The Roles of prM-E Proteins in Historical and Epidemic Zika Virus-mediated Infection and Neurocytotoxicity

 ,
,  , and
, and 
Abstract
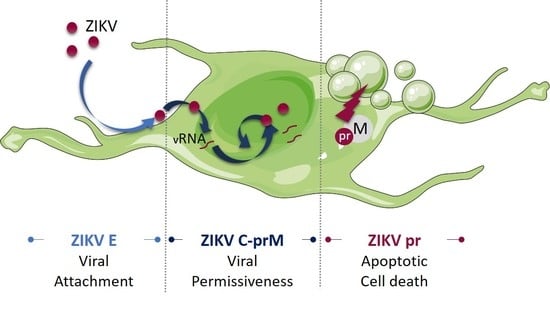
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Zika Virus Molecular Clones and Infection
2.3. Generation and Production of the Chimeric Viruses
2.4. Adenoviral Constructs and Cell Transduction
2.5. Viral Binding Assay
2.6. Immunofluorescence Staining
2.7. Measurement of ZIKV Viral Replication
2.8. MTT Assay
2.9. Measurement of Cellular Necrosis and Apoptosis
2.10. Statistical Analysis
3. Results
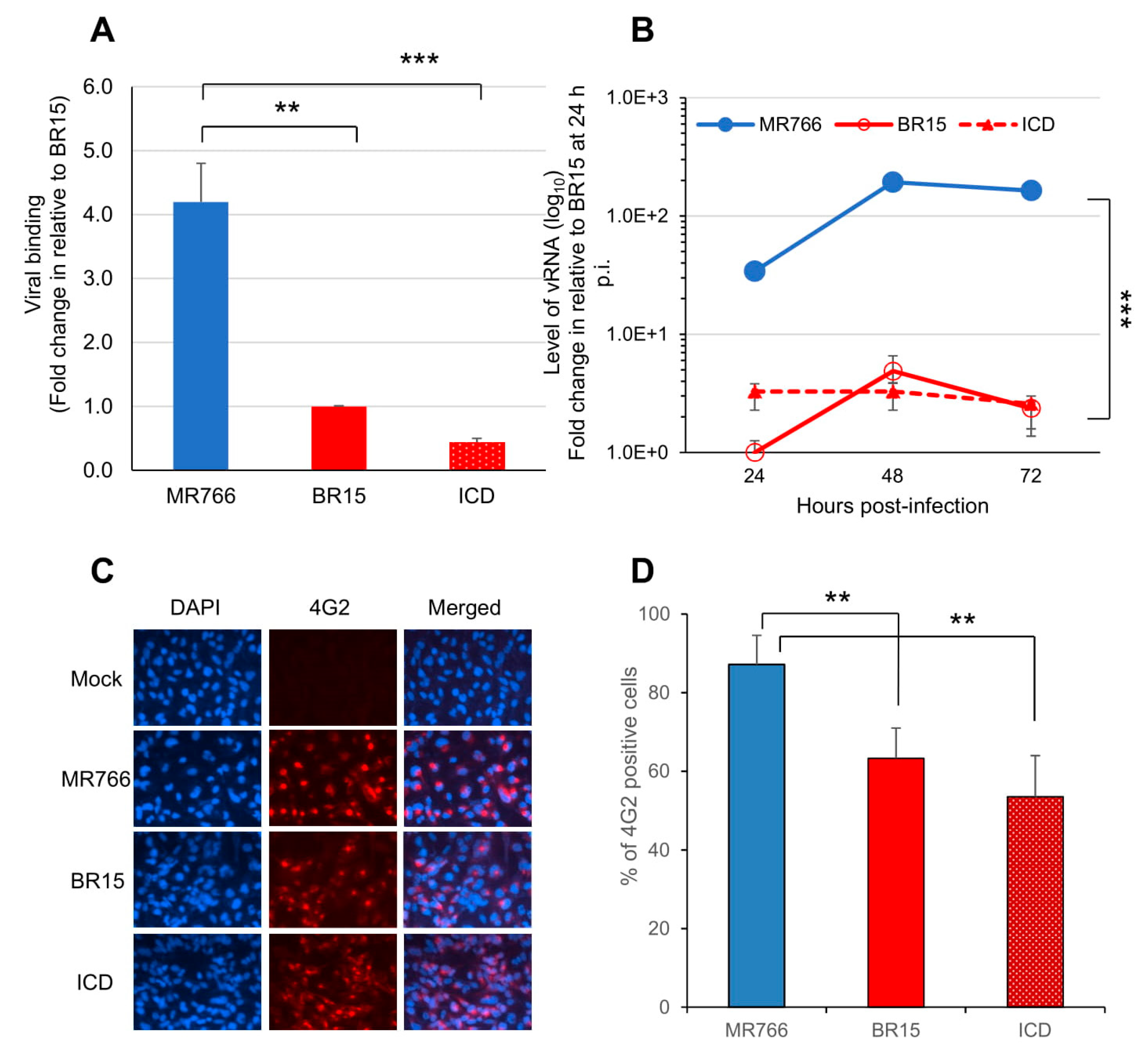
3.1. Comparison of Viral Infectivity Between the Historical African Zika Virus and the Epidemic Brazilian Zika Viruses in Human Brain Glial SNB-19 Cells
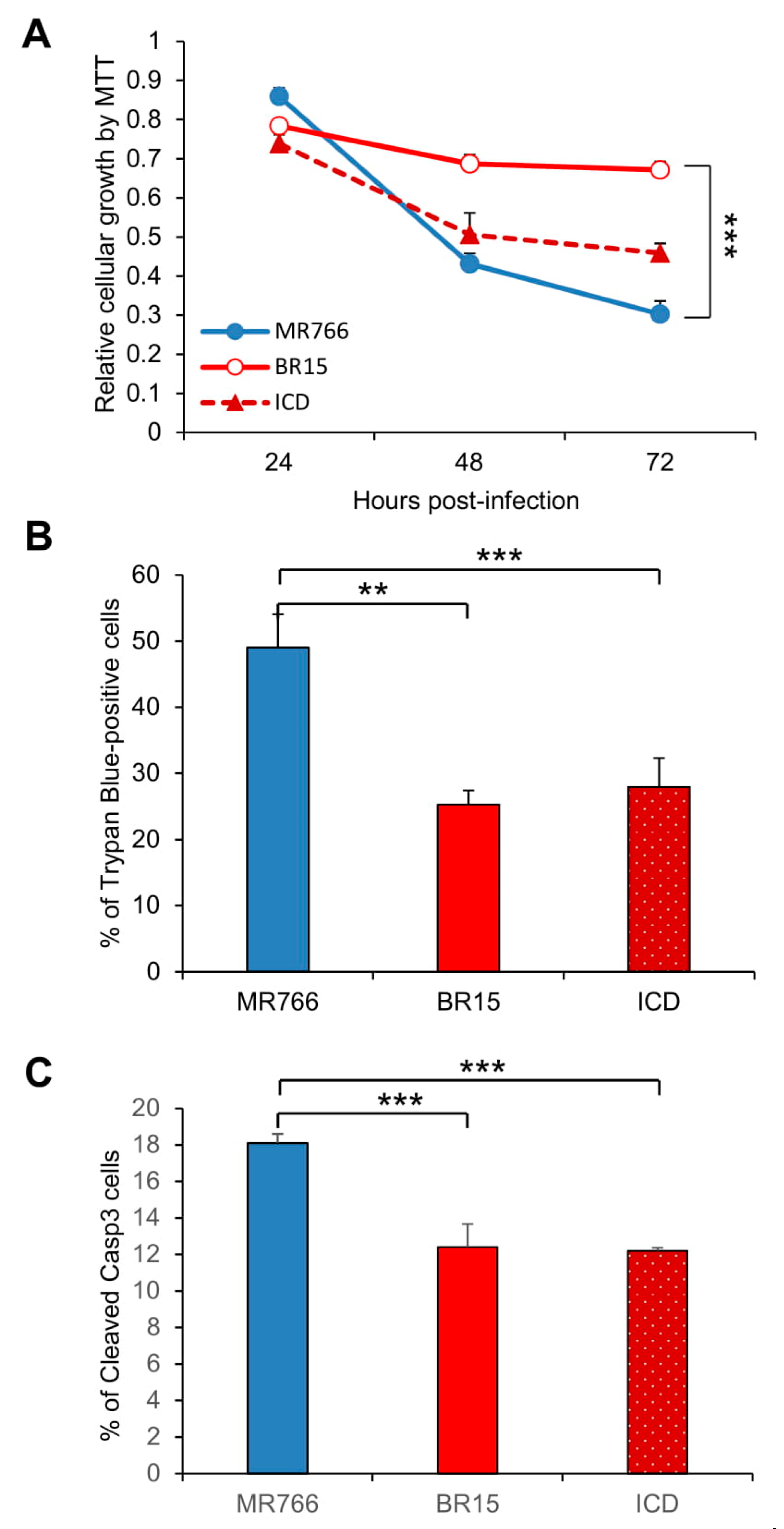
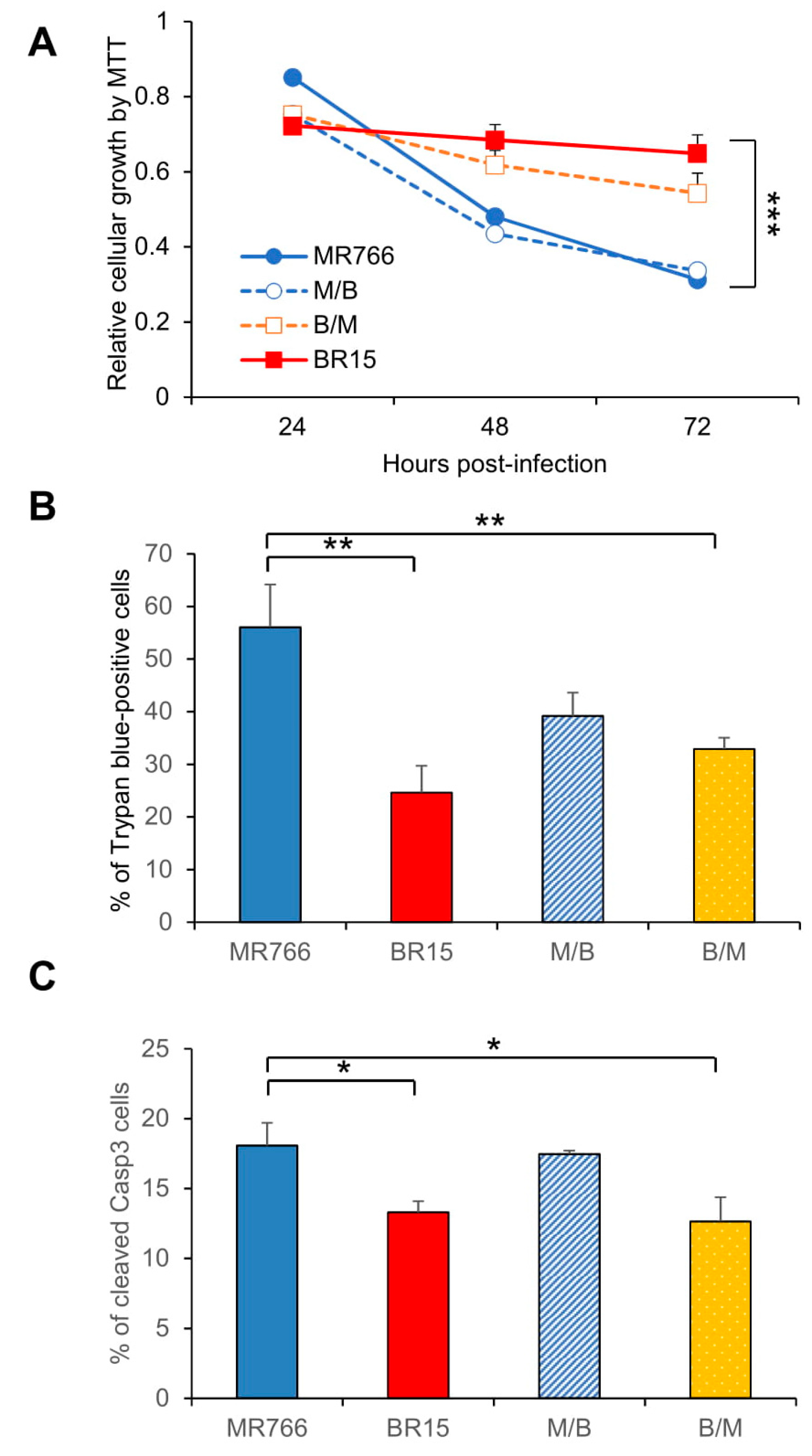
3.2. Comparison of the Historical Zika Virus with the Brazilian Epidemic Zika Viruses in Their Abilities to Induce Cytotoxicity
3.3. Effects of the C-prM Region on Viral Infectivity
3.4. Contribution of the ZIKV C-prM Region to ZIKV-Induced Growth Restriction and Apoptotic Cell Death
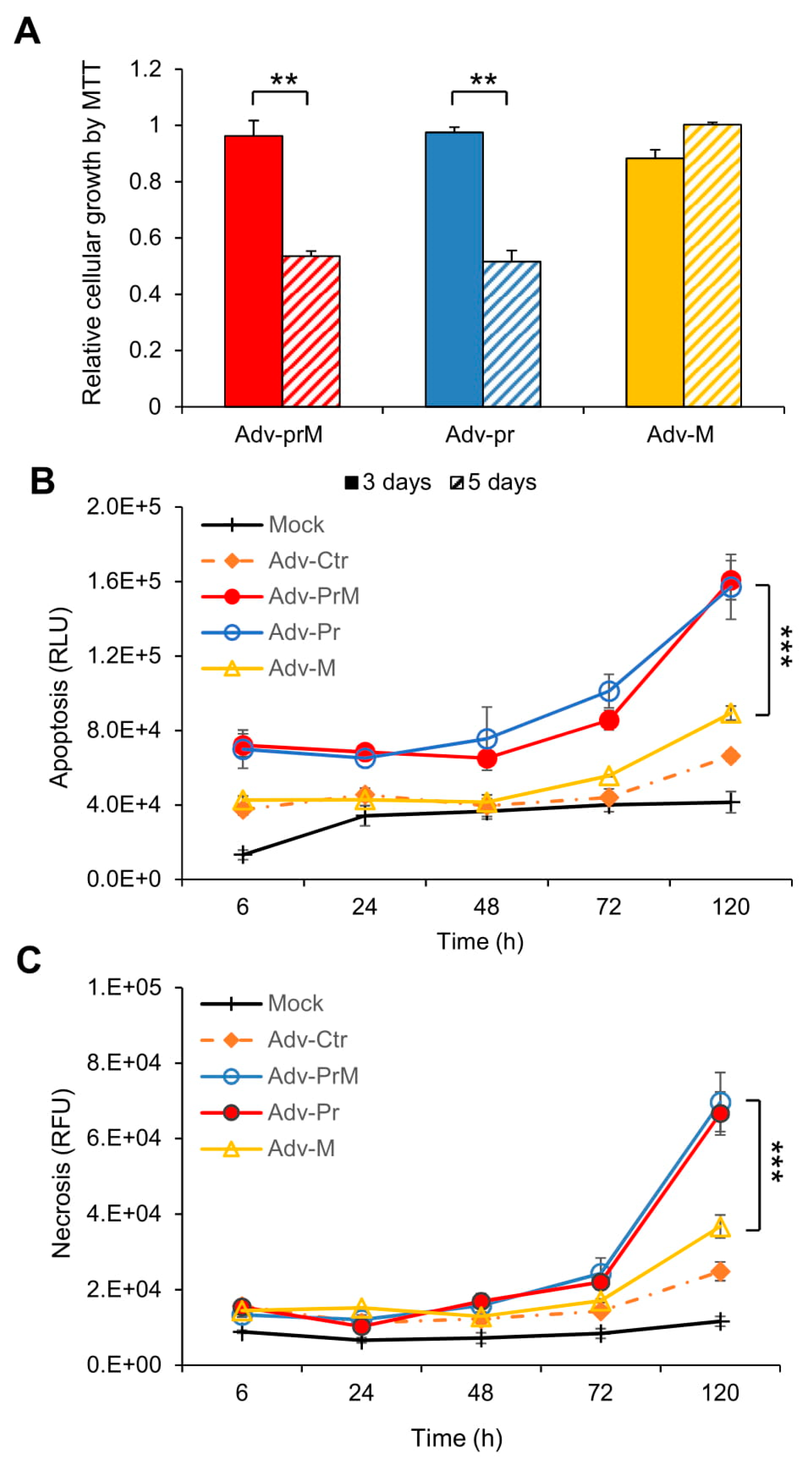
3.5. Effect of prM Protein and Its Processed Protein Products (M and Pr) on ZIKV-Induced Cytotoxicity
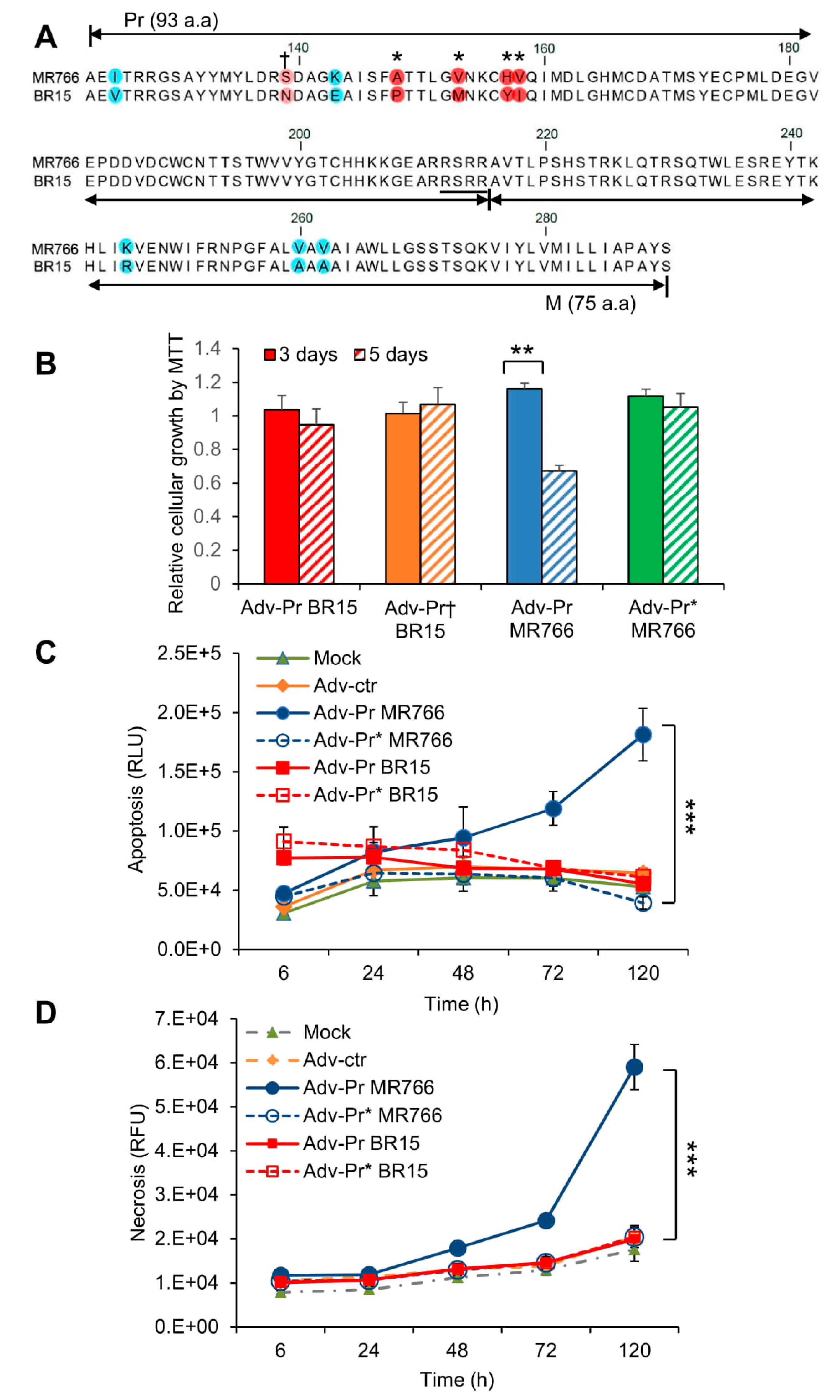
3.6. Mutational Analysis of the Pr Region of the prM Protein and Their Effects on ZIKV-Induced Cytotoxicity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Victora, C.G.; Schuler-Faccini, L.; Matijasevich, A.; Ribeiro, E.; Pessoa, A.; Barros, F.C. Microcephaly in Brazil: How to interpret reported numbers? Lancet 2016, 387, 621–624. [Google Scholar] [CrossRef]
