Influenza Hemagglutinin and Neuraminidase: Yin–Yang Proteins Coevolving to Thwart Immunity


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
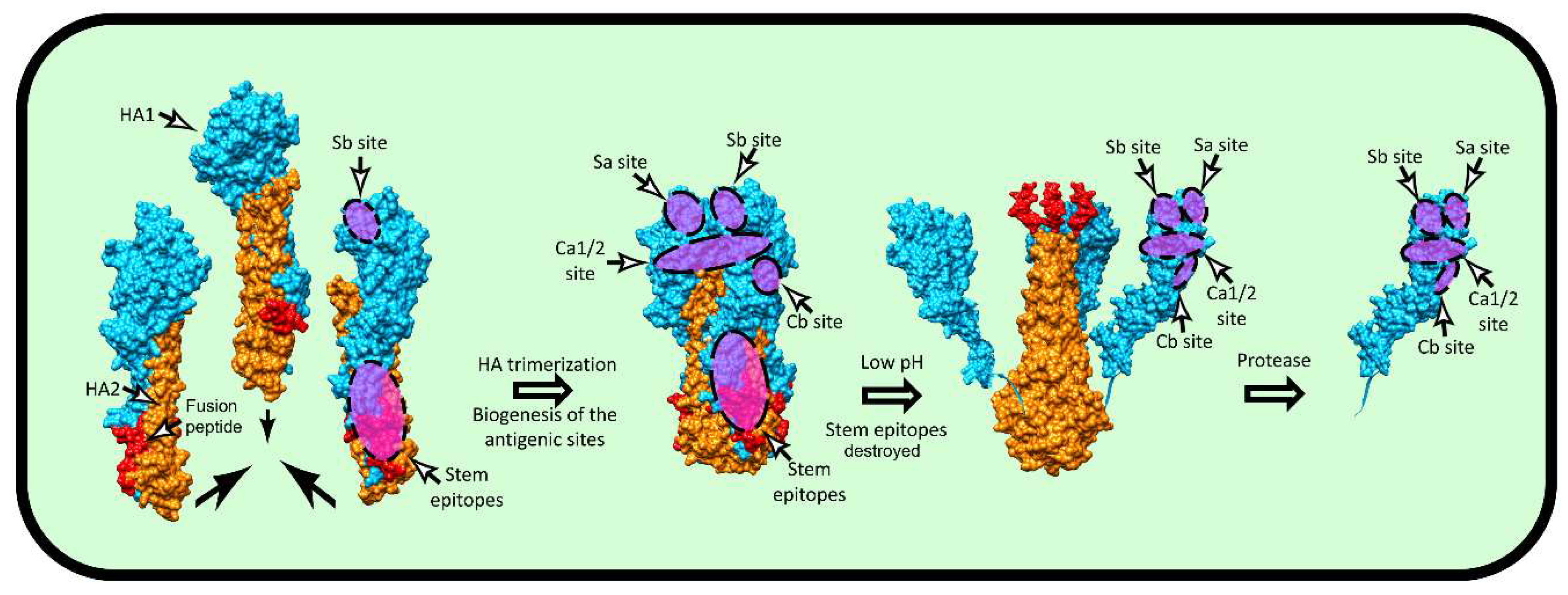
:1. Introduction
2. HA Attaches, NA Releases
3. HA/NA Coevolution: NA Perspective
4. HA/NA Coevolution: HA Perspective
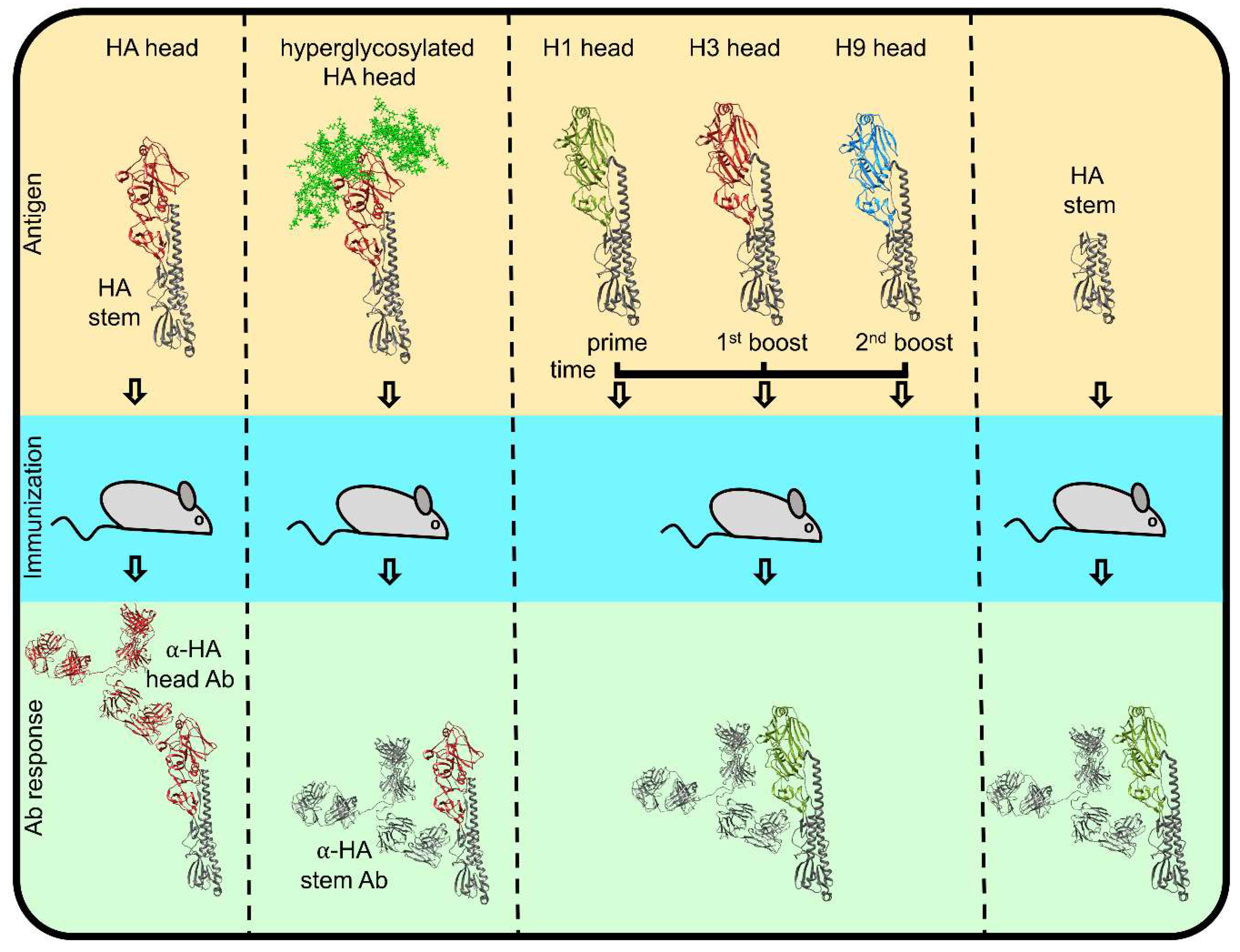
5. Antibody Response to HA and NA
6. Ab-Based NA–HA Cross Talk
7. Future Directions
- (1)
- How does the geometric distribution of HA and NA on the virion effect their functions? Why is NA clustered on virions? Is the extent of clustering variable between IAV strains? How does the relationship between NA and HA alter in filamentous vs. spherical virions? How does the stoichiometry of virion HA and NA influence viral function? What mechanisms does the virus use to control HA–NA virion content?
