Diversity and Evolution of Novel Invertebrate DNA Viruses Revealed by Meta-Transcriptomics

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sample Collection, RNA Extraction, Library Preparation, and Sequencing
2.2. Sequence Assembly and Virus Discovery
2.3. Phylogenetic Analysis
3. Results
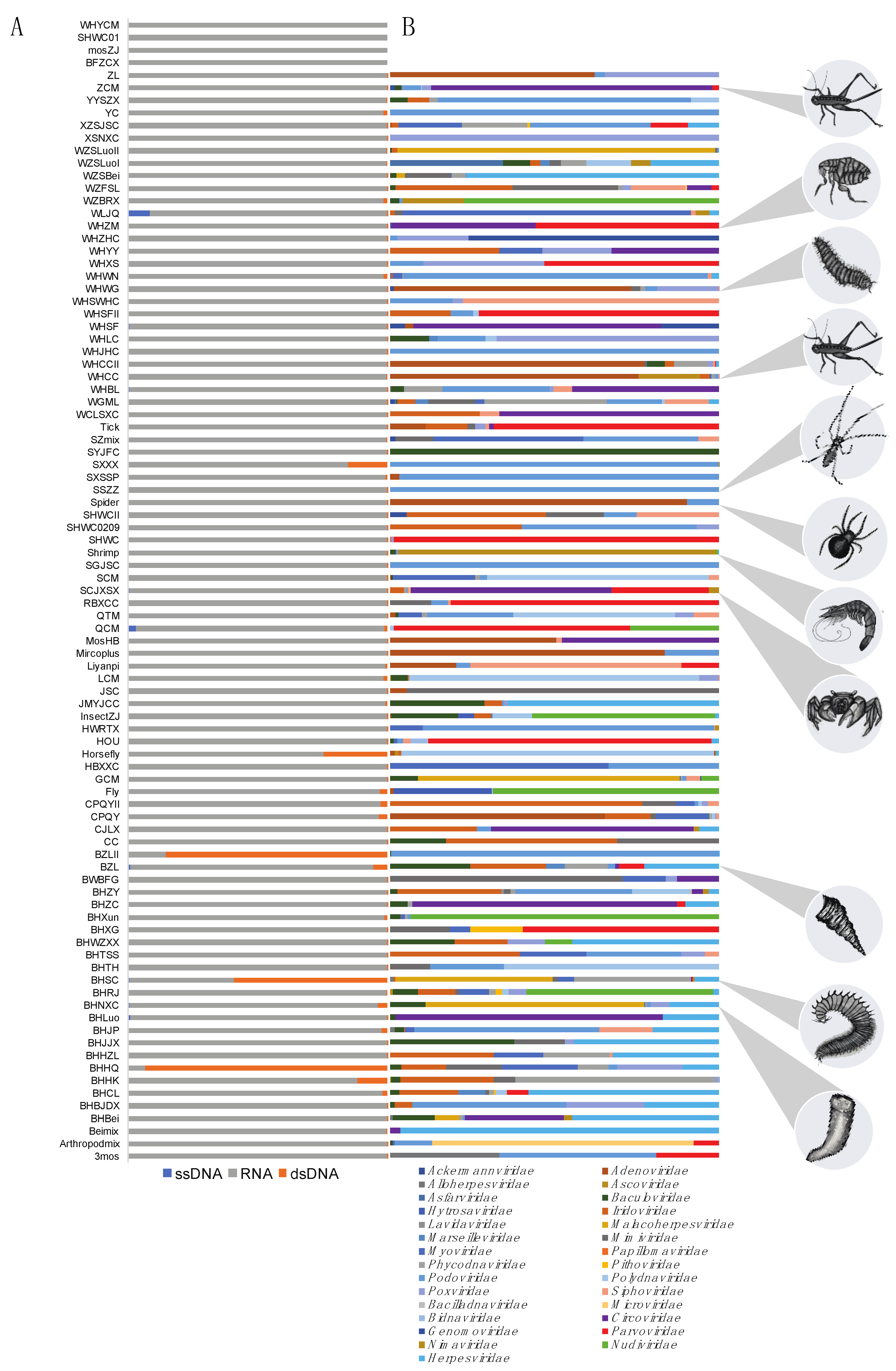
3.1. Data Mining for DNA Viruses
3.2. Diversity and Phylogenetic Relationships of Novel ssDNA Viruses
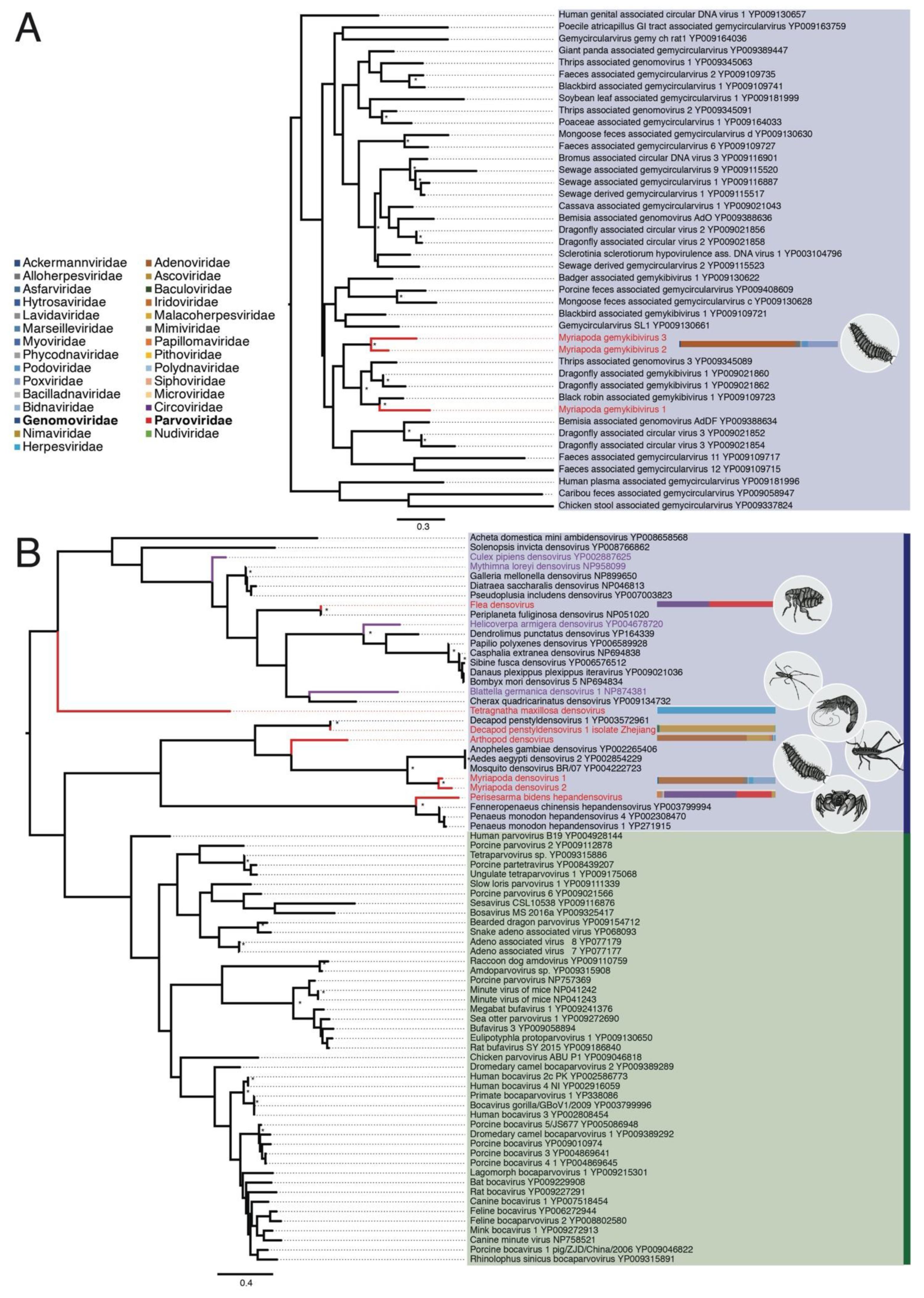
3.2.1. Genomoviridae
3.2.2. Parvoviridae
3.2.3. Circoviridae
3.3. Discovery and Phylogenetic Relationships of Novel dsDNA Viruses
3.3.1. Herpesviridae
3.3.2. Polyomaviridae
3.3.3. Nimaviridae
3.3.4. Nudiviridae
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. eLife 2015, 4, e05378. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E.; Sulakvelidze, A. Bacteriophages: Biology and Applications; CRC Press: Boca Raton, FL, USA, 2004. [Google Scholar]
- Rohwer, F. Global phage diversity. Cell 2003, 113, 141. [Google Scholar] [CrossRef]
- Shi, M.; Zhang, Y.Z.; Holmes, E.C. Meta-transcriptomics and the evolutionary biology of RNA viruses. Virus Res. 2018, 243, 83–90. [Google Scholar] [CrossRef]
- Rosario, K.; Dayaram, A.; Marinov, M.; Ware, J.; Kraberger, S.; Stainton, D.; Breitbart, M.; Varsani, A. Diverse circular ssDNA viruses discovered in dragonflies (Odonata: Epiprocta). J. Gen. Virol. 2012, 93, 2668–2681. [Google Scholar] [CrossRef] [PubMed]
- Dolja, V.V.; Koonin, E.V. Metagenomics reshapes the concepts of RNA virus evolution by revealing extensive horizontal virus transfer. Virus Res. 2018, 244, 36–52. [Google Scholar] [CrossRef]
- Delwart, E.; Li, L.L. Rapidly expanding genetic diversity and host range of the Circoviridae viral family and other Rep encoding small circular ssDNA genomes. Virus Res. 2012, 164, 114–121. [Google Scholar] [CrossRef]
- Rosario, K.; Mettel, K.A.; Benner, B.E.; Johnson, R.; Scott, C.; Yusseff-Vanegas, S.Z.; Baker, C.C.M.; Cassill, D.L.; Storer, C.; Varsani, A.; et al. Virus discovery in all three major lineages of terrestrial arthropods highlights the diversity of single-stranded DNA viruses associated with invertebrates. PeerJ 2018, 6, e5761. [Google Scholar] [CrossRef]
- Kim, K.H.; Chang, H.W.; Nam, Y.D.; Roh, S.W.; Kim, M.S.; Sung, Y.; Jeon, C.O.; Oh, H.M.; Bae, J.W. Amplification of uncultured single-stranded DNA viruses from rice paddy soil. Appl. Environ. Microbiol. 2008, 74, 5975–5985. [Google Scholar] [CrossRef]
- Kim, K.H.; Bae, J.W. Amplification methods bias metagenomic libraries of uncultured single-stranded and double-stranded DNA viruses. Appl. Environ. Microbiol. 2011, 77, 7663–7668. [Google Scholar] [CrossRef]