- Melo, A.S.; Aguiar, R.S.; Amorim, M.M.; Arruda, M.B.; Melo, F.O.; Ribeiro, S.T.; Batista, A.G.; Ferreira, T.; Dos Santos, M.P.; Sampaio, V.V.; et al. Congenital Zika Virus Infection: Beyond Neonatal Microcephaly. JAMA Neurol. 2016, 73, 1407–1416. [Google Scholar] [CrossRef] [PubMed]
- Del Campo, M.; Feitosa, I.M.; Ribeiro, E.M.; Horovitz, D.D.; Pessoa, A.L.; Franca, G.V.; Garcia-Alix, A.; Doriqui, M.J.; Wanderley, H.Y.; Sanseverino, M.V.; et al. The phenotypic spectrum of congenital Zika syndrome. Am. J. Med. Genet. A 2017, 173, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, J.; Korva, M.; Tul, N.; Popovic, M.; Poljsak-Prijatelj, M.; Mraz, J.; Kolenc, M.; Resman Rus, K.; Vesnaver Vipotnik, T.; Fabjan Vodusek, V.; et al. Zika Virus Associated with Microcephaly. N. Engl. J. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Driggers, R.W.; Ho, C.Y.; Korhonen, E.M.; Kuivanen, S.; Jaaskelainen, A.J.; Smura, T.; Rosenberg, A.; Hill, D.A.; DeBiasi, R.L.; Vezina, G.; et al. Zika Virus Infection with Prolonged Maternal Viremia and Fetal Brain Abnormalities. N. Engl. J. Med. 2016. [Google Scholar] [CrossRef] [PubMed]
- Qian, X.; Nguyen, H.N.; Song, M.M.; Hadiono, C.; Ogden, S.C.; Hammack, C.; Yao, B.; Hamersky, G.R.; Jacob, F.; Zhong, C.; et al. Brain-Region-Specific Organoids Using Mini-bioreactors for Modeling ZIKV Exposure. Cell 2016, 165, 1238–1254. [Google Scholar] [CrossRef] [PubMed]
- Dick, G.W. Zika virus. II. Pathogenicity and physical properties. Trans. R Soc. Trop. Med. Hyg. 1952, 46, 521–534. [Google Scholar] [CrossRef]