- (2)
- How do Ab-heavy chains influence the functional activities of anti-HA Abs on NA activity, particularly the larger oligomeric structure of IgM and IgG? Further, what is the functional impact of the effect of Ab binding molecules such as complement?
- (3)
- What is it exactly about HA and NA that enables their rapid antigenic evolution, while analogous proteins on other viruses evolve much more slowly? Is it the freedom for independent mutations afforded by a segmented genome? Is it some inherent resistance to in vivo Ab-mediated neutralization in humans that is not apparent in animal models? Is it something about the immunodominance hierarchy in individuals that enables sequential escape across a population?
Acknowledgments
Conflicts of Interest
References
- Muramoto, Y.; Noda, T.; Kawakami, E.; Akkina, R.; Kawaoka, Y. Identification of novel influenza A virus proteins translated from PA mRNA. J. Virol. 2013, 87, 2455–2462. [Google Scholar] [CrossRef]
- Hutchinson, E.C.; Charles, P.D.; Hester, S.S.; Thomas, B.; Trudgian, D.; Martinez-Alonso, M.; Fodor, E. Conserved and host-specific features of influenza virion architecture. Nat. Commun. 2014, 5, 4816. [Google Scholar] [CrossRef]
- McCrone, J.T.; Woods, R.J.; Martin, E.T.; Malosh, R.E.; Monto, A.S.; Lauring, A.S. Stochastic processes constrain the within and between host evolution of influenza virus. eLife 2018, 7. [Google Scholar] [CrossRef]
- Eichelberger, M.C.; Wan, H. Influenza neuraminidase as a vaccine antigen. Curr. Top. Microbiol. Immunol. 2015, 386, 275–299. [Google Scholar]
- Thyagarajan, B.; Bloom, J.D. The inherent mutational tolerance and antigenic evolvability of influenza hemagglutinin. eLife 2014, 3. [Google Scholar] [CrossRef]
- Visher, E.; Whitefield, S.E.; McCrone, J.T.; Fitzsimmons, W.; Lauring, A.S. The Mutational Robustness of Influenza A Virus. PLoS Pathog. 2016, 12, e1005856. [Google Scholar] [CrossRef]
- Altman, M.O.; Angeletti, D.; Yewdell, J.W. Antibody Immunodominance: The Key to Understanding Influenza Virus Antigenic Drift. Viral Immunol. 2018, 31, 142–149. [Google Scholar] [CrossRef]
- Brooke, C.B.; Ince, W.L.; Wei, J.; Bennink, J.R.; Yewdell, J.W. Influenza A virus nucleoprotein selectively decreases neuraminidase gene-segment packaging while enhancing viral fitness and transmissibility. Proc. Natl. Acad. Sci. USA 2014, 111, 16854–16859. [Google Scholar] [CrossRef] [Green Version]
- Ince, W.L.; Gueye-Mbaye, A.; Bennink, J.R.; Yewdell, J.W. Reassortment complements spontaneous mutation in influenza A virus NP and M1 genes to accelerate adaptation to a new host. J. Virol. 2013, 87, 4330–4338. [Google Scholar] [CrossRef]
- Lowen, A.C. Constraints, Drivers, and Implications of Influenza A Virus Reassortment. Ann. Rev. Virol. 2017, 4, 105–121. [Google Scholar] [CrossRef]
- Phipps, K.L.; Marshall, N.; Tao, H.; Danzy, S.; Onuoha, N.; Steel, J.; Lowen, A.C. Seasonal H3N2 and 2009 Pandemic H1N1 Influenza A Viruses Reassort Efficiently but Produce Attenuated Progeny. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Richard, M.; Herfst, S.; Tao, H.; Jacobs, N.T.; Lowen, A.C. Influenza A virus reassortment is limited by anatomical compartmentalization following co-infection via distinct routes. J. Virol. 2018. [Google Scholar] [CrossRef]
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J.; et al. New world bats harbor diverse influenza A viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef]
- Wu, Y.; Wu, Y.; Tefsen, B.; Shi, Y.; Gao, G.F. Bat-derived influenza-like viruses H17N10 and H18N11. Trends Microbiol. 2014, 22, 183–191. [Google Scholar] [CrossRef]
- Tsai, K.N.; Chen, G.W. Influenza genome diversity and evolution. Microbes Infect. 2011, 13, 479–488. [Google Scholar] [CrossRef]
- Rejmanek, D.; Hosseini, P.R.; Mazet, J.A.; Daszak, P.; Goldstein, T. Evolutionary Dynamics and Global Diversity of Influenza A Virus. J. Virol. 2015, 89, 10993–11001. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Eichelberger, M.C.; Compans, R.W.; Air, G.M. Influenza type A virus neuraminidase does not play a role in viral entry, replication, assembly, or budding. J. Virol. 1995, 69, 1099–1106. [Google Scholar]
- Stray, S.J.; Cummings, R.D.; Air, G.M. Influenza virus infection of desialylated cells. Glycobiology 2000, 10, 649–658. [Google Scholar] [CrossRef] [Green Version]
- Mammen, M.; Choi, S.K.; Whitesides, G.M. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew Chem. Int. Ed. Engl. 1998, 37, 2754–2794. [Google Scholar] [CrossRef]
- Song, H.; Qi, J.; Xiao, H.; Bi, Y.; Zhang, W.; Xu, Y.; Wang, F.; Shi, Y.; Gao, G.F. Avian-to-Human Receptor-Binding Adaptation by Influenza A Virus Hemagglutinin H4. Cell Rep. 2017, 20, 1201–1214. [Google Scholar] [CrossRef]
- Wang, F.; Qi, J.; Bi, Y.; Zhang, W.; Wang, M.; Zhang, B.; Wang, M.; Liu, J.; Yan, J.; Shi, Y.; et al. Adaptation of avian influenza A (H6N1) virus from avian to human receptor-binding preference. EMBO J. 2015, 34, 1661–1673. [Google Scholar] [CrossRef] [Green Version]
- Kosik, I.; Ince, W.L.; Gentles, L.E.; Oler, A.J.; Kosikova, M.; Angel, M.; Magadan, J.G.; Xie, H.; Brooke, C.B.; Yewdell, J.W. Correction: Influenza A virus hemagglutinin glycosylation compensates for antibody escape fitness costs. PLoS Pathog. 2018, 14, e1007141. [Google Scholar] [CrossRef]
- Badham, M.D.; Rossman, J.S. Filamentous Influenza Viruses. Curr. Clin. Microbiol. Rep. 2016, 3, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Calder, L.J.; Wasilewski, S.; Berriman, J.A.; Rosenthal, P.B. Structural organization of a filamentous influenza A virus. Proc. Natl. Acad. Sci. USA 2010, 107, 10685–10690. [Google Scholar] [CrossRef] [Green Version]
