Microtubule-Dependent Trafficking of Alphaherpesviruses in the Nervous System: The Ins and Outs


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Alphaherpesvirinae and the Road Travelled: An Overview
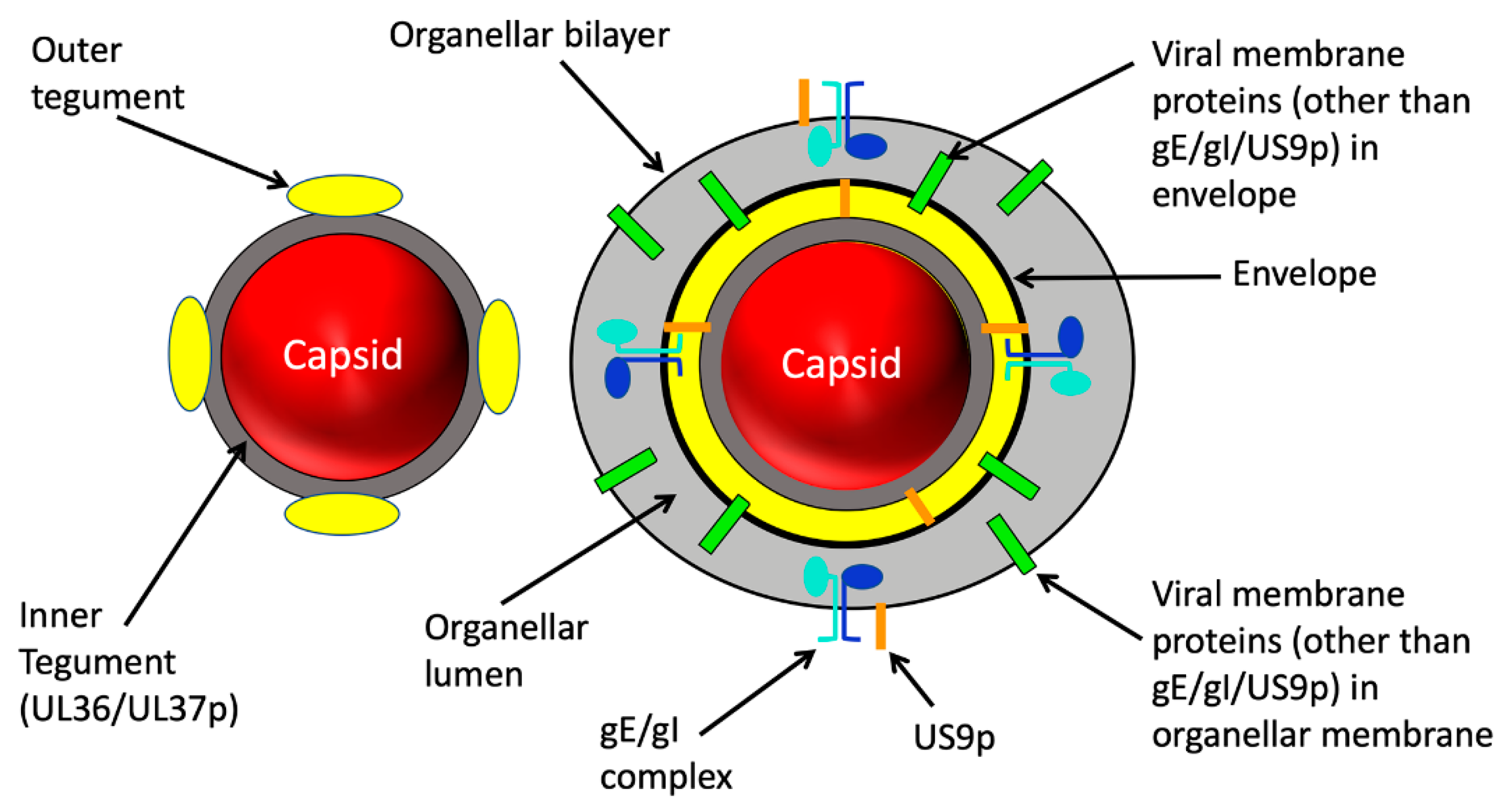
2. Structure of the Trafficking Alphaherpesvirus Particle
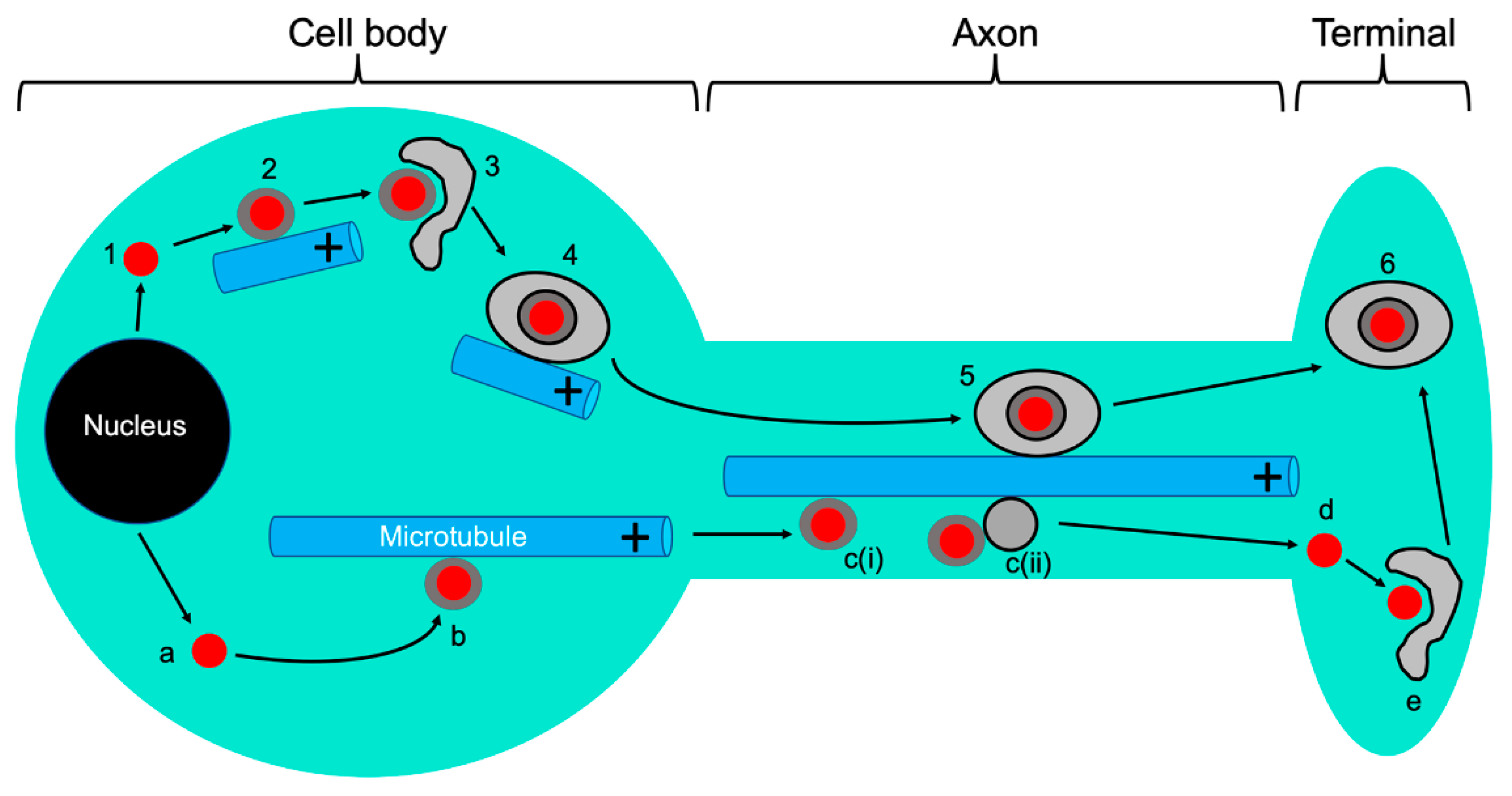
3. Viral Entry into Neurons, Capsid Attachment to Microtubules, and Retrograde Transport
3.1. Entry and Retrograde Transport, an Overview
3.2. Induction of Host Protein Synthesis in the Axon Upon Alphaherpesvirus Infection
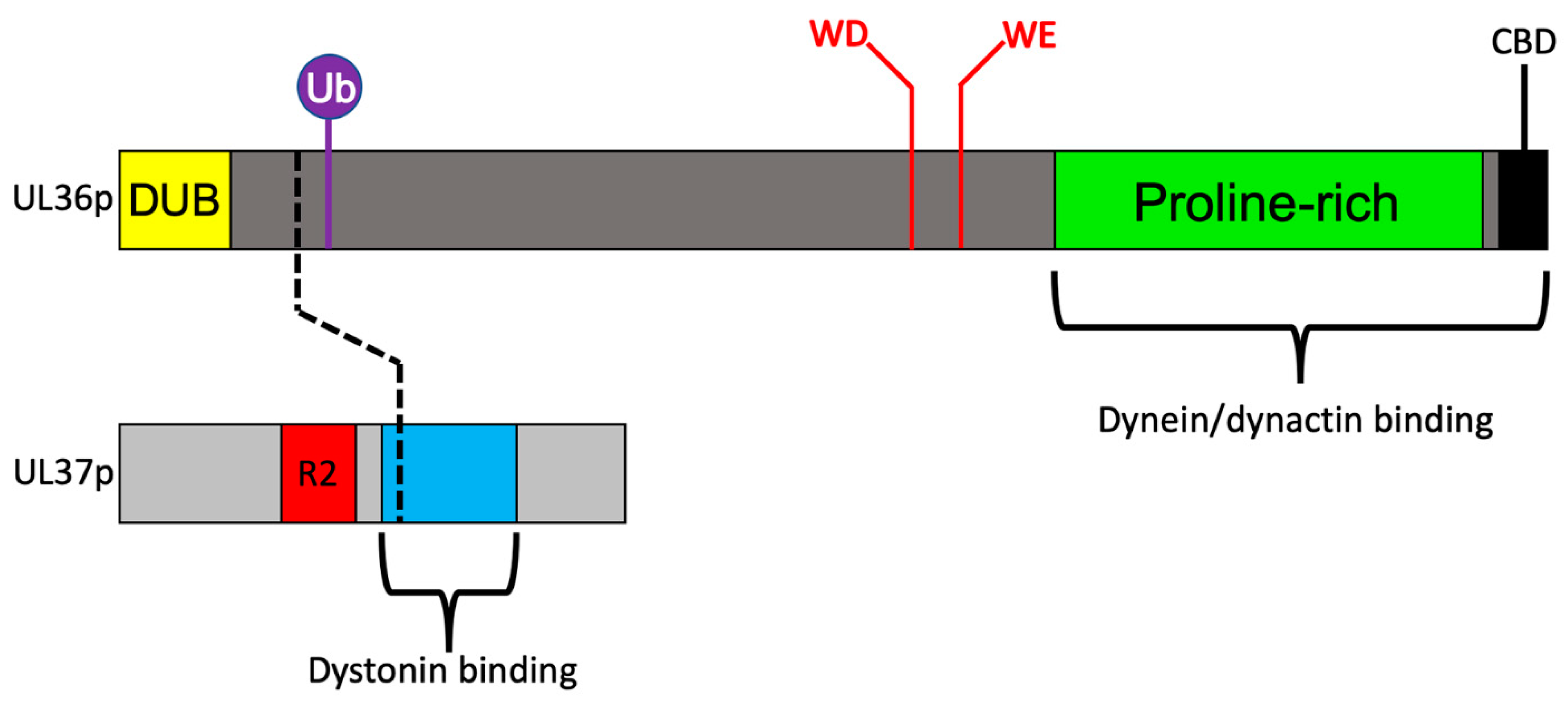
3.3. Recruitment of Dynein and Dynactin by the Inner Tegument Protein UL36p
3.4. A Ubiquitin Switch in UL36p Sustains Processive Retrograde Transport
3.5. Retrograde Transport Functions Provided by the Inner Tegument Protein UL37p
4. Climbing out of a Well: Trafficking from the MTOC to the Nucleus
5. Anterograde Trafficking of Progeny Capsids and their Cytoplasmic Envelopment
5.1. The Emergence of Progeny Naked Capsids from the Nucleus and Their Trafficking in the Cytoplasm
5.2. Reorganization of Microtubules during Alphaherpesvirus Infection
5.3. Coordination of MT-Directed Transport of Capsids with Cytoplasmic Envelopment
5.4. MT-Directed Transport of Enveloping Capsids Is Arrested Until Envelopment Is Complete
6. Anterograde Trafficking Down the Axon: A Multitude of Trafficking Particles and of Models to Account for Them
6.1. The “Married” and “Separate” Models
6.2. Trafficking of Viral Glycoproteins in the Absence of the Viral Capsid
7. The Virally Encoded Membrane Proteins gE, gI, and US9p Play Key Roles in Axonal Trafficking and Spread of Alphaherpesvirus Particles
7.1. The Virally Encoded Membrane Proteins gE, gI, and US9p: An Overview
7.2. Molecular Roles for gE/gI and US9p in Anterograde Transport of Enveloped Virions (the Married Model)
7.3. Molecular Roles for gE/gI and US9p in Anterograde Transport of Naked Capsids and Viral Glycoproteins (the Separate Model)
7.4. Summary of Roles for gE/gI and US9p in Anterograde Transport of Alphaherpesviruses in the Axon
7.5. Motor Choice and Additional Viral Candidates for Kinesin Recruitment
8. Final Steps: Exocytosis of Enveloped Virions at the Nerve Terminal
9. Conclusions
Funding
Conflicts of Interest
References
- Pellett, P.E.; Roizman, B. Herpesviridae. In Fields Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1802–1822. [Google Scholar]
- Smith, G. Herpesvirus transport to the nervous system and back again. Annu. Rev. Microbiol. 2012, 66, 153–176. [Google Scholar] [CrossRef] [PubMed]
- Kramer, T.; Enquist, L.W. Directional spread of alphaherpesviruses in the nervous system. Viruses 2013, 5, 678–707. [Google Scholar] [CrossRef] [PubMed]
- Steiner, I.; Kennedy, P.G.; Pachner, A.R. The neurotropic herpes viruses: Herpes simplex and varicella-zoster. Lancet Neurol. 2007, 6, 1015–1028. [Google Scholar] [CrossRef]
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes simplex viruses. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013; Volume 2, pp. 1824–1898. [Google Scholar]
- Smith, G.A. Assembly and egress of an alphaherpesvirus clockwork. Adv. Anat. Embryol. Cell Biol. 2017, 223, 171–193. [Google Scholar] [PubMed]
- Pomeranz, L.E.; Reynolds, A.E.; Hengartner, C.J. Molecular biology of pseudorabies virus: Impact on neurovirology and veterinary medicine. Microbiol. Mol. Biol. Rev. 2005, 69, 462–500. [Google Scholar] [CrossRef] [PubMed]
- Nicola, A.V.; Hou, J.; Major, E.O.; Straus, S.E. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 2005, 79, 7609–7616. [Google Scholar] [CrossRef]
- Smith, G.A.; Pomeranz, L.; Gross, S.P.; Enquist, L.W. Local modulation of plus-end transport targets herpesvirus entry and egress in sensory axons. Proc. Natl. Acad. Sci. USA 2004, 101, 16034–16039. [Google Scholar] [CrossRef]
- Denes, C.E.; Miranda-Saksena, M.; Cunningham, A.L.; Diefenbach, R.J. Cytoskeletons in the closet-subversion in alphaherpesvirus infections. Viruses 2018, 10, 79. [Google Scholar] [CrossRef]
- Miranda-Saksena, M.; Denes, C.E.; Diefenbach, R.J.; Cunningham, A.L. Infection and transport of herpes simplex virus type 1 in neurons: Role of the cytoskeleton. Viruses 2018, 10, 92. [Google Scholar] [CrossRef]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument assembly and secondary envelopment of alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef]
- Barnes, J.; Wilson, D.W. Seeking closure: How do herpesviruses recruit the cellular ESCRT apparatus? J. Virol. 2019, 93, e00392–19. [Google Scholar] [CrossRef] [PubMed]
- Crump, C. Virus assembly and egress of HSV. Adv. Exp. Med. Biol. 2018, 1045, 23–44. [Google Scholar] [PubMed]
- Heming, J.D.; Conway, J.F.; Homa, F.L. Herpesvirus capsid assembly and DNA packaging. Adv. Anat. Embryol. Cell Biol. 2017, 223, 119–142. [Google Scholar] [PubMed]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C.; Klupp, B.G.; Granzow, H. Herpesvirus assembly: An update. Virus Res. 2009, 143, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Fan, W.H.; Roberts, A.P.; McElwee, M.; Bhella, D.; Rixon, F.J.; Lauder, R. The large tegument protein pUL36 is essential for formation of the capsid vertex-specific component at the capsid-tegument interface of herpes simplex virus 1. J. Virol. 2015, 89, 1502–1511. [Google Scholar] [CrossRef] [PubMed]
- Koenigsberg, A.L.; Heldwein, E.E. The dynamic nature of the conserved tegument protein UL37 of herpesviruses. J. Biol. Chem. 2018, 293, 15827–15839. [Google Scholar] [CrossRef]
- Coller, K.E.; Lee, J.I.; Ueda, A.; Smith, G.A. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J. Virol. 2007, 81, 11790–11797. [Google Scholar] [CrossRef]
- Kelly, B.J.; Mijatov, B.; Fraefel, C.; Cunningham, A.L.; Diefenbach, R.J. Identification of a single amino acid residue which is critical for the interaction between HSV-1 inner tegument proteins pUL36 and pUL37. Virology 2012, 422, 308–316. [Google Scholar] [CrossRef]
- Kelly, B.J.; Bauerfeind, R.; Binz, A.; Sodeik, B.; Laimbacher, A.S.; Fraefel, C.; Diefenbach, R.J. The interaction of the HSV-1 tegument proteins pUL36 and pUL37 is essential for secondary envelopment during viral egress. Virology 2014, 454, 67–77. [Google Scholar] [CrossRef]