- Bell, T.M.; Field, E.J.; Narang, H.K. Zika virus infection of the central nervous system of mice. Arch. Gesamte. Virusforsch. 1971, 35, 183–193. [Google Scholar] [CrossRef] [Green Version]
- Dick, G.W.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R Soc. Trop Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Lee, I.; Bos, S.; Li, G.; Wang, S.; Gadea, G.; Despres, P.; Zhao, R.Y. Probing Molecular Insights into Zika Virus(-)Host Interactions. Viruses 2018, 10. [Google Scholar] [CrossRef]
- Musso, D.; Nhan, T.; Robin, E.; Roche, C.; Bierlaire, D.; Zisou, K.; Shan Yan, A.; Cao-Lormeau, V.M.; Broult, J. Potential for Zika virus transmission through blood transfusion demonstrated during an outbreak in French Polynesia, November 2013 to February 2014. Euro. Surveill. 2014, 19. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Hennessey, M.; Fischer, M.; Staples, J.E. Zika Virus Spreads to New Areas-Region of the Americas, May 2015-January 2016. MMWR Morb. Mortal. Wkly. Rep. 2016, 65, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Talero-Gutierrez, C.; Rivera-Molina, A.; Perez-Pavajeau, C.; Ossa-Ospina, I.; Santos-Garcia, C.; Rojas-Anaya, M.C.; de-la-Torre, A. Zika virus epidemiology: From Uganda to world pandemic, an update. Epidemiol. Infect. 2018, 146, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Poulsen, M.; Fenyvuesvolgyi, C.; Yashiroda, Y.; Yoshida, M.; Simard, J.M.; Gallo, R.C.; Zhao, R.Y. Characterization of cytopathic factors through genome-wide analysis of the Zika viral proteins in fission yeast. Proc. Natl. Acad. Sci. USA 2017, 114, E376–E385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef]
- Harris, E.; Holden, K.L.; Edgil, D.; Polacek, C.; Clyde, K. Molecular biology of flaviviruses. Novartis. Found. Symp. 2006, 277, 23–39, discussion 40, 71–23, 251–253. [Google Scholar]