- Matrosovich, M.; Tuzikov, A.; Bovin, N.; Gambaryan, A.; Klimov, A.; Castrucci, M.R.; Donatelli, I.; Kawaoka, Y. Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J. Virol. 2000, 74, 8502–8512. [Google Scholar] [CrossRef]
- Dadonaite, B.; Vijayakrishnan, S.; Fodor, E.; Bhella, D.; Hutchinson, E.C. Filamentous influenza viruses. J. Gen. Virol. 2016, 97, 1755–1764. [Google Scholar] [CrossRef]
- Chen, L.M.; Blixt, O.; Stevens, J.; Lipatov, A.S.; Davis, C.T.; Collins, B.E.; Cox, N.J.; Paulson, J.C.; Donis, R.O. In vitro evolution of H5N1 avian influenza virus toward human-type receptor specificity. Virology 2012, 422, 105–113. [Google Scholar] [CrossRef]
- Leung, H.S.; Li, O.T.; Chan, R.W.; Chan, M.C.; Nicholls, J.M.; Poon, L.L. Entry of influenza A Virus with a alpha2,6-linked sialic acid binding preference requires host fibronectin. J. Virol. 2012, 86, 10704–10713. [Google Scholar] [CrossRef]
- Chutinimitkul, S.; Herfst, S.; Steel, J.; Lowen, A.C.; Ye, J.; van Riel, D.; Schrauwen, E.J.; Bestebroer, T.M.; Koel, B.; Burke, D.F.; et al. Virulence-associated substitution D222G in the hemagglutinin of 2009 pandemic influenza A(H1N1) virus affects receptor binding. J. Virol. 2010, 84, 11802–11813. [Google Scholar] [CrossRef]
- Watanabe, T.; Kiso, M.; Fukuyama, S.; Nakajima, N.; Imai, M.; Yamada, S.; Murakami, S.; Yamayoshi, S.; Iwatsuki-Horimoto, K.; Sakoda, Y.; et al. Characterization of H7N9 influenza A viruses isolated from humans. Nature 2013, 501, 551–555. [Google Scholar] [CrossRef] [Green Version]
- Belser, J.A.; Gustin, K.M.; Pearce, M.B.; Maines, T.R.; Zeng, H.; Pappas, C.; Sun, X.; Carney, P.J.; Villanueva, J.M.; Stevens, J.; et al. Pathogenesis and transmission of avian influenza A (H7N9) virus in ferrets and mice. Nature 2013, 501, 556–559. [Google Scholar] [CrossRef]
- Gulati, S.; Smith, D.F.; Cummings, R.D.; Couch, R.B.; Griesemer, S.B.; St George, K.; Webster, R.G.; Air, G.M. Human H3N2 Influenza Viruses Isolated from 1968 To 2012 Show Varying Preference for Receptor Substructures with No Apparent Consequences for Disease or Spread. PLoS ONE 2013, 8, e66325. [Google Scholar]
- Yang, H.; Carney, P.J.; Chang, J.C.; Guo, Z.; Villanueva, J.M.; Stevens, J. Structure and receptor binding preferences of recombinant human A(H3N2) virus hemagglutinins. Virology 2015, 477, 18–31. [Google Scholar] [CrossRef] [Green Version]
- De Graaf, M.; Fouchier, R.A. Role of receptor binding specificity in influenza A virus transmission and pathogenesis. EMBO J. 2014, 33, 823–841. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.P.; Xiong, X.L.; Wharton, S.A.; Martin, S.R.; Coombs, P.J.; Vachieri, S.G.; Christodoulou, E.; Walker, P.A.; Liu, J.F.; Skehel, J.J.; et al. Evolution of the receptor binding properties of the influenza A(H3N2) hemagglutinin. Proc. Natl. Acad. Sci. USA 2012, 109, 21474–21479. [Google Scholar] [CrossRef]
- Vigerust, D.J.; Ulett, K.B.; Boyd, K.L.; Madsen, J.; Hawgood, S.; McCullers, J.A. N-linked glycosylation attenuates H3N2 influenza viruses. J. Virol. 2007, 81, 8593–8600. [Google Scholar] [CrossRef]
- Das, S.R.; Hensley, S.E.; David, A.; Schmidt, L.; Gibbs, J.S.; Puigbo, P.; Ince, W.L.; Bennink, J.R.; Yewdell, J.W. Fitness costs limit influenza A virus hemagglutinin glycosylation as an immune evasion strategy. Proc. Natl. Acad. Sci. USA 2011, 108, E1417–E1422. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.J.; de Vries, R.P.; Grant, O.C.; Thompson, A.J.; McBride, R.; Tsogtbaatar, B.; Lee, P.S.; Razi, N.; Wilson, I.A.; Woods, R.J.; et al. Recent H3N2 Viruses Have Evolved Specificity for Extended, Branched Human-type Receptors, Conferring Potential for Increased Avidity. Cell Host Microbe 2017, 21, 23–34. [Google Scholar] [CrossRef]
- Sieczkarski, S.B.; Whittaker, G.R. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 2002, 76, 10455–10464. [Google Scholar] [CrossRef]
- De Vries, E.; Tscherne, D.M.; Wienholts, M.J.; Cobos-Jimenez, V.; Scholte, F.; Garcia-Sastre, A.; Rottier, P.J.; de Haan, C.A. Dissection of the influenza A virus endocytic routes reveals macropinocytosis as an alternative entry pathway. PLoS Pathog. 2011, 7, e1001329. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, C. Influenza A virus H5N1 entry into host cells is through clathrin-dependent endocytosis. Sci. China Life Sci. 2009, 52, 464–469. [Google Scholar] [CrossRef]
- Sun, E.Z.; Liu, A.A.; Zhang, Z.L.; Liu, S.L.; Tian, Z.Q.; Pang, D.W. Real-Time Dissection of Distinct Dynamin-Dependent Endocytic Routes of Influenza A Virus by Quantum Dot-Based Single-Virus Tracking. ACS Nano 2017, 11, 4395–4406. [Google Scholar] [CrossRef]
- Rossman, J.S.; Leser, G.P.; Lamb, R.A. Filamentous influenza virus enters cells via macropinocytosis. J. Virol. 2012, 86, 10950–10960. [Google Scholar] [CrossRef]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 A resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef]
- Bizebard, T.; Gigant, B.; Rigolet, P.; Rasmussen, B.; Diat, O.; Bosecke, P.; Wharton, S.A.; Skehel, J.J.; Knossow, M. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 1995, 376, 92–94. [Google Scholar] [CrossRef]
- Chen, J.; Lee, K.H.; Steinhauer, D.A.; Stevens, D.J.; Skehel, J.J.; Wiley, D.C. Structure of the hemagglutinin precursor cleavage site, a determinant of influenza pathogenicity and the origin of the labile conformation. Cell 1998, 95, 409–417. [Google Scholar] [CrossRef]
- Das, D.K.; Govindan, R.; Nikic-Spiegel, I.; Krammer, F.; Lemke, E.A.; Munro, J.B. Direct Visualization of the Conformational Dynamics of Single Influenza Hemagglutinin Trimers. Cell 2018. [Google Scholar] [CrossRef]
- Leikina, E.; Ramos, C.; Markovic, I.; Zimmerberg, J.; Chernomordik, L.V. Reversible stages of the low-pH-triggered conformational change in influenza virus hemagglutinin. EMBO J. 2002, 21, 5701–5710. [Google Scholar] [CrossRef] [Green Version]