- Kazlauskas, D.; Dayaram, A.; Kraberger, S.; Goldstien, S.; Varsani, A.; Krupovic, M. Evolutionary history of ssDNA bacilladnaviruses features horizontal acquisition of the capsid gene from ssRNA nodaviruses. Virology 2017, 504, 114–121. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Ghabrial, S.A.; Jiang, D.; Varsani, A. Genomoviridae: A new family of widespread single-stranded DNA viruses. Arch. Virol. 2016, 161, 2633–2643. [Google Scholar] [CrossRef] [PubMed]
- Rosario, K.; Breitbart, M.; Harrach, B.; Segales, J.; Delwart, E.; Biagini, P.; Varsani, A. Revisiting the taxonomy of the family Circoviridae: Establishment of the genus Cyclovirus and removal of the genus Gyrovirus. Arch. Virol. 2017, 162, 1447–1463. [Google Scholar] [CrossRef] [PubMed]
- Varsani, A.; Krupovic, M. Smacoviridae: A new family of animal-associated single-stranded DNA viruses. Arch. Virol. 2018, 163, 3213–3214. [Google Scholar] [CrossRef]
- Palacios, G.; Druce, J.; Du, L.; Tran, T.; Birch, C.; Briese, T.; Conlan, S.; Quan, P.L.; Hui, J.; Marshall, J.; et al. A new arenavirus in a cluster of fatal transplant-associated diseases. N. Engl. J. Med. 2008, 358, 991–998. [Google Scholar] [CrossRef]
- Li, C.-X.; Chang, W.-S.; Mitsakos, K.; Rodger, J.; Holmes, E.C.; Hudson, B.J. Identification of a novel equine papillomavirus in semen from a thoroughbred stallion with a penile lesion. Viruses 2019, 11, 713. [Google Scholar] [CrossRef]
- Asplund, M.; Kjartansdottir, K.R.; Mollerup, S.; Vinner, L.; Fridholm, H.; Herrera, J.A.R.; Friis-Nielsen, J.; Hansen, T.A.; Jensen, R.H.; Nielsen, I.B.; et al. Contaminating viral sequences in high-throughput sequencing viromics: A linkage study of 700 sequencing libraries. Clin. Microbiol. Infect. 2019, 25, 1277–1285. [Google Scholar] [CrossRef]
- Holmes, E.C. Reagent contamination in viromics: All that glitters is not gold. Clin. Microbiol. Infect. 2019, 25, 1167–1168. [Google Scholar] [CrossRef]
- Wilson, E.O. The little things that run the world (the importance and conservation of invertebrates). Conserv. Biol. 1987, 1, 344–346. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Krupovic, M. Networks of evolutionary interactions underlying the polyphyletic origin of ssDNA viruses. Curr. Opin. Virol. 2013, 3, 578–586. [Google Scholar] [CrossRef]
- Belyi, V.A.; Levine, A.J.; Skalka, A.M. Sequences from ancestral single-stranded DNA viruses in vertebrate genomes: The parvoviridae and circoviridae are more than 40 to 50 million years old. J. Virol. 2010, 84, 12458–12462. [Google Scholar] [CrossRef]
- Iyer, L.A.; Balaji, S.; Koonin, E.V.; Aravind, L. Evolutionary genomics of nucleo-cytoplasmic large DNA viruses. Virus Res. 2006, 117, 156–184. [Google Scholar] [CrossRef]