- Yu, I.M.; Zhang, W.; Holdaway, H.A.; Li, L.; Kostyuchenko, V.A.; Chipman, P.R.; Kuhn, R.J.; Rossmann, M.G.; Chen, J. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 2008, 319, 1834–1837. [Google Scholar] [CrossRef]
- Mecharles, S.; Herrmann, C.; Poullain, P.; Tran, T.H.; Deschamps, N.; Mathon, G.; Landais, A.; Breurec, S.; Lannuzel, A. Acute myelitis due to Zika virus infection. Lancet 2016. [Google Scholar] [CrossRef]
- Faye, O.; Freire, C.C.; Iamarino, A.; Faye, O.; de Oliveira, J.V.; Diallo, M.; Zanotto, P.M.; Sall, A.A. Molecular evolution of Zika virus during its emergence in the 20(th) century. PLoS Negl. Trop Dis. 2014, 8, e2636. [Google Scholar] [CrossRef]
- Fontes-Garfias, C.R.; Shan, C.; Luo, H.; Muruato, A.E.; Medeiros, D.B.A.; Mays, E.; Xie, X.; Zou, J.; Roundy, C.M.; Wakamiya, M.; et al. Functional Analysis of Glycosylation of Zika Virus Envelope Protein. Cell Rep. 2017, 21, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Lobigs, M.; Lee, E.; Ng, M.L.; Pavy, M.; Lobigs, P. A flavivirus signal peptide balances the catalytic activity of two proteases and thereby facilitates virus morphogenesis. Virology 2010, 401, 80–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amberg, S.M.; Nestorowicz, A.; McCourt, D.W.; Rice, C.M. NS2B-3 proteinase-mediated processing in the yellow fever virus structural region: In vitro and in vivo studies. J. Virol. 1994, 68, 3794–3802. [Google Scholar] [PubMed]