- Fontana, J.; Cardone, G.; Heymann, J.B.; Winkler, D.C.; Steven, A.C. Structural changes in Influenza virus at low pH characterized by cryo-electron tomography. J. Virol. 2012, 86, 2919–2929. [Google Scholar] [CrossRef]
- Ivanovic, T.; Choi, J.L.; Whelan, S.P.; van Oijen, A.M.; Harrison, S.C. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. eLife 2013, 2, e00333. [Google Scholar] [CrossRef]
- Bottcher, E.; Matrosovich, T.; Beyerle, M.; Klenk, H.D.; Garten, W.; Matrosovich, M. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J. Virol. 2006, 80, 9896–9898. [Google Scholar] [CrossRef]
- Steinhauer, D.A. Role of hemagglutinin cleavage for the pathogenicity of influenza virus. Virology 1999, 258, 1–20. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Bennink, J.R.; Hosaka, Y. Cells process exogenous proteins for recognition by cytotoxic T lymphocytes. Science 1988, 239, 637–640. [Google Scholar] [CrossRef]
- Basak, S.; Tomana, M.; Compans, R.W. Sialic-Acid Is Incorporated into Influenza Hemagglutinin Glycoproteins in the Absence of Viral Neuraminidase. Virus Res. 1985, 2, 61–68. [Google Scholar] [CrossRef]
- Romero-Beltran, L.; Baker, S.F.; Puerto-Solis, M.; Gonzalez-Losa, R.; Conde-Ferraez, L.; Alvarez-Sanchez, L.C.; Martinez-Sobrido, L.; Ayora-Talavera, G. Mutations at highly conserved residues in influenza A(H1N1)pdm09 virus affect neuraminidase activity. Virus Res. 2016, 225, 1–9. [Google Scholar] [CrossRef]
- Chockalingam, A.K.; Hickman, D.; Pena, L.; Ye, J.Q.; Ferrero, A.; Echenique, J.R.; Chen, H.J.; Sutton, T.; Perez, D.R. Deletions in the Neuraminidase Stalk Region of H2N2 and H9N2 Avian Influenza Virus Subtypes Do Not Affect Postinfluenza Secondary Bacterial Pneumonia. J. Virol. 2012, 86, 3564–3573. [Google Scholar] [CrossRef] [Green Version]
- Yamayoshi, S.; Uraki, R.; Ito, M.; Kiso, M.; Nakatsu, S.; Yasuhara, A.; Oishi, K.; Sasaki, T.; Ikuta, K.; Kawaoka, Y. A Broadly Reactive Human Anti-hemagglutinin Stem Monoclonal Antibody That Inhibits Influenza A Virus Particle Release. Ebiomedicine 2017, 17, 182–191. [Google Scholar] [CrossRef]
- Yamamoto-Goshima, F.; Maeno, K. Approach to the involvement of influenza B neuraminidase in the cleavage of HA by host cell protease using low pH-induced cell fusion reaction. Microbiol. Immunol. 1994, 38, 819–822. [Google Scholar] [CrossRef]
- Li, S.; Schulman, J.; Itamura, S.; Palese, P. Glycosylation of neuraminidase determines the neurovirulence of influenza A/WSN/33 virus. J. Virol. 1993, 67, 6667–6673. [Google Scholar]
- Baum, L.G.; Paulson, J.C. The N2 neuraminidase of human influenza virus has acquired a substrate specificity complementary to the hemagglutinin receptor specificity. Virology 1991, 180, 10–15. [Google Scholar] [CrossRef]
- Laver, W.G.; Colman, P.M.; Webster, R.G.; Hinshaw, V.S.; Air, G.M. Influenza virus neuraminidase with hemagglutinin activity. Virology 1984, 137, 314–323. [Google Scholar] [CrossRef]
- Lin, Y.P.; Gregory, V.; Collins, P.; Kloess, J.; Wharton, S.; Cattle, N.; Lackenby, A.; Daniels, R.; Hay, A. Neuraminidase Receptor Binding Variants of Human Influenza A(H3N2) Viruses Resulting from Substitution of Aspartic Acid 151 in the Catalytic Site: A Role in Virus Attachment? J. Virol. 2010, 84, 6769–6781. [Google Scholar] [CrossRef]
- Uhlendorff, J.; Matrosovich, T.; Klenk, H.D.; Matrosovich, M. Functional significance of the hemadsorption activity of influenza virus neuraminidase and its alteration in pandemic viruses. Arch. Virol. 2009, 154, 945–957. [Google Scholar] [CrossRef] [Green Version]
- Mögling, R.; Richard, M.J.; Vliet, S.v.d.; Beek, R.v.; Schrauwen, E.J.A.; Spronken, M.I.; Rimmelzwaan, G.F.; Fouchier, R.A.M. Neuraminidase-mediated haemagglutination of recent human influenza A(H3N2) viruses is determined by arginine 150 flanking the neuraminidase catalytic site. J. Gen. Virol. 2017, 98, 1274–1281. [Google Scholar] [CrossRef] [Green Version]
- Air, G.M. Influenza neuraminidase. Influenza Other Respir. Viruses 2012, 6, 245–256. [Google Scholar] [CrossRef]
- Durrant, J.D.; Bush, R.M.; Amaro, R.E. Microsecond Molecular Dynamics Simulations of Influenza Neuraminidase Suggest a Mechanism for the Increased Virulence of Stalk-Deletion Mutants. J. Phys. Chem. B 2016, 120, 8590–8599. [Google Scholar] [CrossRef]
- Blok, J.; Air, G. Block deletions in the neuraminidase genes from some influenza A viruses of the N1 subtype. Virology 1982, 118, 229–234. [Google Scholar] [CrossRef]
- Li, J.; Zu Dohna, H.; Cardona, C.J.; Miller, J.; Carpenter, T.E. Emergence and genetic variation of neuraminidase stalk deletions in avian influenza viruses. PLoS ONE 2011, 6, e14722. [Google Scholar] [CrossRef]
- Sorrell, E.M.; Song, H.; Pena, L.; Perez, D.R. A 27-amino-acid deletion in the neuraminidase stalk supports replication of an avian H2N2 influenza A virus in the respiratory tract of chickens. J. Virol. 2010, 84, 11831–11840. [Google Scholar] [CrossRef]
- Stech, O.; Veits, J.; Abdelwhab el, S.M.; Wessels, U.; Mettenleiter, T.C.; Stech, J. The Neuraminidase Stalk Deletion Serves as Major Virulence Determinant of H5N1 Highly Pathogenic Avian Influenza Viruses in Chicken. Sci. Rep. 2015, 5, 13493. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Chen, S.; Zhang, X.; Fu, Q.; Zhang, Z.; Shi, S.; Zhu, Y.; Gu, M.; Peng, D.; Liu, X. A 20-amino-acid deletion in the neuraminidase stalk and a five-amino-acid deletion in the NS1 protein both contribute to the pathogenicity of H5N1 avian influenza viruses in mallard ducks. PLoS ONE 2014, 9, e95539. [Google Scholar] [CrossRef]
- Gulati, S.; Smith, D.F.; Air, G.M. Deletions of neuraminidase and resistance to oseltamivir may be a consequence of restricted receptor specificity in recent H3N2 influenza viruses. Virol. J. 2009, 6, 22. [Google Scholar] [CrossRef]