- Moens, M.A.J.; Perez-Tris, J.; Cortey, M.; Benitez, L. Identification of two novel CRESS DNA viruses associated with an Avipoxvirus lesion of a blue-and-gray Tanager (Thraupis episcopus). Infect. Genet. Evol. 2018, 60, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Abbas, A.A.; Taylor, L.J.; Dothard, M.I.; Leiby, J.S.; Fitzgerald, A.S.; Khatib, L.A.; Collman, R.G.; Bushman, F.D. Redondoviridae, a family of small, circular DNA Vvruses of the human oro-respiratory tract associated with periodontitis and critical Illness. Cell Host Microbe 2019, 26, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Kerr, M.; Rosario, K.; Baker, C.C.M.; Breitbart, M. Discovery of four novel circular single-stranded DNA viruses in fungus-farming termites. Microbiol. Resour. Announc. 2018, 6, e00318-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kraberger, S.; Hofstetter, R.W.; Potter, K.A.; Farkas, K.; Varsani, A. Genomoviruses associated with mountain and western pine beetles. Virus Res. 2018, 256, 17–20. [Google Scholar] [CrossRef] [PubMed]
- Kraberger, S.; Schmidlin, K.; Fontenele, R.S.; Walters, M.; Varsani, A. Unravelling the single-stranded DNA virome of the New Zealand Blackfly. Viruses 2019, 11, 532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosario, K.; Schenck, R.O.; Harbeitner, R.C.; Lawler, S.N.; Breitbart, M. Novel circular single-stranded DNA viruses identified in marine invertebrates reveal high sequence diversity and consistent predicted intrinsic disorder patterns within putative structural proteins. Front. Microbiol. 2015, 6, 696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labonte, J.M.; Suttle, C.A. Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 2013, 7, 2169–2177. [Google Scholar] [CrossRef]
- Kraberger, S.; Arguello-Astorga, G.R.; Greenfield, L.G.; Galilee, C.; Law, D.; Martin, D.P.; Varsani, A. Characterisation of a diverse range of circular replication-associated protein encoding DNA viruses recovered from a sewage treatment oxidation pond. Infect. Genet. Evol. 2015, 31, 73–86. [Google Scholar] [CrossRef]
- Crowther, R.A.; Berriman, J.A.; Curran, W.L.; Allan, G.M.; Todd, D. Comparison of the structures of three circoviruses: Chicken anemia virus, porcine circovirus type 2, and beak and feather disease virus. J. Virol. 2003, 77, 13036–13041. [Google Scholar] [CrossRef] [Green Version]
- Todd, D. Circoviruses: Immunosuppressive threats to avian species: A review. Avian Pathol. 2000, 29, 373–394. [Google Scholar] [CrossRef]
- Todd, D.; Niagro, F.D.; Ritchie, B.W.; Curran, W.; Allan, G.M.; Lukert, P.D.; Latimer, K.S.; Steffens, W.L., 3rd; McNulty, M.S. Comparison of three animal viruses with circular single-stranded DNA genomes. Arch. Virol. 1991, 117, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, M.; Csagola, A.; Farkas, S.L.; Szekely, C.; Tuboly, T. First detection and analysis of a fish circovirus. J. Gen. Virol. 2011, 92, 1817–1821. [Google Scholar] [CrossRef] [PubMed]