- Elshuber, S.; Allison, S.L.; Heinz, F.X.; Mandl, C.W. Cleavage of protein prM is necessary for infection of BHK-21 cells by tick-borne encephalitis virus. J. Gen. Virol. 2003, 84, 183–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anfasa, F.; Siegers, J.Y.; van der Kroeg, M.; Mumtaz, N.; Stalin Raj, V.; de Vrij, F.M.S.; Widagdo, W.; Gabriel, G.; Salinas, S.; Simonin, Y.; et al. Phenotypic Differences between Asian and African Lineage Zika Viruses in Human Neural Progenitor Cells. mSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Abushouk, A.I.; Negida, A.; Ahmed, H. An updated review of Zika virus. J. Clin. Virol. 2016, 84, 53–58. [Google Scholar] [CrossRef] [PubMed]
- Simonin, Y.; van Riel, D.; Van de Perre, P.; Rockx, B.; Salinas, S. Differential virulence between Asian and African lineages of Zika virus. PLoS Negl. Trop Dis. 2017, 11, e0005821. [Google Scholar] [CrossRef] [PubMed]
- Beaver, J.T.; Lelutiu, N.; Habib, R.; Skountzou, I. Evolution of Two Major Zika Virus Lineages: Implications for Pathology, Immune Response, and Vaccine Development. Front. Immunol. 2018, 9, 1640. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, A.; Dai, L.; Contreras, D.; Sinha, S.; Sun, R.; Arumugaswami, V. Comparative analysis of protein evolution in the genome of pre-epidemic and epidemic Zika virus. Infect. Genet. Evol. 2017, 51, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Bos, S.; Viranaicken, W.; Turpin, J.; El-Kalamouni, C.; Roche, M.; Krejbich-Trotot, P.; Despres, P.; Gadea, G. The structural proteins of epidemic and historical strains of Zika virus differ in their ability to initiate viral infection in human host cells. Virology 2018, 516, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Huang, X.Y.; Liu, Z.Y.; Zhang, F.; Zhu, X.L.; Yu, J.Y.; Ji, X.; Xu, Y.P.; Li, G.; Li, C.; et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017, 