- Eshaghi, A.; Shalhoub, S.; Rosenfeld, P.; Li, A.; Higgins, R.R.; Stogios, P.J.; Savchenko, A.; Bastien, N.; Li, Y.; Rotstein, C.; et al. Multiple influenza A (H3N2) mutations conferring resistance to neuraminidase inhibitors in a bone marrow transplant recipient. Antimicrob. Agents Chemother. 2014, 58, 7188–7197. [Google Scholar] [CrossRef]
- Brooke, C.B. Population Diversity and Collective Interactions during Influenza Virus Infection. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Mitnaul, L.J.; Matrosovich, M.N.; Castrucci, M.R.; Tuzikov, A.B.; Bovin, N.V.; Kobasa, D.; Kawaoka, Y. Balanced hemagglutinin and neuraminidase activities are critical for efficient replication of influenza A virus. J. Virol. 2000, 74, 6015–6020. [Google Scholar] [CrossRef]
- Khatchikian, D.; Orlich, M.; Rott, R. Increased viral pathogenicity after insertion of a 28S ribosomal RNA sequence into the haemagglutinin gene of an influenza virus. Nature 1989, 340, 156–157. [Google Scholar] [CrossRef]
- Hensley, S.E.; Das, S.R.; Gibbs, J.S.; Bailey, A.L.; Schmidt, L.M.; Bennink, J.R.; Yewdell, J.W. Influenza A virus hemagglutinin antibody escape promotes neuraminidase antigenic variation and drug resistance. PLoS ONE 2011, 6, e15190. [Google Scholar] [CrossRef]
- Das, S.R.; Hensley, S.E.; Ince, W.L.; Brooke, C.B.; Subba, A.; Delboy, M.G.; Russ, G.; Gibbs, J.S.; Bennink, J.R.; Yewdell, J.W. Defining influenza A virus hemagglutinin antigenic drift by sequential monoclonal antibody selection. Cell Host Microbe 2013, 13, 314–323. [Google Scholar]
- Brooke, C.B. Biological activities of ‘noninfectious’ influenza A virus particles. Future Virol. 2014, 9, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Brooke, C.B.; Ince, W.L.; Wrammert, J.; Ahmed, R.; Wilson, P.C.; Bennink, J.R.; Yewdell, J.W. Most influenza a virions fail to express at least one essential viral protein. J. Virol. 2013, 87, 3155–3162. [Google Scholar] [CrossRef]
- Hughes, M.T.; Matrosovich, M.; Rodgers, M.E.; McGregor, M.; Kawaoka, Y. Influenza A viruses lacking sialidase activity can undergo multiple cycles of replication in cell culture, eggs, or mice. J. Virol. 2000, 74, 5206–5212. [Google Scholar] [CrossRef]
- Moules, V.; Ferraris, O.; Terrier, O.; Giudice, E.; Yver, M.; Rolland, J.P.; Bouscambert-Duchamp, M.; Bergeron, C.; Ottmann, M.; Fournier, E.; et al. In vitro characterization of naturally occurring influenza H3NA- viruses lacking the NA gene segment: Toward a new mechanism of viral resistance? Virology 2010, 404, 215–224. [Google Scholar] [CrossRef]
- Neverov, A.D.; Kryazhimskiy, S.; Plotkin, J.B.; Bazykin, G.A. Coordinated Evolution of Influenza A Surface Proteins. PLoS Genetics 2015, 11, e1005404. [Google Scholar] [CrossRef]
- Staschke, K.A.; Colacino, J.M.; Baxter, A.J.; Air, G.M.; Bansal, A.; Hornback, W.J.; Munroe, J.E.; Laver, W.G. Molecular basis for the resistance of influenza viruses to 4-guanidino-Neu5Ac2en. Virology 1995, (2), 642–646. [Google Scholar] [CrossRef]
- Carr, J.; Ives, J.; Kelly, L.; Lambkin, R.; Oxford, J.; Mendel, D.; Tai, L.; Roberts, N. Influenza virus carrying neuraminidase with reduced sensitivity to oseltamivir carboxylate has altered properties in vitro and is compromised for infectivity and replicative ability in vivo. Antiviral Res. 2002, 54, 79–88. [Google Scholar] [CrossRef]
- Ives, J.A.; Carr, J.A.; Mendel, D.B.; Tai, C.Y.; Lambkin, R.; Kelly, L.; Oxford, J.S.; Hayden, F.G.; Roberts, N.A. The H274Y mutation in the influenza A/H1N1 neuraminidase active site following oseltamivir phosphate treatment leave virus severely compromised both in vitro and in vivo. Antiviral Res. 2002, 55, 307–317. [Google Scholar] [CrossRef]
- Pizzorno, A.; Abed, Y.; Plante, P.L.; Carbonneau, J.; Baz, M.; Hamelin, M.E.; Corbeil, J.; Boivin, G. Evolution of oseltamivir resistance mutations in Influenza A(H1N1) and A(H3N2) viruses during selection in experimentally infected mice. Antimicrob. Agents Chemother. 2014, 58, 6398–6405. [Google Scholar] [CrossRef]
- McKimm-Breschkin, J.L.; Blick, T.J.; Sahasrabudhe, A.; Tiong, T.; Marshall, D.; Hart, G.J.; Bethell, R.C.; Penn, C.R. Generation and characterization of variants of NWS/G70C influenza virus after in vitro passage in 4-amino-Neu5Ac2en and 4-guanidino-Neu5Ac2en. Antimicrob. Agents Chemother. 1996, 40, 40–46. [Google Scholar] [CrossRef]
- Blick, T.J.; Sahasrabudhe, A.; McDonald, M.; Owens, I.J.; Morley, P.J.; Fenton, R.J.; McKimm-Breschkin, J.L. The interaction of neuraminidase and hemagglutinin mutations in influenza virus in resistance to 4-guanidino-Neu5Ac2en. Virology 1998, 246, 95–103. [Google Scholar] [CrossRef]
- Barnett, J.M.; Cadman, A.; Burrell, F.M.; Madar, S.H.; Lewis, A.P.; Tisdale, M.; Bethell, R. In vitro selection and characterisation of influenza B/Beijing/1/87 isolates with altered susceptibility to zanamivir. Virology 1999, 265, 286–295. [Google Scholar] [CrossRef]
- Altman, M.O.; Bennink, J.R.; Yewdell, J.W.; Herrin, B.R. Lamprey VLRB response to influenza virus supports universal rules of immunogenicity and antigenicity. eLife 2015, 4. [Google Scholar] [CrossRef]
- Gerhard, W.; Yewdell, J.; Frankel, M.E.; Webster, R. Antigenic structure of influenza virus haemagglutinin defined by hybridoma antibodies. Nature 1981, 290, 713–717. [Google Scholar] [CrossRef]
- Xu, R.; Ekiert, D.C.; Krause, J.C.; Hai, R.; Crowe, J.E., Jr.; Wilson, I.A. Structural basis of preexisting immunity to the 2009 H1N1 pandemic influenza virus. Science 2010, 328, 357–360. [Google Scholar] [CrossRef]
- Wiley, D.C.; Wilson, I.A.; Skehel, J.J. Structural identification of the antibody-binding sites of Hong Kong influenza haemagglutinin and their involvement in antigenic variation. Nature 1981, 289, 373–378. [Google Scholar] [CrossRef]