- Mijatov, B.; Cunningham, A.L.; Diefenbach, R.J. Residues F593 and E596 of HSV-1 tegument protein pUL36 (VP1/2) mediate binding of tegument protein pUL37. Virology 2007, 368, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Klupp, B.G.; Fuchs, W.; Granzow, H.; Nixdorf, R.; Mettenleiter, T.C. Pseudorabies virus UL36 tegument protein physically interacts with the UL37 protein. J. Virol. 2002, 76, 3065–3071. [Google Scholar] [CrossRef] [PubMed]
- Vittone, V.; Diefenbach, E.; Triffett, D.; Douglas, M.W.; Cunningham, A.L.; Diefenbach, R.J. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 2005, 79, 9566–9571. [Google Scholar] [CrossRef] [PubMed]
- Pitts, J.D.; Klabis, J.; Richards, A.L.; Smith, G.A.; Heldwein, E.E. Crystal structure of the herpesvirus inner tegument protein UL37 supports its essential role in control of viral trafficking. J. Virol. 2014, 88, 5462–5473. [Google Scholar] [CrossRef] [PubMed]
- Koenigsberg, A.L.; Heldwein, E.E. Crystal Structure of the N-terminal half of the traffic controller UL37 from Herpes simplex virus 1. J. Virol. 2017, 91, e01244-17. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.J. A null mutation in the UL36 gene of herpes simplex virus type 1 results in accumulation of unenveloped DNA-filled capsids in the cytoplasm of infected cells. J. Virol. 2000, 74, 11608–11618. [Google Scholar] [CrossRef] [PubMed]
- Desai, P.; Sexton, G.L.; McCaffery, J.M.; Person, S. A null mutation in the gene encoding the herpes simplex virus type 1 UL37 polypeptide abrogates virus maturation. J. Virol. 2001, 75, 10259–10271. [Google Scholar] [CrossRef]
- Newcomb, W.W.; Brown, J.C. Structure and capsid association of the herpesvirus large tegument protein UL36. J. Virol. 2010, 84, 9408–9414. [Google Scholar] [CrossRef]
- Ko, D.H.; Cunningham, A.L.; Diefenbach, R.J. The major determinant for addition of tegument protein pUL48 (VP16) to capsids in herpes simplex virus type 1 is the presence of the major tegument protein pUL36 (VP1/2). J. Virol. 2010, 84, 1397–1405. [Google Scholar] [CrossRef]
- Svobodova, S.; Bell, S.; Crump, C.M. Analysis of the interaction between the essential herpes simplex virus 1 tegument proteins VP16 and VP1/2. J. Virol. 2012, 86, 473–483. [Google Scholar] [CrossRef]
- Chi, J.H.; Harley, C.A.; Mukhopadhyay, A.; Wilson, D.W. The cytoplasmic tail of herpes simplex virus envelope glycoprotein D binds to the tegument protein VP22 and to capsids. J. Gen. Virol. 2005, 86, 253–261. [Google Scholar] [CrossRef]
- Gross, S.T.; Harley, C.A.; Wilson, D.W. The cytoplasmic tail of Herpes simplex virus glycoprotein H binds to the tegument protein VP16 in vitro and in vivo. Virology 2003, 317, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Kamen, D.E.; Gross, S.T.; Girvin, M.E.; Wilson, D.W. Structural basis for the physiological temperature dependence of the association of VP16 with the cytoplasmic tail of herpes simplex virus glycoprotein H. J. Virol. 2005, 79, 6134–6141. [Google Scholar] [CrossRef] [PubMed]
- O’Regan, K.J.; Bucks, M.A.; Murphy, M.A.; Wills, J.W.; Courtney, R.J. A conserved region of the herpes simplex virus type 1 tegument protein VP22 facilitates interaction with the cytoplasmic tail of glycoprotein E (gE). Virology 2007, 358, 192–200. [Google Scholar] [CrossRef] [PubMed]
- Spear, P.G.; Longnecker, R. Herpesvirus entry: An update. J. Virol. 2003, 77, 10179–10185. [Google Scholar] [CrossRef]
- Frankel, E.B.; Audhya, A. ESCRT-dependent cargo sorting at multivesicular endosomes. Semin. Cell Dev. Biol. 2018, 74, 4–10. [Google Scholar] [CrossRef]
- Nicola, A.V.; McEvoy, A.M.; Straus, S.E. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 2003, 77, 5324–5332. [Google Scholar] [CrossRef]
- Piccinotti, S.; Whelan, S.P. Rabies internalizes into primary peripheral neurons via clathrin coated pits and requires fusion at the cell body. PLoS Pathog. 2016, 12, e1005753. [Google Scholar] [CrossRef]
- Ohka, S.; Nomoto, A. Recent insights into poliovirus pathogenesis. Trends Microbiol. 2001, 9, 501–506. [Google Scholar] [CrossRef]
- Koyuncu, O.O.; Hogue, I.B.; Enquist, L.W. Virus infections in the nervous system. Cell Host Microbe 2013, 13, 379–393. [Google Scholar] [CrossRef]
- Sodeik, B. Mechanisms of viral transport in the cytoplasm. Trends Microbiol. 2000, 8, 465–472. [Google Scholar] [CrossRef]
- Antinone, S.E.; Smith, G.A. Retrograde axon transport of herpes simplex virus and pseudorabies virus: A live-cell comparative analysis. J. Virol. 2010, 84, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Olenick, M.A.; Holzbaur, E.L.F. Dynein activators and adaptors at a glance. J. Cell Sci. 2019, 132. [Google Scholar] [CrossRef] [PubMed]
- Gross, S.P. Dynactin: Coordinating motors with opposite inclinations. Curr. Biol. 2003, 13, R320–R322. [Google Scholar] [CrossRef]
- Muller, M.J.; Klumpp, S.; Lipowsky, R. Bidirectional transport by molecular motors: Enhanced processivity and response to external forces. Biophys. J. 2010, 98, 2610–2618. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; Perlman, D.H.; Enquist, L.W. Efficient retrograde transport of pseudorabies virus within neurons requires local protein synthesis in axons. Cell Host Microbe 2013, 13, 54–66. [Google Scholar] [CrossRef]
- Hanz, S.; Perlson, E.; Willis, D.; Zheng, J.Q.; Massarwa, R.; Huerta, J.J.; Koltzenburg, M.; Kohler, M.; van-Minnen, J.; Twiss, J.L.; et al. Axoplasmic importins enable retrograde injury signaling in lesioned nerve. Neuron 2003, 40, 1095–1104. [Google Scholar] [CrossRef]
- Melemedjian, O.K.; Asiedu, M.N.; Tillu, D.V.; Peebles, K.A.; Yan, J.; Ertz, N.; Dussor, G.O.; Price, T.J. IL-6- and NGF-induced rapid control of protein synthesis and nociceptive plasticity via convergent signaling to the eIF4F complex. J. Neurosci. 2010, 30, 15113–15123. [Google Scholar] [CrossRef]
- Yudin, D.; Hanz, S.; Yoo, S.; Iavnilovitch, E.; Willis, D.; Gradus, T.; Vuppalanchi, D.; Segal-Ruder, Y.; Ben-Yaakov, K.; Hieda, M.; et al. Localized regulation of axonal RanGTPase controls retrograde injury signaling in peripheral nerve. Neuron 2008, 59, 241–252. [Google Scholar] [CrossRef]
- Faulkner, N.E.; Dujardin, D.L.; Tai, C.Y.; Vaughan, K.T.; O’Connell, C.B.; Wang, Y.; Vallee, R.B. A role for the lissencephaly gene LIS1 in mitosis and cytoplasmic dynein function. Nat. Cell Biol. 2000, 2, 784–791. [Google Scholar] [CrossRef]