- Lorincz, M.; Dan, A.; Lang, M.; Csaba, G.; Toth, A.G.; Szekely, C.; Csagola, A.; Tuboly, T. Novel circovirus in European catfish (Silurus glanis). Arch. Virol. 2012, 157, 1173–1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Shan, T.; Soji, O.B.; Alam, M.M.; Kunz, T.H.; Zaidi, S.Z.; Delwart, E. Possible cross-species transmission of circoviruses and cycloviruses among farm animals. J. Gen. Virol. 2011, 92, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; McGraw, S.; Zhu, K.; Leutenegger, C.M.; Marks, S.L.; Kubiski, S.; Gaffney, P.; Dela Cruz, F.N., Jr.; Wang, C.; Delwart, E.; et al. Circovirus in tissues of dogs with vasculitis and hemorrhage. Emerg. Infect. Dis. 2013, 19, 534–541. [Google Scholar] [CrossRef]
- Lian, H.; Liu, Y.; Li, N.; Wang, Y.; Zhang, S.; Hu, R. Novel circovirus from mink, China. Emerg. Infect. Dis. 2014, 20, 1548–1550. [Google Scholar] [CrossRef]
- Lima, F.E.; Cibulski, S.P.; Dall Bello, A.G.; Mayer, F.Q.; Witt, A.A.; Roehe, P.M.; d’Azevedo, P.A. A Novel chiropteran circovirus genome recovered from a Brazilian insectivorous bat species. Genome Announc. 2015, 3, e01393-15. [Google Scholar] [CrossRef] [Green Version]
- Wu, P.C.; Chen, T.Y.; Chi, J.N.; Chien, M.S.; Huang, C. Efficient expression and purification of porcine circovirus type 2 virus-like particles in Escherichia coli. J. Biotechnol. 2016, 220, 78–85. [Google Scholar] [CrossRef]
- Ge, X.; Li, J.; Peng, C.; Wu, L.; Yang, X.; Wu, Y.; Zhang, Y.; Shi, Z. Genetic diversity of novel circular ssDNA viruses in bats in China. J. Gen. Virol. 2011, 92, 2646–2653. [Google Scholar] [CrossRef]
- Meehan, B.M.; McNeilly, F.; Todd, D.; Kennedy, S.; Jewhurst, V.A.; Ellis, J.A.; Hassard, L.E.; Clark, E.G.; Haines, D.M.; Allan, G.M. Characterization of novel circovirus DNAs associated with wasting syndromes in pigs. J. Gen. Virol. 1998, 79, 2171–2179. [Google Scholar] [CrossRef]
- Rosario, K.; Marinov, M.; Stainton, D.; Kraberger, S.; Wiltshire, E.J.; Collings, D.A.; Walters, M.; Martin, D.P.; Breitbart, M.; Varsani, A. Dragonfly cyclovirus, a novel single-stranded DNA virus discovered in dragonflies (Odonata: Anisoptera). J. Gen. Virol. 2011, 92, 1302–1308. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.U.; Lin, W.; Wu, R.; Lin, C.; Islam, W.; Arif, M.; Du, Z.; Wu, Z. Complete genome sequences of three novel cycloviruses identified in a dragonfly (Odonata: Anisoptera) from China. Arch. Virol. 2018, 163, 2569–2573. [Google Scholar] [CrossRef] [PubMed]
- Naccache, S.N.; Greninger, A.L.; Lee, D.; Coffey, L.L.; Phan, T.; Rein-Weston, A.; Aronsohn, A.; Hackett, J., Jr.; Delwart, E.L.; Chiu, C.Y. The perils of pathogen discovery: Origin of a novel parvovirus-like hybrid genome traced to nucleic acid extraction spin columns. J. Virol. 2013, 87, 11966–11977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Fu, Y.; Xie, J.; Jiang, D.; Li, G.; Yi, X. A novel virus that infecting hypovirulent strain XG36-1 of plant fungal pathogen Sclerotinia sclerotiorum. Virol. J. 2009, 6, 96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidlin, K.; Sepp, T.; Khalifeh, A.; Smith, K.; Fontenele, R.S.; McGraw, K.J.; Varsani, A. Diverse genomoviruses representing eight new and one known species identified in feces and nests of house finches (Haemorhous mexicanus). Arch. Virol. 2019, 164, 2345–2350. [Google Scholar] [CrossRef] [PubMed]