- Suntronwong, N.; Klinfueng, S.; Vichiwattana, P.; Korkong, S.; Thongmee, T.; Vongpunsawad, S.; Poovorawan, Y. Genetic and antigenic divergence in the influenza A(H3N2) virus circulating between 2016 and 2017 in Thailand. PLoS ONE 2017, 12, e0189511. [Google Scholar] [CrossRef]
- Caton, A.J.; Brownlee, G.G.; Yewdell, J.W.; Gerhard, W. The antigenic structure of the influenza virus A/PR/8/34 hemagglutinin (H1 subtype). Cell 1982, 31, 417–427. [Google Scholar] [CrossRef]
- Magadan, J.G.; Khurana, S.; Das, S.R.; Frank, G.M.; Stevens, J.; Golding, H.; Bennink, J.R.; Yewdell, J.W. Influenza A Virus Hemagglutinin Trimerization Completes Monomer Folding and Antigenicity. J. Virol. 2013. [Google Scholar] [CrossRef]
- Yewdell, J.W.; Gerhard, W.; Bachi, T. Monoclonal anti-hemagglutinin antibodies detect irreversible antigenic alterations that coincide with the acid activation of influenza virus A/PR/834-mediated hemolysis. J. Virol. 1983, 48, 239–248. [Google Scholar]
- Vareckova, E.; Mucha, V.; Ciampor, F.; Betakova, T.; Russ, G. Monoclonal antibodies demonstrate accessible HA2 epitopes in minor subpopulation of native influenza virus haemagglutinin molecules. Arch. Virol. 1993, 130, 45–56. [Google Scholar] [CrossRef]
- Kostolansky, F.; Styk, B.; Russ, G. Inhibition of influenza virus haemolytic and haemagglutination activities by monoclonal antibodies to haemagglutinin glycopolypeptides HA1 and HA2. Acta Virol. 1989, 33, 504–512. [Google Scholar]
- Russ, G.; Polakova, K.; Kostolansky, F.; Styk, B.; Vancikova, M. Monoclonal antibodies to glycopolypeptides HA1 and HA2 of influenza virus haemagglutinin. Acta Virol. 1987, 31, 374–386. [Google Scholar]
- Styk, B.; Russ, G.; Polakova, K. Antigenic glycopolypeptides HA1 and HA2 of influenza virus haemagglutinin. IV. Immunogenic properties of separated haemagglutinin glycopolypeptides. Acta Virol. 1979, 23, 9–20. [Google Scholar]
- Russ, G.; Styk, B.; Polakova, K. Antigenic glycopolypeptides HA1 and HA2 of influenza virus haemagglutinin. II. Reactivity with rabbit sera against intact virus and purified undissociated haemagglutinin. Acta Virol. 1978, 22, 371–382. [Google Scholar]
- Polakova, K.; Russ, G.; Styk, B. Antigenic glycopolypeptides HA1 and HA2 of influenza virus haemagglutinin. I. Gel filtration in 6 M guanidine hydrochloride. Acta Virol. 1978, 22, 362–370. [Google Scholar]
- Okuno, Y.; Isegawa, Y.; Sasao, F.; Ueda, S. A common neutralizing epitope conserved between the hemagglutinins of influenza A virus H1 and H2 strains. J. Virol. 1993, 67, 2552–2558. [Google Scholar]
- Okuno, Y.; Matsumoto, K.; Isegawa, Y.; Ueda, S. Protection against the mouse-adapted A/FM/1/47 strain of influenza A virus in mice by a monoclonal antibody with cross-neutralizing activity among H1 and H2 strains. J. Virol. 1994, 68, 517–520. [Google Scholar]
- Sui, J.; Sheehan, J.; Hwang, W.C.; Bankston, L.A.; Burchett, S.K.; Huang, C.Y.; Liddington, R.C.; Beigel, J.H.; Marasco, W.A. Wide prevalence of heterosubtypic broadly neutralizing human anti-influenza A antibodies. Clin. Infect. Dis. 2011, 52, 1003–1009. [Google Scholar] [CrossRef]
- Yassine, H.M.; Boyington, J.C.; McTamney, P.M.; Wei, C.J.; Kanekiyo, M.; Kong, W.P.; Gallagher, J.R.; Wang, L.; Zhang, Y.; Joyce, M.G.; et al. Hemagglutinin-stem nanoparticles generate heterosubtypic influenza protection. Nat. Med. 2015, 21, 1065–1070. [Google Scholar] [CrossRef]
- Mallajosyula, V.V.; Citron, M.; Ferrara, F.; Lu, X.; Callahan, C.; Heidecker, G.J.; Sarma, S.P.; Flynn, J.A.; Temperton, N.J.; Liang, X.; et al. Influenza hemagglutinin stem-fragment immunogen elicits broadly neutralizing antibodies and confers heterologous protection. Proc. Natl. Acad. Sci. USA 2014, 111, E2514–E2523. [Google Scholar] [CrossRef]
- Impagliazzo, A.; Milder, F.; Kuipers, H.; Wagner, M.V.; Zhu, X.; Hoffman, R.M.; van Meersbergen, R.; Huizingh, J.; Wanningen, P.; Verspuij, J.; et al. A stable trimeric influenza hemagglutinin stem as a broadly protective immunogen. Science 2015, 349, 1301–1306. [Google Scholar] [CrossRef]
- Eggink, D.; Goff, P.H.; Palese, P. Guiding the immune response against influenza virus hemagglutinin toward the conserved stalk domain by hyperglycosylation of the globular head domain. J. Virol. 2014, 88, 699–704. [Google Scholar] [CrossRef]
- Wrammert, J.; Koutsonanos, D.; Li, G.M.; Edupuganti, S.; Sui, J.; Morrissey, M.; McCausland, M.; Skountzou, I.; Hornig, M.; Lipkin, W.I.; et al. Broadly cross-reactive antibodies dominate the human B cell response against 2009 pandemic H1N1 influenza virus infection. J. Exp. Med. 2011, 208, 181–193. [Google Scholar] [CrossRef] [Green Version]
- Pica, N.; Hai, R.; Krammer, F.; Wang, T.T.; Maamary, J.; Eggink, D.; Tan, G.S.; Krause, J.C.; Moran, T.; Stein, C.R.; et al. Hemagglutinin stalk antibodies elicited by the 2009 pandemic influenza virus as a mechanism for the extinction of seasonal H1N1 viruses. Proc. Natl. Acad. Sci. USA 2012, 109, 2573–2578. [Google Scholar] [CrossRef] [Green Version]
- Tete, S.M.; Krammer, F.; Lartey, S.; Bredholt, G.; Wood, J.; Skrede, S.; Cox, R.J. Dissecting the hemagglutinin head and stalk-specific IgG antibody response in healthcare workers following pandemic H1N1 vaccination. Npj Vaccines 2016, 1. [Google Scholar] [CrossRef]
- Tan, G.S.; Lee, P.S.; Hoffman, R.M.; Mazel-Sanchez, B.; Krammer, F.; Leon, P.E.; Ward, A.B.; Wilson, I.A.; Palese, P. Characterization of a broadly neutralizing monoclonal antibody that targets the fusion domain of group 2 influenza A virus hemagglutinin. J. Virol. 2014, 88, 13580–13592. [Google Scholar] [CrossRef]
- Chai, N.; Swem, L.R.; Reichelt, M.; Chen-Harris, H.; Luis, E.; Park, S.; Fouts, A.; Lupardus, P.; Wu, T.D.; Li, O.; et al. Two Escape Mechanisms of Influenza A Virus to a Broadly Neutralizing Stalk-Binding Antibody. PLoS Pathog. 2016, 12, e1005702. [Google Scholar] [CrossRef]