- Smith, D.S.; Niethammer, M.; Ayala, R.; Zhou, Y.; Gambello, M.J.; Wynshaw-Boris, A.; Tsai, L.H. Regulation of cytoplasmic dynein behaviour and microtubule organization by mammalian Lis1. Nat. Cell Biol. 2000, 2, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.Y.; Ori-McKenney, K.M.; McKenney, R.J.; Vershinin, M.; Gross, S.P.; Vallee, R.B. High-resolution imaging reveals indirect coordination of opposite motors and a role for LIS1 in high-load axonal transport. J. Cell Biol. 2011, 195, 193–201. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, M.E.; Cianfrocco, M.A.; Htet, Z.M.; Tran, P.T.; Reck-Peterson, S.L.; Leschziner, A.E. Lis1 has two opposing modes of regulating cytoplasmic dynein. Cell 2017, 170, 1197–1208. [Google Scholar] [CrossRef] [PubMed]
- Hol, E.M.; Capetanaki, Y. Type III Intermediate filaments desmin, glial fibrillary acidic protein (GFAP), vimentin, and peripherin. Cold Spring Harb. Perspect. Biol. 2017, 9, a021642. [Google Scholar] [CrossRef] [PubMed]
- Hayes, M.J.; Rescher, U.; Gerke, V.; Moss, S.E. Annexin-actin interactions. Traffic 2004, 5, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Gauthier-Kemper, A.; Suarez Alonso, M.; Sundermann, F.; Niewidok, B.; Fernandez, M.P.; Bakota, L.; Heinisch, J.J.; Brandt, R. Annexins A2 and A6 interact with the extreme N terminus of tau and thereby contribute to tau’s axonal localization. J. Biol. Chem. 2018, 293, 8065–8076. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, A.; Miranda-Saksena, M.; Boadle, R.A.; Kelly, B.J.; Diefenbach, R.J.; Alam, W.; Cunningham, A.L. Ultrastructural visualization of individual tegument protein dissociation during entry of herpes simplex virus 1 into human and rat dorsal root ganglion neurons. J. Virol. 2012, 86, 6123–6137. [Google Scholar] [CrossRef]
- Granzow, H.; Klupp, B.G.; Mettenleiter, T.C. Entry of pseudorabies virus: An immunogold-labeling study. J. Virol. 2005, 79, 3200–3205. [Google Scholar] [CrossRef]
- Luxton, G.W.; Haverlock, S.; Coller, K.E.; Antinone, S.E.; Pincetic, A.; Smith, G.A. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 5832–5837. [Google Scholar] [CrossRef]
- Coller, K.E.; Smith, G.A. Two viral kinases are required for sustained long distance axon transport of a neuroinvasive herpesvirus. Traffic 2008, 9, 1458–1470. [Google Scholar] [CrossRef]
- Sodeik, B.; Ebersold, M.W.; Helenius, A. Microtubule-mediated transport of incoming herpes simplex virus 1 capsids to the nucleus. J. Cell Biol. 1997, 136, 1007–1021. [Google Scholar] [CrossRef] [PubMed]
- Dohner, K.; Wolfstein, A.; Prank, U.; Echeverri, C.; Dujardin, D.; Vallee, R.; Sodeik, B. Function of dynein and dynactin in herpes simplex virus capsid transport. Mol. Biol. Cell 2002, 13, 2795–2809. [Google Scholar] [CrossRef] [PubMed]
- Dohner, K.; Radtke, K.; Schmidt, S.; Sodeik, B. Eclipse phase of herpes simplex virus type 1 infection: Efficient dynein-mediated capsid transport without the small capsid protein VP26. J. Virol. 2006, 80, 8211–8224. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J. Regulation of dynein-dynactin-driven vesicular transport. Traffic 2017, 18, 336–347. [Google Scholar] [CrossRef] [PubMed]
- Wolfstein, A.; Nagel, C.H.; Radtke, K.; Dohner, K.; Allan, V.J.; Sodeik, B. The inner tegument promotes herpes simplex virus capsid motility along microtubules in vitro. Traffic 2006, 7, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Radtke, K.; Kieneke, D.; Wolfstein, A.; Michael, K.; Steffen, W.; Scholz, T.; Karger, A.; Sodeik, B. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 2010, 6, e1000991. [Google Scholar] [CrossRef]
- Zaichick, S.V.; Bohannon, K.P.; Hughes, A.; Sollars, P.J.; Pickard, G.E.; Smith, G.A. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 2013, 13, 193–203. [Google Scholar] [CrossRef]
- Douglas, M.W.; Diefenbach, R.J.; Homa, F.L.; Miranda-Saksena, M.; Rixon, F.J.; Vittone, V.; Byth, K.; Cunningham, A.L. Herpes simplex virus type 1 capsid protein VP26 interacts with dynein light chains RP3 and Tctex1 and plays a role in retrograde cellular transport. J. Biol. Chem. 2004, 279, 28522–28530. [Google Scholar] [CrossRef]
- Reck-Peterson, S.L.; Redwine, W.B.; Vale, R.D.; Carter, A.P. The cytoplasmic dynein transport machinery and its many cargoes. Nat. Rev. Mol. Cell Biol. 2018, 19, 382–398. [Google Scholar] [CrossRef]
- Jovasevic, V.; Naghavi, M.H.; Walsh, D. Microtubule plus end-associated CLIP-170 initiates HSV-1 retrograde transport in primary human cells. J. Cell Biol. 2015, 211, 323–337. [Google Scholar] [CrossRef]
- Naghavi, M.H.; Walsh, D. Microtubule Regulation and Function during Virus Infection. J. Virol. 2017, 91, e00538–17. [Google Scholar] [CrossRef] [PubMed]
- Jiang, K.; Akhmanova, A. Microtubule tip-interacting proteins: A view from both ends. Curr. Opin. Cell Biol. 2011, 23, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Lansbergen, G.; Grigoriev, I.; Mimori-Kiyosue, Y.; Ohtsuka, T.; Higa, S.; Kitajima, I.; Demmers, J.; Galjart, N.; Houtsmuller, A.B.; Grosveld, F.; et al. CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5beta. Dev. Cell 2006, 11, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Moughamian, A.J.; Osborn, G.E.; Lazarus, J.E.; Maday, S.; Holzbaur, E.L. Ordered recruitment of dynactin to the microtubule plus-end is required for efficient initiation of retrograde axonal transport. J. Neurosci. 2013, 33, 13190–13203. [Google Scholar] [CrossRef] [PubMed]
- Huffmaster, N.J.; Sollars, P.J.; Richards, A.L.; Pickard, G.E.; Smith, G.A. Dynamic ubiquitination drives herpesvirus neuroinvasion. Proc. Natl. Acad. Sci. USA 2015, 112, 12818–12823. [Google Scholar] [CrossRef]
- Kattenhorn, L.M.; Korbel, G.A.; Kessler, B.M.; Spooner, E.; Ploegh, H.L. A deubiquitinating enzyme encoded by HSV-1 belongs to a family of cysteine proteases that is conserved across the family Herpesviridae. Mol. Cell 2005, 19, 547–557. [Google Scholar] [CrossRef]
- Schlieker, C.; Korbel, G.A.; Kattenhorn, L.M.; Ploegh, H.L. A deubiquitinating activity is conserved in the large tegument protein of the herpesviridae. J. Virol. 2005, 79, 15582–15585. [Google Scholar] [CrossRef]
- Schlieker, C.; Weihofen, W.A.; Frijns, E.; Kattenhorn, L.M.; Gaudet, R.; Ploegh, H.L. Structure of a herpesvirus-encoded cysteine protease reveals a unique class of deubiquitinating enzymes. Mol. Cell 2007, 25, 677–687. [Google Scholar] [CrossRef]