- Cotmore, S.F.; Agbandje-McKenna, M.; Chiorini, J.A.; Mukha, D.V.; Pintel, D.J.; Qiu, J.; Soderlund-Venermo, M.; Tattersall, P.; Tijssen, P.; Gatherer, D.; et al. The family Parvoviridae. Arch. Virol. 2014, 159, 1239–1247. [Google Scholar] [CrossRef] [PubMed]
- Halder, S.; Nam, H.J.; Govindasamy, L.; Vogel, M.; Dinsart, C.; Salome, N.; McKenna, R.; Agbandje-McKenna, M. Production, purification, crystallization and structure determination of H-1 Parvovirus. Acta Cryst. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 1571–1576. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Tattersall, P. Parvovirus diversity and DNA damage responses. Cold Spring Harb. Perspect. Biol. 2013, 5, a012989. [Google Scholar] [CrossRef] [Green Version]
- Cotmore, S.F.; Tattersall, P. Parvoviral host range and cell entry mechanisms. Adv. Virus Res. 2007, 70, 183–232. [Google Scholar] [CrossRef]
- Agbandje-McKenna, M.; Kleinschmidt, J. AAV capsid structure and cell interactions. Methods Mol. Biol. 2011, 807, 47–92. [Google Scholar] [CrossRef]
- Cotmore, S.F.; Agbandje-McKenna, M.; Canuti, M.; Chiorini, J.A.; Eis-Hubinger, A.M.; Hughes, J.; Mietzsch, M.; Modha, S.; Ogliastro, M.; Penzes, J.J.; et al. ICTV Virus Taxonomy Profile: Parvoviridae. J. Gen. Virol. 2019, 100, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.; Bergoin, M.; van Oers, M.M. Diversity of large DNA viruses of invertebrates. J. Invertebr. Pathol. 2017, 147, 4–22. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Paz, A. White spot syndrome virus: An overview on an emergent concern. Vet. Res. 2010, 41, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hameed, A.S.S.; Yoganadhan, K.; Sathish, S.; Rasheed, M.; Murugan, V.; Jayaraman, K. White spot syndrome virus (WSSV) in two species of freshwater crabs (Paratelphusa hydrodomous and P. pulvinata). Aquaculture 2001, 201, 179–186. [Google Scholar] [CrossRef]
- Savin, K.W.; Cocks, B.G.; Wong, F.; Sawbridge, T.; Cogan, N.; Savage, D.; Warner, S. A neurotropic herpesvirus infecting the gastropod, abalone, shares ancestry with oyster herpesvirus and a herpesvirus associated with the amphioxus genome. Virol. J. 2010, 7, 308. [Google Scholar] [CrossRef] [Green Version]
- Madinda, N.F.; Ehlers, B.; Wertheim, J.O.; Akoua-Koffi, C.; Bergl, R.A.; Boesch, C.; Akonkwa, D.B.M.; Eckardt, W.; Fruth, B.; Gillespie, T.R.; et al. Assessing host-virus codivergence for close relatives of Merkel Cell polyomavirus infecting African Great Apes. J. Virol. 2016, 90, 8531–8541. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Family | Virus Name | Library | Length | NR * Protein Hit | NR * Protein Hit Name and Species | NR * Hit Percentage (%) Amino Acid Identity | NR * Hit Length | NR * e-Value | Taxonomy as Identified by Taxonomist (Family, Order, Genus) | Conserved Domain ID | Conserved Domain Name | Conserved Domain e-Value | Abundance in Library (%) ** | |