- Magadan, J.G.; Altman, M.O.; Ince, W.L.; Hickman, H.D.; Stevens, J.; Chevalier, A.; Baker, D.; Wilson, P.C.; Ahmed, R.; Bennink, J.R.; et al. Biogenesis of influenza a virus hemagglutinin cross-protective stem epitopes. PLoS Pathog. 2014, 10, e1004204. [Google Scholar] [CrossRef]
- Seok, J.H.; Kim, J.; Lee, D.B.; Cho, K.J.; Lee, J.H.; Bae, G.; Chung, M.S.; Kim, K.H. Conformational modulation of influenza virus hemagglutinin: Characterization and in vivo efficacy of monomeric form. Sci. Rep. 2017, 7, 7540. [Google Scholar] [CrossRef]
- Harris, A.K.; Myerson, J.R.; Matsuoka, Y.; Kuybeda, O.; Moran, A.; Bliss, D.; Das, S.R.; Yewdell, J.W.; Sapiro, G.; Subbarao, K.; et al. Structure and accessibility of HA trimers on intact 2009 H1N1 pandemic influenza virus to stem region-specific neutralizing antibodies. Proc. Natl. Acad. Sci USA 2013, in press. [Google Scholar] [CrossRef]
- Dreyfus, C.; Ekiert, D.C.; Wilson, I.A. Structure of a classical broadly neutralizing stem antibody in complex with a pandemic H2 influenza virus hemagglutinin. J. Virol. 2013, 87, 7149–7154. [Google Scholar] [CrossRef]
- Angeletti, D.; Gibbs, J.S.; Angel, M.; Kosik, I.; Hickman, H.D.; Frank, G.M.; Das, S.R.; Wheatley, A.K.; Prabhakaran, M.; Leggat, D.J.; et al. Defining B cell immunodominance to viruses. Nat. Immunol. 2017, 18, 456–463. [Google Scholar] [CrossRef]
- Kavaler, J.; Caton, A.J.; Staudt, L.M.; Schwartz, D.; Gerhard, W. A set of closely related antibodies dominates the primary antibody response to the antigenic site CB of the A/PR/8/34 influenza virus hemagglutinin. J. Immunol. 1990, 145, 2312–2321. [Google Scholar] [PubMed]
- Staudt, L.M.; Gerhard, W. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. I. Significant variation in repertoire expression between individual mice. J. Exp. Med. 1983, 157, 687–704. [Google Scholar] [CrossRef] [Green Version]
- Kosik, I.; Yewdell, J.W. Influenza A virus hemagglutinin specific antibodies interfere with virion neuraminidase activity via two distinct mechanisms. Virology 2017, 500, 178–183. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.J.; Dimmock, N.J. Two influenza A virus-specific Fabs neutralize by inhibiting virus attachment to target cells, while neutralization by their IgGs is complex and occurs simultaneously through fusion inhibition and attachment inhibition. Virology 2000, 278, 423–435. [Google Scholar] [CrossRef]
- Edwards, M.J.; Dimmock, N.J. A haemagglutinin (HA1)-specific FAb neutralizes influenza A virus by inhibiting fusion activity. J. Gen. Virol. 2001, 82, 1387–1395. [Google Scholar] [CrossRef] [Green Version]
- Edwards, M.J.; Dimmock, N.J. Hemagglutinin 1-specific immunoglobulin G and Fab molecules mediate postattachment neutralization of influenza A virus by inhibition of an early fusion event. J. Virol. 2001, 75, 10208–10218. [Google Scholar] [CrossRef]
- Kilbourne, E.D.; Laver, W.G.; Schulman, J.L.; Webster, R.G. Antiviral activity of antiserum specific for an influenza virus neuraminidase. J. Virol. 1968, 2, 281–288. [Google Scholar]
- Ogra, P.L.; Chow, T.; Beutner, K.R.; Rubi, E.; Strussenberg, J.; DeMello, S.; Rizzone, C. Clinical and immunologic evaluation of neuraminidase-specific influenza A virus vaccine in humans. J. Infect. Dis. 1977, 135, 499–506. [Google Scholar] [CrossRef]
- Beutner, K.R.; Chow, T.; Rubi, E.; Strussenberg, J.; Clement, J.; Ogra, P.L. Evaluation of a neuraminidase-specific influenza A virus vaccine in children: Antibody responses and effects on two successive outbreaks of natural infection. J. Infect. Dis. 1979, 140, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Murphy, B.R.; Kasel, J.A.; Chanock, R.M. Association of serum anti-neuraminidase antibody with resistance to influenza in man. N. Engl. J. Med. 1972, 286, 1329–1332. [Google Scholar] [CrossRef] [PubMed]
- Air, G.M.; Els, M.C.; Brown, L.E.; Laver, W.G.; Webster, R.G. Location of antigenic sites on the three-dimensional structure of the influenza N2 virus neuraminidase. Virology 1985, 145, 237–248. [Google Scholar] [CrossRef]
- Webster, R.G.; Brown, L.E.; Laver, W.G. Antigenic and biological characterization of influenza virus neuraminidase (N2) with monoclonal antibodies. Virology 1984, 135, 30–42. [Google Scholar] [CrossRef]
- Webster, R.G.; Hinshaw, V.S.; Laver, W.G. Selection and analysis of antigenic variants of the neuraminidase of N2 influenza viruses with monoclonal antibodies. Virology 1982, 117, 93–104. [Google Scholar] [CrossRef]
- Laver, W.G.; Air, G.M.; Webster, R.G.; Markoff, L.J. Amino acid sequence changes in antigenic variants of type A influenza virus N2 neuraminidase. Virology 1982, 122, 450–460. [Google Scholar] [CrossRef]
- Wan, H.; Gao, J.; Xu, K.; Chen, H.; Couzens, L.K.; Rivers, K.H.; Easterbrook, J.D.; Yang, K.; Zhong, L.; Rajabi, M.; et al. Molecular basis for broad neuraminidase immunity: Conserved epitopes in seasonal and pandemic H1N1 as well as H5N1 influenza viruses. J. Virol. 2013, 87, 9290–9300. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Fantoni, G.; Couzens, L.; Gao, J.; Plant, E.; Ye, Z.; Eichelberger, M.C.; Wan, H. Comparative Efficacy of Monoclonal Antibodies That Bind to Different Epitopes of the 2009 Pandemic H1N1 Influenza Virus Neuraminidase. J. Virol. 2016, 90, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Johansson, B.E.; Kilbourne, E.D. Dissociation of influenza virus hemagglutinin and neuraminidase eliminates their intravirionic antigenic competition. J. Virol. 1993, 67, 5721–5723. [Google Scholar] [PubMed]
- Johansson, B.E.; Bucher, D.J.; Kilbourne, E.D. Purified influenza virus hemagglutinin and neuraminidase are equivalent in stimulation of antibody response but induce contrasting types of immunity to infection. J. Virol. 1989, 63, 1239–1246. [Google Scholar]