- Krautwald, M.; Fuchs, W.; Klupp, B.G.; Mettenleiter, T.C. Translocation of incoming pseudorabies virus capsids to the cell nucleus is delayed in the absence of tegument protein pUL37. J. Virol. 2009, 83, 3389–3396. [Google Scholar] [CrossRef]
- Richards, A.L.; Sollars, P.J.; Pitts, J.D.; Stults, A.M.; Heldwein, E.E.; Pickard, G.E.; Smith, G.A. The pUL37 tegument protein guides alpha-herpesvirus retrograde axonal transport to promote neuroinvasion. PLoS Pathog. 2017, 13, e1006741. [Google Scholar] [CrossRef]
- Ferrier, A.; Boyer, J.G.; Kothary, R. Cellular and molecular biology of neuronal dystonin. Int Rev. Cell Mol. Biol. 2013, 300, 85–120. [Google Scholar] [PubMed]
- Pasdeloup, D.; McElwee, M.; Beilstein, F.; Labetoulle, M.; Rixon, F.J. Herpesvirus tegument protein pUL37 interacts with dystonin/BPAG1 to promote capsid transport on microtubules during egress. J. Virol. 2013, 87, 2857–2867. [Google Scholar] [CrossRef] [PubMed]
- Ryan, S.D.; Bhanot, K.; Ferrier, A.; De Repentigny, Y.; Chu, A.; Blais, A.; Kothary, R. Microtubule stability, Golgi organization, and transport flux require dystonin-a2-MAP1B interaction. J. Cell Biol. 2012, 196, 727–742. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Ding, J.; Kowal, A.S.; Nardine, T.; Allen, E.; Delcroix, J.D.; Wu, C.; Mobley, W.; Fuchs, E.; Yang, Y. BPAG1n4 is essential for retrograde axonal transport in sensory neurons. J. Cell Biol. 2003, 163, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Ding, J.; Wu, C.; Bhagavatula, P.; Cui, B.; Chu, S.; Mobley, W.C.; Yang, Y. Retrolinkin, a membrane protein, plays an important role in retrograde axonal transport. Proc. Natl. Acad. Sci. USA 2007, 104, 2223–2228. [Google Scholar] [CrossRef] [PubMed]
- McElwee, M.; Beilstein, F.; Labetoulle, M.; Rixon, F.J.; Pasdeloup, D. Dystonin/BPAG1 promotes plus-end-directed transport of herpes simplex virus 1 capsids on microtubules during entry. J. Virol. 2013, 87, 11008–11018. [Google Scholar] [CrossRef]
- Zhao, J.; Zeng, Y.; Xu, S.; Chen, J.; Shen, G.; Yu, C.; Knipe, D.; Yuan, W.; Peng, J.; Xu, W.; et al. A Viral deamidase targets the helicase domain of RIG-I to block RNA-induced activation. Cell Host Microbe 2016, 20, 770–784. [Google Scholar] [CrossRef]
- Zhang, J.; Zhao, J.; Xu, S.; Li, J.; He, S.; Zeng, Y.; Xie, L.; Xie, N.; Liu, T.; Lee, K.; et al. Species-specific deamidation of cGAS by herpes simplex virus UL37 protein facilitates viral replication. Cell Host Microbe 2018, 24, 234–248. [Google Scholar] [CrossRef]
- Stults, A.M.; Smith, G.A. The herpes simplex virus type I deamidase enhances propagation but is dispensable for retrograde axonal transport into the nervous system. J. Virol. 2019, 93, e01172-19. [Google Scholar] [CrossRef]
- Ojala, P.M.; Sodeik, B.; Ebersold, M.W.; Kutay, U.; Helenius, A. Herpes simplex virus type 1 entry into host cells: Reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol. Cell. Biol. 2000, 20, 4922–4931. [Google Scholar] [CrossRef]
- Copeland, A.M.; Newcomb, W.W.; Brown, J.C. Herpes simplex virus replication: Roles of viral proteins and nucleoporins in capsid-nucleus attachment. J. Virol. 2009, 83, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Abaitua, F.; Hollinshead, M.; Bolstad, M.; Crump, C.M.; O’Hare, P. A Nuclear localization signal in herpesvirus protein VP1-2 is essential for infection via capsid routing to the nuclear pore. J. Virol. 2012, 86, 8998–9014. [Google Scholar] [CrossRef] [PubMed]
- Schipke, J.; Pohlmann, A.; Diestel, R.; Binz, A.; Rudolph, K.; Nagel, C.H.; Bauerfeind, R.; Sodeik, B. The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 2012, 86, 3682–3700. [Google Scholar] [CrossRef] [PubMed]
- Abaitua, F.; Daikoku, T.; Crump, C.M.; Bolstad, M.; O’Hare, P. A single mutation responsible for temperature-sensitive entry and assembly defects in the VP1-2 protein of herpes simplex virus. J. Virol. 2011, 85, 2024–2036. [Google Scholar] [CrossRef] [PubMed]
- Jovasevic, V.; Liang, L.; Roizman, B. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J. Virol. 2008, 82, 3311–3319. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Zhou, Z.H. Structure of the herpes simplex virus 1 capsid with associated tegument protein complexes. Science 2018, 360, eaao7298. [Google Scholar] [CrossRef]
- Mettenleiter, T.C. Vesicular nucleo-cytoplasmic transport-herpesviruses as pioneers in cell biology. Viruses 2016, 8, 266. [Google Scholar] [CrossRef]
- Bigalke, J.M.; Heldwein, E.E. Have nec coat, will travel: Structural basis of membrane budding during nuclear egress in herpesviruses. Adv. Virus Res. 2017, 97, 107–141. [Google Scholar]
- Bigalke, J.M.; Heldwein, E.E. Nuclear exodus: Herpesviruses lead the way. Annu. Rev. Virol. 2016, 3, 387–409. [Google Scholar] [CrossRef]
- Arii, J.; Watanabe, M.; Maeda, F.; Tokai-Nishizumi, N.; Chihara, T.; Miura, M.; Maruzuru, Y.; Koyanagi, N.; Kato, A.; Kawaguchi, Y. ESCRT-III mediates budding across the inner nuclear membrane and regulates its integrity. Nat. Commun. 2018, 9, 3379. [Google Scholar] [CrossRef]
- Banfield, B.W. Beyond the NEC: Modulation of herpes simplex virus nuclear egress by viral and cellular components. Curr. Clin. Microbiol. Rep. 2019, 6, 1–9. [Google Scholar] [CrossRef]
- Bucks, M.A.; O’Regan, K.J.; Murphy, M.A.; Wills, J.W.; Courtney, R.J. Herpes simplex virus type 1 tegument proteins VP1/2 and UL37 are associated with intranuclear capsids. Virology 2007, 361, 316–324. [Google Scholar] [CrossRef] [PubMed]
- Mohl, B.S.; Bottcher, S.; Granzow, H.; Kuhn, J.; Klupp, B.G.; Mettenleiter, T.C. Intracellular localization of the pseudorabies virus large tegument protein pUL36. J. Virol. 2009, 83, 9641–9651. [Google Scholar] [CrossRef] [PubMed]
- Leelawong, M.; Lee, J.I.; Smith, G.A. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J. Virol. 2012, 86, 6303–6314. [Google Scholar] [CrossRef]
- Sandbaumhuter, M.; Dohner, K.; Schipke, J.; Binz, A.; Pohlmann, A.; Sodeik, B.; Bauerfeind, R. Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell. Microbiol. 2013, 15, 248–269. [Google Scholar] [CrossRef]
- Buch, A.; Muller, O.; Ivanova, L.; Dohner, K.; Bialy, D.; Bosse, J.B.; Pohlmann, A.; Binz, A.; Hegemann, M.; Nagel, C.H.; et al. Inner tegument proteins of herpes simplex virus are sufficient for intracellular capsid motility in neurons but not for axonal targeting. PLoS Pathog. 2017, 13, e1006813. [Google Scholar] [CrossRef]