| ssDNA | Parvoviridae | Tetragnatha maxillosa densovirus | SSZZ | 1087 | YP_164339.1 | Nonstructural protein 1 [Dendrolimus punctatus densovirus] | 28.7 | 157 | 4.40 × 10−11 | Parvoviridae, Densovirinae, Iteradensovirus | 329234 | Parvo_NS1 | 2.50 × 10−15 | 0.002 |
| Perisesarma bidens hepandensovirus | SCJXSX | 940 | YP_003799994.1 | Non-structural protein 1 [Fenneropenaeus chinensis hepandensovirus] | 59 | 156 | 5.10 × 10−48 | Parvoviridae, Densovirinae, Hepandensovirus | 0.0002 | |||||
| Periplaneta fuliginosa densovirus variant Hubei | WHZM | 1658 | NP_051017.1 | Structural protein [Periplaneta fuliginosa densovirus] | 100 | 353 | 1.50 × 10−204 | Parvoviridae, Densovirinae, Ambidensovirus | 280494 | Denso_VP4 | 0 | 0.27 | ||
| Arthropod densovirus | WHCC | 970 | AJD76764.1 | NS1-like protein 2 [Penaeus stylirostris densovirus] | 41.8 | 170 | 2.30 × 10−27 | Parvoviridae, Densovirinae | 329234 | Parvo_NS1 | 8.91 × 10−14 | 0.0001 | ||
| Decapod penstyldensovirus 1 variant Zhejiang | Shrimp | 3137 | AGO44085.1 | Nonstructural protein 1 [Decapod penstyldensovirus 1] | 94.6 | 723 | 0 | Parvoviridae, Densovirinae, Penstyldensovirus | 329234 | Parvo_NS1 | 9.36 × 10−09 | 0.02 | ||
| Mydriapoda densovirus 1 | WGML | 739 | AAT35223.1 | Nonstructural protein 1 [Haemagogus equinus densovirus] | 46.6 | 249 | 7.30 × 10−58 | Parvoviridae, Densovirinae, Brevidensovirus | 0.00006 | |||||
| Myridapoda densovirus 2 | WGML | 2460 | AAT35223.1 | Nonstructural protein 1 [Haemagogus equinus densovirus] | 43.5 | 481 | 2.50 × 10−102 | Parvoviridae, Densovirinae, Brevidensovirus | 329234 | Parvo_NS1 | 2.55 × 10−05 | 0.001 | ||
| Flea cyclovirus | WHZM | 1602 | AEL87792.1 | Putative replication-associated protein [Bat circovirus ZS/China/2011] | 97.9 | 286 | 8.40 × 10−168 | Circoviridae, Cyclovirus | 280553 | Viral_Rep | 7.27 × 10−16 | 0.87 | ||
| ssDNA | Circoviridae | Sandworm circovirus | BHSC | 1063 | YP_009116906.1 | Replication-associated protein [Sewage-associated circular DNA virus-21] | 41.5 | 236 | 5.80 × 10−40 | NA | 280553 | Viral_Rep | 1.02 × 10−09 | 0.0004 |
| Arthropod cyclovirus | ZCM | 1911 | AIZ46815.1 | Replication associated protein [Swine cyclovirus] | 77.4 | 221 | 3.80 × 10−98 | Circoviridae, Cyclovirus | 280553 | Viral_Rep | 0.00136865 | 0.02 | ||
| Family | Virus name | Library | Length | NR * protein hit | NR * protein hit name and species | NR * hit percentage | NR * hit length | NR * e-value | Taxonomy as identified by Taxonomist (Family, Order, Genus) | Conserved domain ID | Conserved domain name | Conserved domain e-value | Abundance in library (%) ** | |
| Genomoviridae | Myriapoda gemycircularvirus 1 | WGML | 4487 | YP_009109722.1 | Capsid protein [Faeces-associated gemycircularvirus 8] | 66.3 | 264 | 5.00 × 10−101 | Genomoviridae, Gemykibivirus | Gemini_AL1 | 6.57 × 10−10 | 0.003 | ||