358, 933–936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, H.; Hammack, C.; Ogden, S.C.; Wen, Z.; Qian, X.; Li, Y.; Yao, B.; Shin, J.; Zhang, F.; Lee, E.M.; et al. Zika Virus Infects Human Cortical Neural Progenitors and Attenuates Their Growth. Cell Stem. Cell 2016, 18, 587–590. [Google Scholar] [CrossRef] [PubMed]
- Gadea, G.; Bos, S.; Krejbich-Trotot, P.; Clain, E.; Viranaicken, W.; El-Kalamouni, C.; Mavingui, P.; Despres, P. A robust method for the rapid generation of recombinant Zika virus expressing the GFP reporter gene. Virology 2016, 497, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Faria, N.R.; Azevedo Rdo, S.; Kraemer, M.U.; Souza, R.; Cunha, M.S.; Hill, S.C.; Theze, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I.; et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Widman, D.G.; Young, E.; Yount, B.L.; Plante, K.S.; Gallichotte, E.N.; Carbaugh, D.L.; Peck, K.M.; Plante, J.; Swanstrom, J.; Heise, M.T.; et al. A Reverse Genetics Platform That Spans the Zika Virus Family Tree. MBio 2017, 8. [Google Scholar] [CrossRef]
- Tsetsarkin, K.A.; Kenney, H.; Chen, R.; Liu, G.; Manukyan, H.; Whitehead, S.S.; Laassri, M.; Chumakov, K.; Pletnev, A.G. A Full-Length Infectious cDNA Clone of Zika Virus from the 2015 Epidemic in Brazil as a Genetic Platform for Studies of Virus-Host Interactions and Vaccine Development. MBio 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.Y.; Liu, S.Q.; Deng, C.L.; Zhang, Q.Y.; Zhang, B. Detection of Zika virus by SYBR green one-step real-time RT-PCR. J. Virol. Methods 2016, 236, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Frumence, E.; Roche, M.; Krejbich-Trotot, P.; El-Kalamouni, C.; Nativel, B.; Rondeau, P.; Misse, D.; Gadea, G.; Viranaicken, W.; Despres, P. The South Pacific epidemic strain of Zika virus replicates efficiently in human epithelial A549 cells leading to IFN-beta production and apoptosis induction. Virology 2016, 493, 217–226. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Rodriguez, R.A.; Pepper, I.L.; Gerba, C.P. Application of PCR-based methods to assess the infectivity of enteric viruses in environmental samples. Appl. Environ. Microbiol. 2009, 75, 297–307. [Google Scholar] [CrossRef]
- Zheng, A.; Umashankar, M.; Kielian, M. In vitro and in vivo studies identify important features of dengue virus pr-E protein interactions. PLoS Pathog. 2010, 6, e1001157. [Google Scholar] [CrossRef]
- Shao, Q.; Herrlinger, S.; Zhu, Y.N.; Yang, M.; Goodfellow, F.; Stice, S.L.; Qi, X.P.; Brindley, M.A.; Chen, J.F. The African Zika virus MR-766 is more virulent and causes more severe brain damage than current Asian lineage and dengue virus. Development 2017, 144, 4114–4124. [Google Scholar] [CrossRef] [PubMed]
- Simonin, Y.; Loustalot, F.; Desmetz, C.; Foulongne, V.; Constant, O.; Fournier-Wirth, C.; Leon, F.; Moles, J.P.; Goubaud, A.; Lemaitre, J.M.; et al. Zika Virus Strains Potentially Display Different Infectious Profiles in Human Neural Cells. EBioMedicine 2016, 12, 161–169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGrath, E.L.; Rossi, S.L.; Gao, J.; Widen, S.G.; Grant, A.C.; Dunn, T.J.; Azar, S.R.; Roundy, C.M.; Xiong, Y.; Prusak, D.J.; et al. Differential Responses of Human Fetal Brain Neural Stem Cells to Zika