- Lee, G.E.; Murray, J.W.; Wolkoff, A.W.; Wilson, D.W. Reconstitution of herpes simplex virus microtubule-dependent trafficking in vitro. J. Virol. 2006, 80, 4264–4275. [Google Scholar] [CrossRef]
- Shanda, S.K.; Wilson, D.W. UL36p is required for efficient transport of membrane-associated herpes simplex virus type 1 along microtubules. J. Virol. 2008, 82, 7388–7394. [Google Scholar] [CrossRef]
- Luxton, G.W.; Lee, J.I.; Haverlock-Moyns, S.; Schober, J.M.; Smith, G.A. The pseudorabies virus VP1/2 tegument protein is required for intracellular capsid transport. J. Virol. 2006, 80, 201–209. [Google Scholar] [CrossRef]
- Elliott, G.; O’Hare, P. Herpes simplex virus type 1 tegument protein VP22 induces the stabilization and hyperacetylation of microtubules. J. Virol. 1998, 72, 6448–6455. [Google Scholar]
- Kotsakis, A.; Pomeranz, L.E.; Blouin, A.; Blaho, J.A. Microtubule reorganization during herpes simplex virus type 1 infection facilitates the nuclear localization of VP22, a major virion tegument protein. J. Virol. 2001, 75, 8697–8711. [Google Scholar] [CrossRef] [PubMed]
- Naghavi, M.H.; Gundersen, G.G.; Walsh, D. Plus-end tracking proteins, CLASPs, and a viral Akt mimic regulate herpesvirus-induced stable microtubule formation and virus spread. Proc. Natl. Acad. Sci. USA 2013, 110, 18268–18273. [Google Scholar] [CrossRef] [PubMed]
- Pasdeloup, D.; Labetoulle, M.; Rixon, F.J. Differing effects of herpes simplex virus 1 and pseudorabies virus infections on centrosomal function. J. Virol. 2013, 87, 7102–7112. [Google Scholar] [CrossRef] [PubMed]
- Wloga, D.; Joachimiak, E.; Fabczak, H. Tubulin post-translational modifications and microtubule dynamics. Int. J. Mol. Sci. 2017, 18, 2207. [Google Scholar] [CrossRef]
- Reed, N.A.; Cai, D.; Blasius, T.L.; Jih, G.T.; Meyhofer, E.; Gaertig, J.; Verhey, K.J. Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 2006, 16, 2166–2172. [Google Scholar] [CrossRef]
- Hollinshead, M.; Johns, H.L.; Sayers, C.L.; Gonzalez-Lopez, C.; Smith, G.L.; Elliott, G. Endocytic tubules regulated by Rab GTPases 5 and 11 are used for envelopment of herpes simplex virus. EMBO J. 2012, 31, 4204–4220. [Google Scholar] [CrossRef]
- Johns, H.L.; Gonzalez-Lopez, C.; Sayers, C.L.; Hollinshead, M.; Elliott, G. Rab6 dependent post-Golgi trafficking of HSV1 envelope proteins to sites of virus envelopment. Traffic 2014, 15, 157–178. [Google Scholar] [CrossRef]
- Turcotte, S.; Letellier, J.; Lippe, R. Herpes simplex virus type 1 capsids transit by the trans-Golgi network, where viral glycoproteins accumulate independently of capsid egress. J. Virol. 2005, 79, 8847–8860. [Google Scholar] [CrossRef]
- Harley, C.A.; Dasgupta, A.; Wilson, D.W. Characterization of herpes simplex virus-containing organelles by subcellular fractionation: Role for organelle acidification in assembly of infectious particles. J. Virol. 2001, 75, 1236–1251. [Google Scholar] [CrossRef]
- Granzow, H.; Klupp, B.G.; Fuchs, W.; Veits, J.; Osterrieder, N.; Mettenleiter, T.C. Egress of alphaherpesviruses: Comparative ultrastructural study. J. Virol. 2001, 75, 3675–3684. [Google Scholar] [CrossRef]
- Henaff, D.; Radtke, K.; Lippe, R. Herpesviruses exploit several host compartments for envelopment. Traffic 2012, 13, 1443–1449. [Google Scholar] [CrossRef] [PubMed]
- Hambleton, S.; Gershon, M.D.; Gershon, A.A. The role of the trans-Golgi network in varicella zoster virus biology. Cell Mol. Life Sci. 2004, 61, 3047–3056. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Saksena, M.; Boadle, R.A.; Armati, P.; Cunningham, A.L. In rat dorsal root ganglion neurons, herpes simplex virus type 1 tegument forms in the cytoplasm of the cell body. J. Virol. 2002, 76, 9934–9951. [Google Scholar] [CrossRef] [PubMed]
- Henaff, D.; Remillard-Labrosse, G.; Loret, S.; Lippe, R. Analysis of the early steps of herpes simplex virus 1 capsid tegumentation. J. Virol. 2013, 87, 4895–4906. [Google Scholar] [CrossRef]
- Fuchs, W.; Klupp, B.G.; Granzow, H.; Mettenleiter, T.C. Essential function of the pseudorabies virus UL36 gene product is independent of its interaction with the UL37 protein. J. Virol. 2004, 78, 11879–11889. [Google Scholar] [CrossRef]
- Klupp, B.G.; Granzow, H.; Mundt, E.; Mettenleiter, T.C. Pseudorabies virus UL37 gene product is involved in secondary envelopment. J. Virol. 2001, 75, 8927–8936. [Google Scholar] [CrossRef]
- Ivanova, L.; Buch, A.; Dohner, K.; Pohlmann, A.; Binz, A.; Prank, U.; Sandbaumhuter, M.; Bauerfeind, R.; Sodeik, B. Conserved tryptophan motifs in the large tegument protein pUL36 are required for efficient secondary envelopment of herpes simplex virus capsids. J. Virol. 2016, 90, 5368–5383. [Google Scholar] [CrossRef]
- Dodding, M.P.; Way, M. Coupling viruses to dynein and kinesin-1. EMBO J. 2011, 30, 3527–3539. [Google Scholar] [CrossRef]
- Dodding, M.P.; Mitter, R.; Humphries, A.C.; Way, M. A kinesin-1 binding motif in vaccinia virus that is widespread throughout the human genome. EMBO J. 2011, 30, 4523–4538. [Google Scholar] [CrossRef]
- Pernigo, S.; Lamprecht, A.; Steiner, R.A.; Dodding, M.P. Structural basis for kinesin-1:cargo recognition. Science 2013, 340, 356–359. [Google Scholar] [CrossRef]
- Kharkwal, H.; Furgiuele, S.S.; Smith, C.G.; Wilson, D.W. Herpes simplex virus capsid-organelle association in the absence of the large tegument protein UL36p. J. Virol. 2015, 89, 11372–11382. [Google Scholar] [CrossRef] [PubMed]
- Kharkwal, H.; Smith, C.G.; Wilson, D.W. HSV capsid localization to ESCRT-VPS4 complexes in the presence and absence of the large tegument protein UL36p. J. Virol. 2016, 90, 7257–7267. [Google Scholar] [CrossRef] [PubMed]
- Jambunathan, N.; Chouljenko, D.; Desai, P.; Charles, A.S.; Subramanian, R.; Chouljenko, V.N.; Kousoulas, K.G. Herpes simplex virus 1 protein UL37 interacts with viral glycoprotein gK and membrane protein UL20 and functions in cytoplasmic virion envelopment. J. Virol. 2014, 88, 5927–5935. [Google Scholar] [CrossRef] [PubMed]
- Chouljenko, D.V.; Jambunathan, N.; Chouljenko, V.N.; Naderi, M.; Brylinski, M.; Caskey, J.R.; Kousoulas, K.G. Herpes Simplex Virus 1 UL37 Protein Tyrosine Residues Conserved among All Alphaherpesviruses Are Required for Interactions with Glycoprotein K, Cytoplasmic Virion Envelopment, and Infectious Virus Production. J. Virol. 2016, 90, 10351–10361. [Google Scholar] [CrossRef]