| Myriapoda gemycircularvirus 2 | WGML | 4571 | YP_009259554.1 | Replication initiation associated protein [Gemycircularvirus HV-GcV2] | 49.3 | 343 | 1.50 × 10−84 | Genomoviridae, Gemykibivirus | Gemini_AL1 | 3.30 × 10−18 | 0.0005 | |||
| Myriapoda gemycircularvirus 3 | WGML | 4648 | YP_009259554.1 | Replication initiation associated protein [Gemycircularvirus HV-GcV2] | 46.9 | 356 | 1.30 × 10−83 | Genomoviridae, Gemykibivirus | Gemini_AL1 | 2.90 × 10−13 | 0.005 | |||
| dsDNA | Polyomaviridae | Spider polyomavirus | Spider | 2281 | AID54935.1 | Large T antigen [Trichodysplasia spinulosa-associated polyomavirus] | 35.2 | 244 | 1.20 × 10−26 | Polyomaviridae, Alphapolyomavirus | 333065 | PHA03102 | 1.92 × 10−40 | N/A |
| Nimaviridae | White spot syndrome virus variant Zhejiang | Shrimp | 837 | YP_009220600.1 | Anti-apoptosis protein [White spot syndrome virus] | 88.5 | 139 | 2.90 × 10−63 | Nimaviridae, Whispovirus | 329062 | TK | 3.83 × 10−17 | 0.0001 | |
| Nudiviridae | Charybdis crab nudivirus 1 | BHXun | 1054 | YP_009051843.1 | DNA polymerase [Penaeus monodon nudivirus] | 48.5 | 342 | 4.70 × 10−90 | Nudiviridae | 324564 | POLBc | 7.10 × 10−10 | 0.000553059 | |
| Nudiviridae | Charybdis crab nudivirus 2 | BHXun | 1168 | YP_009051843.1 | DNA polymerase [Penaeus monodon nudivirus] | 52.6 | 371 | 6.90 × 10−103 | Nudiviridae | 332123 | POLBc | 3.63 × 10−12 | 0.000721381 | |
| Herpesviridae | Sandworm herpesvirus | BHSC | 1971 | AMW36220.1 | Putative DNA packaging terminase [Abalone herpesvirus Taiwan/2005] | 33.9 | 254 | 9.50 × 10−28 | Herpesvirales | 333071 | AddB | 0.011 | 0.000222584 | |
| Peanut worm herpesvirus | BHNXC | 13115 | YP_009229862.1 | DNA packaging terminase subunit 1 [Myotis gammaherpesvirus 8] | 25.6 | 429 | 9.40 × 10−23 | Herpesviridae, Gammaherpesvirinae | 330553 | SMC_N | 0.000744176 | 0.018654163 |
| Library | Name | Host Classification | Species of Host (No. Individuals) | Sample Type | Habitat | Data Generated | Library Accession |
|---|---|---|---|---|---|---|---|
| Hubei Myriapoda mix | WGML | Arthropoda: Myriapoda | Diplopoda sp. (7), Otostigmus scaber (4), Scolopocryptops sp.(3), Otostigmus scaber (1), Myriapoda sp. (1). | Whole individual | Hubei (Terrestrial) | 7,337,861,800 | SRX1029951 |
| Hubei flea and ant mix | WHZM | Arthropoda: Insecta | Ctenocephalides felis (10-15), Tetramorium tsushimae (N/A) | Whole individual | Hubei (Terrestrial) | 1,680,999,000 | SRR3400840 |
| Spiders | Spider | Arthropoda: Chelicerata | Neoscona nautica (14), Parasteatoda tepidariorum (3), Plexippus setipes (3), Pirata sp. (1), Araneae sp. (8) | Whole individual | Hubei (Terrestrial) | 11,361,912,300 | SRX833697 |
| Insect Mix 4 (described previously) | WHCC | Arthropoda | Pseudothemis zonata (2), Nepidae sp. (2 species, 3), Camponotus japonicas (~5), Diplonychus sp. (2), Asellus sp. (N/A) | Whole individual | Hubei (Terrestrial) | 11,973,368,200 | SRX833688 |