Virus Infection. Stem Cell Rep. 2017, 8, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Shan, C.; Xie, X.; Muruato, A.E.; Rossi, S.L.; Roundy, C.M.; Azar, S.R.; Yang, Y.; Tesh, R.B.; Bourne, N.; Barrett, A.D.; et al. An Infectious cDNA Clone of Zika Virus to Study Viral Virulence, Mosquito Transmission, and Antiviral Inhibitors. Cell Host Microbe 2016, 19, 891–900. [Google Scholar] [CrossRef]
- Stadler, K.; Allison, S.L.; Schalich, J.; Heinz, F.X. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 1997, 71, 8475–8481. [Google Scholar]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, G.; Bos, S.; Tsetsarkin, K.A.; Pletnev, A.G.; Desprès, P.; Gadea, G.; Zhao, R.Y. The Roles of prM-E Proteins in Historical and Epidemic Zika Virus-mediated Infection and Neurocytotoxicity. Viruses 2019, 11, 157. https://doi.org/10.3390/v11020157
Li G, Bos S, Tsetsarkin KA, Pletnev AG, Desprès P, Gadea G, Zhao RY. The Roles of prM-E Proteins in Historical and Epidemic Zika Virus-mediated Infection and Neurocytotoxicity. Viruses. 2019; 11(2):157. https://doi.org/10.3390/v11020157
Chicago/Turabian StyleLi, Ge, Sandra Bos, Konstantin A. Tsetsarkin, Alexander G. Pletnev, Philippe Desprès, Gilles Gadea, and Richard Y. Zhao. 2019. "The Roles of prM-E Proteins in Historical and Epidemic Zika Virus-mediated Infection and Neurocytotoxicity" Viruses 11, no. 2: 157. https://doi.org/10.3390/v11020157
APA StyleLi, G., Bos, S., Tsetsarkin, K. A., Pletnev, A. G., Desprès, P., Gadea, G., & Zhao, R. Y. (2019). The Roles of prM-E Proteins in Historical and Epidemic Zika Virus-mediated Infection and Neurocytotoxicity. Viruses, 11(2), 157. https://doi.org/10.3390/v11020157