- Kendal, A.P.; Noble, G.R.; Dowdle, W.R. Neuraminidase content of influenza vaccines and neuraminidase antibody responses after vaccination of immunologically primed and unprimed populations. J. Infect. Dis. 1977, 136 (Suppl. 3), S415–S424. [Google Scholar] [CrossRef]
- Kilbourne, E.D.; Cerini, C.P.; Khan, M.W.; Mitchell, J.W., Jr.; Ogra, P.L. Immunologic response to the influenza virus neuraminidase is influenced by prior experience with the associated viral hemagglutinin. I. Studies in human vaccinees. J. Immunol. 1987, 138, 3010–3013. [Google Scholar]
- Johansson, B.E.; Moran, T.M.; Bona, C.A.; Popple, S.W.; Kilbourne, E.D. Immunologic response to influenza virus neuraminidase is influenced by prior experience with the associated viral hemagglutinin. II. Sequential infection of mice simulates human experience. J. Immunol. 1987, 139, 2010–2014. [Google Scholar]
- Rajendran, M.; Nachbagauer, R.; Ermler, M.E.; Bunduc, P.; Amanat, F.; Izikson, R.; Cox, M.; Palese, P.; Eichelberger, M.; Krammer, F. Analysis of Anti-Influenza Virus Neuraminidase Antibodies in Children, Adults, and the Elderly by ELISA and Enzyme Inhibition: Evidence for Original Antigenic Sin. mBio 2017, 8. [Google Scholar] [CrossRef]
- Monto, A.S.; Petrie, J.G.; Cross, R.T.; Johnson, E.; Liu, M.; Zhong, W.; Levine, M.; Katz, J.M.; Ohmit, S.E. Antibody to Influenza Virus Neuraminidase: An Independent Correlate of Protection. J. Infect. Dis. 2015, 212, 1191–1199. [Google Scholar] [CrossRef] [Green Version]
- Couch, R.B.; Kasel, J.A.; Gerin, J.L.; Schulman, J.L.; Kilbourne, E.D. Induction of partial immunity to influenza by a neuraminidase-specific vaccine. J. Infect Dis. 1974, 129, 411–420. [Google Scholar] [CrossRef]
- Couzens, L.; Gao, J.; Westgeest, K.; Sandbulte, M.; Lugovtsev, V.; Fouchier, R.; Eichelberger, M. An optimized enzyme-linked lectin assay to measure influenza A virus neuraminidase inhibition antibody titers in human sera. J. Virol. Methods 2014, 210, 7–14. [Google Scholar] [CrossRef] [Green Version]
- Harris, A.; Cardone, G.; Winkler, D.C.; Heymann, J.B.; Brecher, M.; White, J.M.; Steven, A.C. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2006, 103, 19123–19127. [Google Scholar] [CrossRef] [Green Version]
- Murti, K.G.; Webster, R.G. Distribution of hemagglutinin and neuraminidase on influenza virions as revealed by immunoelectron microscopy. Virology 1986, 149, 36–43. [Google Scholar] [CrossRef]
- Fontana, J.; Steven, A.C. Influenza virus-mediated membrane fusion: Structural insights from electron microscopy. Arch. Biochem. Biophys. 2015, 581, 86–97. [Google Scholar] [CrossRef] [Green Version]
- Paniker, C.K. Serological relationships between the neuraminidases in influenza viruses. J. Gen. Virol. 1968, 2, 385–394. [Google Scholar] [CrossRef]
- Russ, G.; Varekova, E.; Styk, B. Steric effects in the reaction of influenza virus neuraminidases with antibodies. Acta Virologica. 1974, 18, 299–306. [Google Scholar]
- Chen, Y.-Q.; Lan, L.Y.-L.; Huang, M.; Henry, C.; Wilson, P.C. Hemagglutinin stalk-reactive antibodies interfere with influenza virus neuraminidase activity by steric hindrance. bioRxiv 2018. [Google Scholar] [CrossRef] [PubMed]
- Kosik, I.; Angeletti, D.; Gibbs, J.S.; Angel, M.; Takeda, K.; Kosikova, M.; Nair, V.; Hickman, H.D.; Xie, H.; Brooke, C.B.; et al. Neuraminidase inhibition contributes to influenza A virus neutralization by anti-hemagglutinin stem antibodies. J. Exp. Med. 2019, 216, 304–316. [Google Scholar] [CrossRef]
- DiLillo, D.J.; Palese, P.; Wilson, P.C.; Ravetch, J.V. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Invest. 2016, 126, 605–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DiLillo, D.J.; Tan, G.S.; Palese, P.; Ravetch, J.V. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcgammaR interactions for protection against influenza virus in vivo. Nat. Med. 2014, 20, 143–151. [Google Scholar] [CrossRef]
- Bar-On, Y.; Seidel, E.; Tsukerman, P.; Mandelboim, M.; Mandelboim, O. Influenza Virus Uses Its Neuraminidase Protein to Evade the Recognition of Two Activating NK Cell Receptors. J. Infect. Dis. 2014, 210, 410–418. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Tan, G.S.; Mullarkey, C.E.; Lee, A.J.; Lam, M.M.; Krammer, F.; Henry, C.; Wilson, P.C.; Ashkar, A.A.; Palese, P.; et al. Epitope specificity plays a critical role in regulating antibody-dependent cell-mediated cytotoxicity against influenza A virus. Proc. Natl. Acad. Sci. USA 2016, 113, 11931–11936. [Google Scholar] [CrossRef] [Green Version]
- Leon, P.E.; He, W.; Mullarkey, C.E.; Bailey, M.J.; Miller, M.S.; Krammer, F.; Palese, P.; Tan, G.S. Optimal activation of Fc-mediated effector functions by influenza virus hemagglutinin antibodies requires two points of contact. Proc. Natl. Acad. Sci. USA 2016, 113, E5944–E5951. [Google Scholar] [CrossRef] [PubMed] [Green Version]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosik, I.; Yewdell, J.W. Influenza Hemagglutinin and Neuraminidase: Yin–Yang Proteins Coevolving to Thwart Immunity. Viruses 2019, 11, 346. https://doi.org/10.3390/v11040346
Kosik I, Yewdell JW. Influenza Hemagglutinin and Neuraminidase: Yin–Yang Proteins Coevolving to Thwart Immunity. Viruses. 2019; 11(4):346. https://doi.org/10.3390/v11040346
Chicago/Turabian StyleKosik, Ivan, and Jonathan W. Yewdell. 2019. "Influenza Hemagglutinin and Neuraminidase: Yin–Yang Proteins Coevolving to Thwart Immunity" Viruses 11, no. 4: 346. https://doi.org/10.3390/v11040346
APA StyleKosik, I., & Yewdell, J. W. (2019). Influenza Hemagglutinin and Neuraminidase: Yin–Yang Proteins Coevolving to Thwart Immunity. Viruses, 11(4), 346. https://doi.org/10.3390/v11040346