar] [CrossRef]
- Bourgade, K.; Garneau, H.; Giroux, G.; Le Page, A.Y.; Bocti, C.; Dupuis, G.; Frost, E.H.; Fülöp, T. β-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology 2015, 16, 85–98. [Google Scholar] [CrossRef]
- Bourgade, K.; Le Page, A.; Bocti, C.; Witkowski, J.M.; Dupuis, G.; Frost, E.H.; Fueloep, T. Protective Effect of Amyloid-beta Peptides Against Herpes Simplex Virus-1 Infection in a Neuronal Cell Culture Model. J. Alzheimer’s Dis. 2016, 50, 1227–1241. [Google Scholar] [CrossRef]
- Rathore, N.; Ramani, S.R.; Pantua, H.; Payandeh, J.; Bhangale, T.; Wuster, A.; Kapoor, M.; Sun, Y.; Kapadia, S.B.; Gonzalez, L.; et al. Paired Immunoglobulin-like Type 2 Receptor Alpha G78R variant alters ligand binding and confers protection to Alzheimer’s disease. PLoS Genet. 2018, 14, e1007427. [Google Scholar] [CrossRef]
- Dubin, G.; Socolof, E.; Frank, I.; Friedman, H.M. Herpes simplex virus type 1 Fc receptor protects infected cells from antibody-dependent cellular cytotoxicity. J. Virol. 1991, 65, 7046–7050. [Google Scholar] [Green Version]
- Dubin, G.; Frank, I.; Friedman, H.M. Herpes simplex virus type 1 encodes two Fc receptors which have different binding characteristics for monomeric immunoglobulin G (IgG) and IgG complexes. J. Virol. 1990, 64, 2725–2731. [Google Scholar] [Green Version]
- Atherton, A.; Armour, K.L.; Bell, S.; Minson, A.C.; Clark, M.R. The herpes simplex virus type 1 Fc receptor discriminates between IgG1 allotypes. Eur. J. Immunol. 2000, 30, 2540–2547. [Google Scholar] [CrossRef]
- Pandey, J.P. Immunoglobulin Genes and Immunity to Herpes Simplex Virus Type 1. J. Infect. Dis 2012, 206, 143–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandey, J.P.; Kothera, R.T.; Liu, S.; Costa, A.S.; Mancuso, R.; Agostini, S. Immunoglobulin Genes and Immunity to HSV1 in Alzheimer’s Disease. J. Alzheimer’s Dis. 2019, 70, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.J.; Lukiw, W.J.; Kammerman, E.M.; Hill, J.M. Intracerebral propagation of Alzheimer’s disease: Strengthening evidence of a herpes simplex virus etiology. Alzheimer’s Dement. 2013, 9, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Castellano, J.M.; Kim, J.; Stewart, F.R.; Jiang, H.; DeMattos, R.B.; Patterson, B.W.; Fagan, A.M.; Morris, J.C.; Mawuenyega, K.G.; Cruchaga, C.; et al. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 2011, 3, 89ra57. [Google Scholar] [CrossRef] [PubMed]
- Safieh, M.; Korczyn, A.D.; Michaelson, D.M. ApoE4: An emerging therapeutic target for Alzheimer’s disease. BMC Med. 2019, 17, 64. [Google Scholar] [CrossRef] [PubMed]
- Lövheim, H.; Norman, T.; Weidung, B.; Olsson, J.; Josefsson, M.; Adolfsson, R.; Nyberg, L.; Elgh, F. Herpes Simplex Virus, APOEε4, and Cognitive Decline in Old Age: Results from the Betula Cohort Study. J. Alzheimers Dis. 2019, 67, 211–220. [Google Scholar] [CrossRef]
- Burgos, J.S.; Ramirez, C.; Sastre, I.; Bullido, M.J.; Valdivieso, F. ApoE4 is more efficient than E3 in brain access by herpes simplex virus type 1. Neuroreport 2003, 14, 1825–1827. [Google Scholar] [CrossRef]
- Burgos, J.S.; Ramirez, C.; Sastre, I.; Valdivieso, F. Effect of apolipoprotein E on the cerebral load of latent herpes simplex virus type 1 DNA. J. Virol. 2006, 80, 5383–5387. [Google Scholar] [CrossRef]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mangold, C.A.; Szpara, M.L. Persistent Infection with Herpes Simplex Virus 1 and Alzheimer’s Disease—A Call to Study How Variability in Both Virus and Host may Impact Disease. Viruses 2019, 11, 966. https://doi.org/10.3390/v11100966
Mangold CA, Szpara ML. Persistent Infection with Herpes Simplex Virus 1 and Alzheimer’s Disease—A Call to Study How Variability in Both Virus and Host may Impact Disease. Viruses. 2019; 11(10):966. https://doi.org/10.3390/v11100966
Chicago/Turabian StyleMangold, Colleen A., and Moriah L. Szpara. 2019. "Persistent Infection with Herpes Simplex Virus 1 and Alzheimer’s Disease—A Call to Study How Variability in Both Virus and Host may Impact Disease" Viruses 11, no. 10: 966. https://doi.org/10.3390/v11100966
APA StyleMangold, C. A., & Szpara, M. L. (2019). Persistent Infection with Herpes Simplex Virus 1 and Alzheimer’s Disease—A Call to Study How Variability in Both Virus and Host may Impact Disease. Viruses, 11(10), 966. https://doi.org/10.3390/v11100966