- Crump, C.M.; Yates, C.; Minson, T. Herpes simplex virus type 1 cytoplasmic envelopment requires functional Vps4. J. Virol. 2007, 81, 7380–7387. [Google Scholar] [CrossRef] [PubMed]
- Ibiricu, I.; Huiskonen, J.T.; Dohner, K.; Bradke, F.; Sodeik, B.; Grunewald, K. Cryo electron tomography of herpes simplex virus during axonal transport and secondary envelopment in primary neurons. PLoS Pathog. 2011, 7, e1002406. [Google Scholar] [CrossRef] [PubMed]
- Schimert, K.I.; Budaitis, B.G.; Reinemann, D.N.; Lang, M.J.; Verhey, K.J. Intracellular cargo transport by single-headed kinesin motors. Proc. Natl. Acad. Sci. USA 2019, 116, 6152–6161. [Google Scholar] [CrossRef] [PubMed]
- Hirokawa, N.; Noda, Y.; Tanaka, Y.; Niwa, S. Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 2009, 10, 682–696. [Google Scholar] [CrossRef]
- Hirokawa, N.; Noda, Y. Intracellular transport and kinesin superfamily proteins, KIFs: Structure, function, and dynamics. Physiol. Rev. 2008, 88, 1089–1118. [Google Scholar] [CrossRef]
- Kharkwal, H.; Smith, C.G.; Wilson, D.W. Blocking ESCRT-mediated envelopment inhibits microtubule-dependent trafficking of alphaherpesviruses in vitro. J. Virol. 2014, 88, 14467–14478. [Google Scholar] [CrossRef]
- McCullough, J.; Frost, A.; Sundquist, W.I. Structures, functions, and dynamics of ESCRT-III/Vps4 membrane remodeling and fission complexes. Annu. Rev. Cell Dev. Biol. 2018, 34, 85–109. [Google Scholar] [CrossRef] [PubMed]
- McCullough, J.; Clippinger, A.K.; Talledge, N.; Skowyra, M.L.; Saunders, M.G.; Naismith, T.V.; Colf, L.A.; Afonine, P.; Arthur, C.; Sundquist, W.I.; et al. Structure and membrane remodeling activity of ESCRT-III helical polymers. Science 2015, 350, 1548–1551. [Google Scholar] [CrossRef]
- Christ, L.; Raiborg, C.; Wenzel, E.M.; Campsteijn, C.; Stenmark, H. Cellular functions and molecular mechanisms of the ESCRT membrane-scission machinery. Trends Biochem. Sci. 2017, 42, 42–56. [Google Scholar] [CrossRef]
- Kratchmarov, R.; Taylor, M.P.; Enquist, L.W. Making the case: Married versus separate models of alphaherpes virus anterograde transport in axons. Rev. Med. Virol. 2012, 22, 378–391. [Google Scholar] [CrossRef]
- Cunningham, A.; Miranda-Saksena, M.; Diefenbach, R.; Johnson, D. Letter in response to: Making the case: Married versus separate models of alphaherpes virus anterograde transport in axons. Rev. Med. Virol. 2013, 23, 414–418. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Miranda-Saksena, M.; Douglas, M.W.; Cunningham, A.L. Transport and egress of herpes simplex virus in neurons. Rev. Med. Virol. 2008, 18, 35–51. [Google Scholar] [CrossRef]
- Scherer, J.; Yaffe, Z.A.; Vershinin, M.; Enquist, L.W. Dual-color herpesvirus capsids discriminate inoculum from progeny and reveal axonal transport dynamics. J. Virol. 2016, 90, 9997–10006. [Google Scholar] [CrossRef]
- Huang, J.; Lazear, H.M.; Friedman, H.M. Completely assembled virus particles detected by transmission electron microscopy in proximal and mid-axons of neurons infected with herpes simplex virus type 1, herpes simplex virus type 2 and pseudorabies virus. Virology 2011, 409, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Miranda-Saksena, M.; Wakisaka, H.; Tijono, B.; Boadle, R.A.; Rixon, F.; Takahashi, H.; Cunningham, A.L. Herpes simplex virus type 1 accumulation, envelopment, and exit in growth cones and varicosities in mid-distal regions of axons. J. Virol. 2006, 80, 3592–3606. [Google Scholar] [CrossRef]
- Miranda-Saksena, M.; Boadle, R.A.; Aggarwal, A.; Tijono, B.; Rixon, F.J.; Diefenbach, R.J.; Cunningham, A.L. Herpes simplex virus utilizes the large secretory vesicle pathway for anterograde transport of tegument and envelope proteins and for viral exocytosis from growth cones of human fetal axons. J. Virol. 2009, 83, 3187–3199. [Google Scholar] [CrossRef] [PubMed]
- Penfold, M.E.; Armati, P.; Cunningham, A.L. Axonal transport of herpes simplex virions to epidermal cells: Evidence for a specialized mode of virus transport and assembly. Proc. Natl. Acad. Sci. USA 1994, 91, 6529–6533. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Wisner, T.W.; Johnson, D.C. Herpes simplex virus capsids are transported in neuronal axons without an envelope containing the viral glycoproteins. J. Virol. 2006, 80, 11165–11177. [Google Scholar] [CrossRef] [PubMed]
- Snyder, A.; Bruun, B.; Browne, H.M.; Johnson, D.C. A herpes simplex virus gD-YFP fusion glycoprotein is transported separately from viral capsids in neuronal axons. J. Virol. 2007, 81, 8337–8340. [Google Scholar] [CrossRef] [PubMed]
- LaVail, J.H.; Tauscher, A.N.; Sucher, A.; Harrabi, O.; Brandimarti, R. Viral regulation of the long distance axonal transport of herpes simplex virus nucleocapsid. Neuroscience 2007, 146, 974–985. [Google Scholar] [CrossRef]
- Qi, Y.; Wang, J.K.; McMillian, M.; Chikaraishi, D.M. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J. Neurosci. 1997, 17, 1217–1225. [Google Scholar] [CrossRef]
- Li, Y.; Hou, L.X.; Aktiv, A.; Dahlstrom, A. Studies of the central nervous system-derived CAD cell line, a suitable model for intraneuronal transport studies? J. Neurosci. Res. 2007, 85, 2601–2609. [Google Scholar] [CrossRef]
- Antinone, S.E.; Zaichick, S.V.; Smith, G.A. Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J. Virol. 2010, 84, 13019–13030. [Google Scholar] [CrossRef]
- Negatsch, A.; Granzow, H.; Maresch, C.; Klupp, B.G.; Fuchs, W.; Teifke, J.P.; Mettenleiter, T.C. Ultrastructural analysis of virion formation and intraaxonal transport of herpes simplex virus type 1 in primary rat neurons. J. Virol. 2010, 84, 13031–13035. [Google Scholar] [CrossRef]
- DuRaine, G.; Wisner, T.W.; Howard, P.; Williams, M.; Johnson, D.C. Herpes simplex virus gE/gI and US9 promote both envelopment and sorting of virus particles in the cytoplasm of neurons, two processes that precede anterograde transport in axons. J. Virol. 2017, 91, e00050–17. [Google Scholar] [CrossRef]
- Wisner, T.W.; Sugimoto, K.; Howard, P.W.; Kawaguchi, Y.; Johnson, D.C. Anterograde transport of herpes simplex virus capsids in neurons by both separate and married mechanisms. J. Virol. 2011, 85, 5919–5928. [Google Scholar] [CrossRef]
- Holland, D.J.; Miranda-Saksena, M.; Boadle, R.A.; Armati, P.; Cunningham, A.L. Anterograde transport of herpes simplex virus proteins in axons of peripheral human fetal neurons: An immunoelectron microscopy study. J. Virol. 1999, 73, 8503–8511. [Google Scholar] [PubMed]