| Shrimps | Shrimp | Arthropoda: Crustacea | Exopalaemon carinicauda (12), Metapenaeus sp. (6), Solenocera crassicornis (6), Penaeus monodon (12), Litopenaeus vannamei (12) | Representative tissues from gill, various glands, muscle, and gut | Zhejiang (Costal/Marine) | 5,365,359,900 | SRX833698 |
| Tetragnatha maxillosa mix Hubei (subset of arthropodmix) | SSZZ | Arthropoda: Chelicerata | Tetragnatha maxillosa (2) | Whole individual | Hubei (Land/Freshwater) | 2,933,255,400 | SRR3401144 |
| Sesarmid crab mix Beihai | SCJXSX | Arthropoda: Crustacea | Perisesarma bidens (8) | Representative tissues from gill, various glands, muscle, and stomach (cleared) | Guangxi (Costal/Marine) | 6,176,296,400 | SRR3401386 |
| Turritella sea snails mix Beihai | BZL | Mollusca | Turritella sp.(12) | Visceral mass with gut content removed | Guangxi (Costal/Marine) | 5,294,621,400 | SRX1712841 |
| Orthoptera mix Hubei | ZCM | Arthropoda: Insecta | Orthoptera sp. (3), Conocephalus sp. (4), Gryllidae sp. (3) | Whole individual | Hubei (Terrestrial) | 7,042,417,800 | SRX1029952 |
| Sandworms mix Beihai | BHSC | Annelida | Marphysa sp. (12), Nereis aibuhitensis (12) | Whole individual | Guangxi (Costal/Marine) | 6,114,388,200 | SRX1712659 |
| Millipedes | WHWG | Arthropoda: Myriapoda | Polydesmidae sp. (2 species, 12 individuals) | Whole individual | Hubei, Beijing (Terrestrial) | 7,176,702,400 | SRX833700 |
| Charybdis crab mix Beihai | BHXun | Arthropoda: Crustacea | Charybdis sp. (12) | Representative tissues from gill, various glands, muscle, and stomach (cleaned) | Guangxi (Costal/Marine) | 6,439,342,400 | SRR3401303 |
| Arthopod mix Hubei | Arthropodmix | Arthropoda | Cicadellidae sp. (10), Tetragnatha maxillosa (1), Heteroptera sp. (10), Scutigeridae sp. (2), Ctenocephalides felis (10-15), Tetramorium tsushimae (N\A) | Whole individual | Hubei (Terrestrial) | 7,768,505,600 | SRX1029954 |
| Peanut worms mix Beihai | BHNXC | Sipuncula | Phascolosoma esculenta (12), Sipunculus nudus (12) | Whole individual | Guangxi (Costal/Marine) | 7,041,903,800 | SRR3401570 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Porter, A.F.; Shi, M.; Eden, J.-S.; Zhang, Y.-Z.; Holmes, E.C. Diversity and Evolution of Novel Invertebrate DNA Viruses Revealed by Meta-Transcriptomics. Viruses 2019, 11, 1092. https://doi.org/10.3390/v11121092
Porter AF, Shi M, Eden J-S, Zhang Y-Z, Holmes EC. Diversity and Evolution of Novel Invertebrate DNA Viruses Revealed by Meta-Transcriptomics. Viruses. 2019; 11(12):1092. https://doi.org/10.3390/v11121092
Chicago/Turabian StylePorter, Ashleigh F., Mang Shi, John-Sebastian Eden, Yong-Zhen Zhang, and Edward C. Holmes. 2019. "Diversity and Evolution of Novel Invertebrate DNA Viruses Revealed by Meta-Transcriptomics" Viruses 11, no. 12: 1092. https://doi.org/10.3390/v11121092