- Lo, K.Y.; Kuzmin, A.; Unger, S.M.; Petersen, J.D.; Silverman, M.A. KIF1A is the primary anterograde motor protein required for the axonal transport of dense-core vesicles in cultured hippocampal neurons. Neurosci. Lett. 2011, 491, 168–173. [Google Scholar] [CrossRef] [PubMed]
- Szilagyi, J.F.; Cunningham, C. Identification and characterization of a novel non-infectious herpes simplex virus-related particle. J. Gen. Virol. 1991, 72, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Heilingloh, C.S.; Krawczyk, A. Role of L-particles during herpes simplex virus infection. Front. Microbiol. 2017, 8, 2565. [Google Scholar] [CrossRef]
- Rixon, F.J.; Addison, C.; McLauchlan, J. Assembly of enveloped tegument structures (L particles) can occur independently of virion maturation in herpes simplex virus type 1-infected cells. J. Gen. Virol. 1992, 73, 277–284. [Google Scholar] [CrossRef]
- McLauchlan, J.; Rixon, F.J. Characterization of enveloped tegument structures (L particles) produced by alphaherpesviruses: Integrity of the tegument does not depend on the presence of capsid or envelope. J. Gen. Virol. 1992, 73, 269–276. [Google Scholar] [CrossRef]
- Russell, T.; Bleasdale, B.; Hollinshead, M.; Elliott, G. Qualitative differences in capsidless L-particles released as a by-product of bovine herpesvirus 1 and herpes simplex virus 1 infections. J. Virol. 2018, 92, e01259–18. [Google Scholar] [CrossRef]
- Taylor, M.P.; Kramer, T.; Lyman, M.G.; Kratchmarov, R.; Enquist, L.W. Visualization of an alphaherpesvirus membrane protein that is essential for anterograde axonal spread of infection in neurons. MBio 2012, 3, e00063-12. [Google Scholar] [CrossRef]
- Aleman, N.; Quiroga, M.I.; Lopez-Pena, M.; Vazquez, S.; Guerrero, F.H.; Nieto, J.M. L-particle production during primary replication of pseudorabies virus in the nasal mucosa of swine. J. Virol. 2003, 77, 5657–5667. [Google Scholar] [CrossRef]
- Kratchmarov, R.; Enquist, L.W.; Taylor, M.P. Us9-independent axonal sorting and transport of the pseudorabies virus glycoprotein gM. J. Virol. 2015, 89, 6511–6514. [Google Scholar] [CrossRef]
- Ibiricu, I.; Maurer, U.E.; Grunewald, K. Characterization of herpes simplex virus type 1 L-particle assembly and egress in hippocampal neurones by electron cryo-tomography. Cell. Microbiol. 2013, 15, 285–291. [Google Scholar] [CrossRef] [PubMed]
- Enquist, L.W.; Husak, P.J.; Banfield, B.W.; Smith, G.A. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 1998, 51, 237–347. [Google Scholar] [PubMed]
- Husak, P.J.; Kuo, T.; Enquist, L.W. Pseudorabies virus membrane proteins gI and gE facilitate anterograde spread of infection in projection-specific neurons in the rat. J. Virol. 2000, 74, 10975–10983. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tirabassi, R.S.; Townley, R.A.; Eldridge, M.G.; Enquist, L.W. Characterization of pseudorabies virus mutants expressing carboxy-terminal truncations of gE: Evidence for envelope incorporation, virulence, and neurotropism domains. J. Virol. 1997, 71, 6455–6464. [Google Scholar]
- Ch’ng, T.H.; Enquist, L.W. Efficient axonal localization of alphaherpesvirus structural proteins in cultured sympathetic neurons requires viral glycoprotein E. J. Virol. 2005, 79, 8835–8846. [Google Scholar] [CrossRef]
- Brideau, A.D.; Card, J.P.; Enquist, L.W. Role of pseudorabies virus Us9, a type II membrane protein, in infection of tissue culture cells and the rat nervous system. J. Virol. 2000, 74, 834–845. [Google Scholar] [CrossRef]
- Lyman, M.G.; Kemp, C.D.; Taylor, M.P.; Enquist, L.W. Comparison of the pseudorabies virus Us9 protein with homologs from other veterinary and human alphaherpesviruses. J. Virol. 2009, 83, 6978–6986. [Google Scholar] [CrossRef]
- Brideau, A.D.; Banfield, B.W.; Enquist, L.W. The Us9 gene product of pseudorabies virus, an alphaherpesvirus, is a phosphorylated, tail-anchored type II membrane protein. J. Virol. 1998, 72, 4560–4570. [Google Scholar]
- Brideau, A.D.; Eldridge, M.G.; Enquist, L.W. Directional transneuronal infection by pseudorabies virus is dependent on an acidic internalization motif in the Us9 cytoplasmic tail. J. Virol. 2000, 74, 4549–4561. [Google Scholar] [CrossRef]
- Lyman, M.G.; Curanovic, D.; Enquist, L.W. Targeting of pseudorabies virus structural proteins to axons requires association of the viral Us9 protein with lipid rafts. PLoS Pathog. 2008, 4, e1000065. [Google Scholar] [CrossRef]
- Johnson, D.C.; Webb, M.; Wisner, T.W.; Brunetti, C. Herpes simplex virus gE/gI sorts nascent virions to epithelial cell junctions, promoting virus spread. J. Virol. 2001, 75, 821–833. [Google Scholar] [CrossRef] [PubMed]
- Dingwell, K.S.; Doering, L.C.; Johnson, D.C. Glycoproteins E and I facilitate neuron-to-neuron spread of herpes simplex virus. J. Virol. 1995, 69, 7087–7098. [Google Scholar] [PubMed]
- Wang, F.; Zumbrun, E.E.; Huang, J.; Si, H.; Makaroun, L.; Friedman, H.M. Herpes simplex virus type 2 glycoprotein E is required for efficient virus spread from epithelial cells to neurons and for targeting viral proteins from the neuron cell body into axons. Virology 2010, 405, 269–279. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Polcicova, K.; Biswas, P.S.; Banerjee, K.; Wisner, T.W.; Rouse, B.T.; Johnson, D.C. Herpes keratitis in the absence of anterograde transport of virus from sensory ganglia to the cornea. Proc. Natl. Acad. Sci. USA 2005, 102, 11462–11467. [Google Scholar] [CrossRef]
- McGraw, H.M.; Awasthi, S.; Wojcechowskyj, J.A.; Friedman, H.M. Anterograde spread of herpes simplex virus type 1 requires glycoprotein E and glycoprotein I but not Us9. J. Virol. 2009, 83, 8315–8326. [Google Scholar] [CrossRef][Green Version]
- Snyder, A.; Polcicova, K.; Johnson, D.C. Herpes simplex virus gE/gI and US9 proteins promote transport of both capsids and virion glycoproteins in neuronal axons. J. Virol. 2008, 82, 10613–10624. [Google Scholar] [CrossRef]
- Howard, P.W.; Howard, T.L.; Johnson, D.C. Herpes simplex virus membrane proteins gE/gI and US9 act cooperatively to promote transport of capsids and glycoproteins from neuron cell bodies into initial axon segments. J. Virol. 2013, 87, 403–414. [Google Scholar] [CrossRef][Green Version]
- Lyman, M.G.; Feierbach, B.; Curanovic, D.; Bisher, M.; Enquist, L.W. Pseudorabies virus Us9 directs axonal sorting of viral capsids. J. Virol. 2007, 81, 11363–11371. [Google Scholar] [CrossRef]
- Ch’ng, T.H.; Enquist, L.W. Neuron-to-cell spread of pseudorabies virus in a compartmented neuronal culture system. J. Virol. 2005, 79, 10875–10889. [Google Scholar] [CrossRef]
- Kramer, T.; Greco, T.M.; Taylor, M.P.; Ambrosini, A.E.; Cristea, I.M.; Enquist, L.W. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 2012, 12, 806–814. [Google Scholar] [CrossRef]
- Kratchmarov, R.; Kramer, T.; Greco, T.M.; Taylor, M.P.; Ch’ng, T.H.; Cristea, I.M.; Enquist, L.W. Glycoproteins gE and gI are required for efficient KIF1A-dependent anterograde axonal transport of alphaherpesvirus particles in neurons. J. Virol. 2013, 87, 9431–9440. [Google Scholar] [CrossRef] [PubMed]
- Daniel, G.R.; Sollars, P.J.; Pickard, G.E.; Smith, G.A. Pseudorabies virus fast-axonal transport occurs by a pUS9-independent mechanism. J. Virol. 2015, 89, 8088–8091. [Google Scholar] [CrossRef] [PubMed]
- Leterrier, C. The Axon Initial Segment: An Updated Viewpoint. J. Neurosci 2018, 38, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Song, A.H.; Wang, D.; Chen, G.; Li, Y.; Luo, J.; Duan, S.; Poo, M.M. A selective filter for cytoplasmic transport at the axon initial segment. Cell 2009, 136, 1148–1160. [Google Scholar] [CrossRef] [PubMed]
- DuRaine, G.; Wisner, T.W.; Howard, P.; Johnson, D.C. Kinesin-1 Proteins KIF5A, -5B, and -5C promote anterograde transport of herpes simplex virus enveloped virions in axons. J. Virol. 2018, 92, e01269-18. [Google Scholar] [CrossRef] [PubMed]
- Satpute-Krishnan, P.; DeGiorgis, J.A.; Bearer, E.L. Fast anterograde transport of herpes simplex virus: Role for the amyloid precursor protein of alzheimer’s disease. Aging Cell 2003, 2, 305–318. [Google Scholar] [CrossRef]
- Muller, U.; Kins, S. APP on the move. Trends Mol. Med. 2002, 8, 152–155. [Google Scholar] [CrossRef]
- Lazarov, O.; Morfini, G.A.; Lee, E.B.; Farah, M.H.; Szodorai, A.; DeBoer, S.R.; Koliatsos, V.E.; Kins, S.; Lee, V.M.; Wong, P.C.; et al. Axonal transport, amyloid precursor protein, kinesin-1, and the processing apparatus: Revisited. J. Neurosci. 2005, 25, 2386–2395. [Google Scholar] [CrossRef]
- Diefenbach, R.J.; Davis, A.; Miranda-Saksena, M.; Fernandez, M.A.; Kelly, B.J.; Jones, C.A.; LaVail, J.H.; Xue, J.; Lai, J.; Cunningham, A.L. The Basic domain of herpes simplex virus 1 pUS9 Recruits kinesin-1 To facilitate egress from neurons. J. Virol. 2016, 90, 2102–2111. [Google Scholar] [CrossRef]
- Miranda-Saksena, M.; Boadle, R.A.; Diefenbach, R.J.; Cunningham, A.L. Dual role of herpes simplex virus 1 pUS9 in virus anterograde axonal transport and final assembly in growth cones in distal axons. J. Virol. 2016, 90, 2653–2663. [Google Scholar] [CrossRef]
- Hirokawa, N. mRNA transport in dendrites: RNA granules, motors, and tracks. J. Neurosci. 2006, 26, 7139–7142. [Google Scholar] [CrossRef] [PubMed]
- Nakata, T.; Hirokawa, N. Microtubules provide directional cues for polarized axonal transport through interaction with kinesin motor head. J. Cell Biol. 2003, 162, 1045–1055. [Google Scholar] [CrossRef] [PubMed]
- Poppers, J.; Mulvey, M.; Khoo, D.; Mohr, I. Inhibition of PKR activation by the proline-rich RNA binding domain of the herpes simplex virus type 1 Us11 protein. J. Virol. 2000, 74, 11215–11221. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, R.J.; Miranda-Saksena, M.; Diefenbach, E.; Holland, D.J.; Boadle, R.A.; Armati, P.J.; Cunningham, A.L. Herpes simplex virus tegument protein US11 interacts with conventional kinesin heavy chain. J. Virol. 2002, 76, 3282–3291. [Google Scholar] [CrossRef] [PubMed]
- Benboudjema, L.; Mulvey, M.; Gao, Y.; Pimplikar, S.W.; Mohr, I. Association of the herpes simplex virus type 1 Us11 gene product with the cellular kinesin light-chain-related protein PAT1 results in the redistribution of both polypeptides. J. Virol. 2003, 77, 9192–9203. [Google Scholar] [CrossRef]
- Koshizuka, T.; Kawaguchi, Y.; Nishiyama, Y. Herpes simplex virus type 2 membrane protein UL56 associates with the kinesin motor protein KIF1A. J. Gen. Virol. 2005, 86, 527–533. [Google Scholar] [CrossRef]
- Ushijima, Y.; Goshima, F.; Kimura, H.; Nishiyama, Y. Herpes simplex virus type 2 tegument protein UL56 relocalizes ubiquitin ligase Nedd4 and has a role in transport and/or release of virions. Virol. J. 2009, 6, 168–180. [Google Scholar] [CrossRef]
- Kehm, R.; Rosen-Wolff, A.; Darai, G. Restitution of the UL56 gene expression of HSV-1 HFEM led to restoration of virulent phenotype; deletion of the amino acids 217 to 234 of the UL56 protein abrogates the virulent phenotype. Virus Res. 1996, 40, 17–31. [Google Scholar] [CrossRef]
- Xiong, R.; Rao, P.; Kim, S.; Li, M.; Wen, X.; Yuan, W. Herpes Simplex Virus 1 US3 Phosphorylates Cellular KIF3A To Downregulate CD1d Expression. J. Virol. 2015, 89, 6646–6655. [Google Scholar] [CrossRef]
- Hogue, I.B.; Scherer, J.; Enquist, L.W. Exocytosis of alphaherpesvirus virions, light particles, and glycoproteins uses constitutive secretory mechanisms. MBio 2016, 7, e00820–16. [Google Scholar] [CrossRef]
- Hogue, I.B.; Bosse, J.B.; Hu, J.R.; Thiberge, S.Y.; Enquist, L.W. Cellular mechanisms of alpha herpesvirus egress: Live cell fluorescence microscopy of pseudorabies virus exocytosis. PLoS Pathog. 2014, 10, e1004535. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.P.; Kobiler, O.; Enquist, L.W. Alphaherpesvirus axon-to-cell spread involves limited virion transmission. Proc. Natl. Acad. Sci. USA 2012, 109, 17046–17051. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Diwaker, D.; Wilson, D.W. Microtubule-Dependent Trafficking of Alphaherpesviruses in the Nervous System: The Ins and Outs. Viruses 2019, 11, 1165. https://doi.org/10.3390/v11121165
Diwaker D, Wilson DW. Microtubule-Dependent Trafficking of Alphaherpesviruses in the Nervous System: The Ins and Outs. Viruses. 2019; 11(12):1165. https://doi.org/10.3390/v11121165
Chicago/Turabian StyleDiwaker, Drishya, and Duncan W. Wilson. 2019. "Microtubule-Dependent Trafficking of Alphaherpesviruses in the Nervous System: The Ins and Outs" Viruses 11, no. 12: 1165. https://doi.org/10.3390/v11121165
APA StyleDiwaker, D., & Wilson, D. W. (2019). Microtubule-Dependent Trafficking of Alphaherpesviruses in the Nervous System: The Ins and Outs. Viruses, 11(12), 1165. https://doi.org/10.3390/v11121165