Conserved Secondary Structures in Viral mRNAs

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
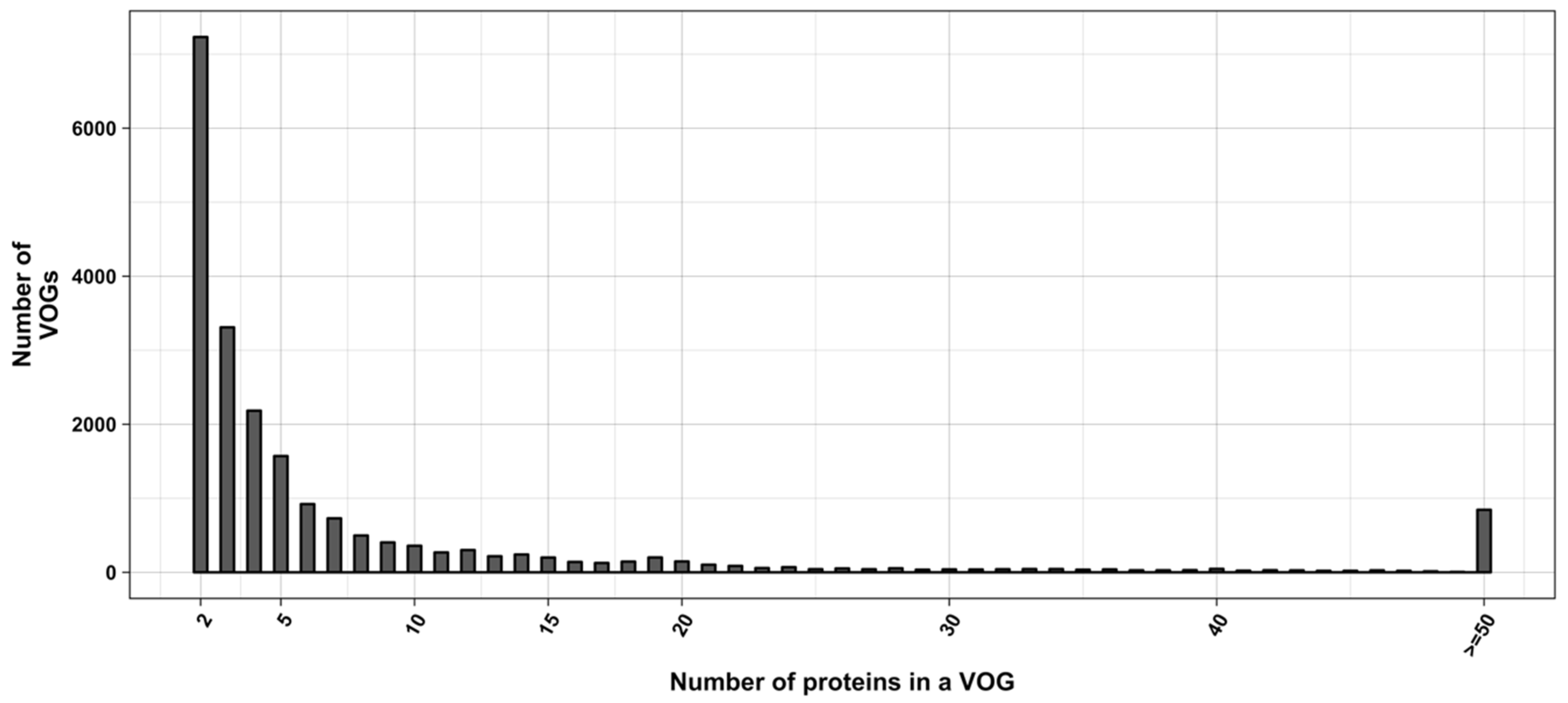
2.1. Viral Orthologous Groups (VOGs)
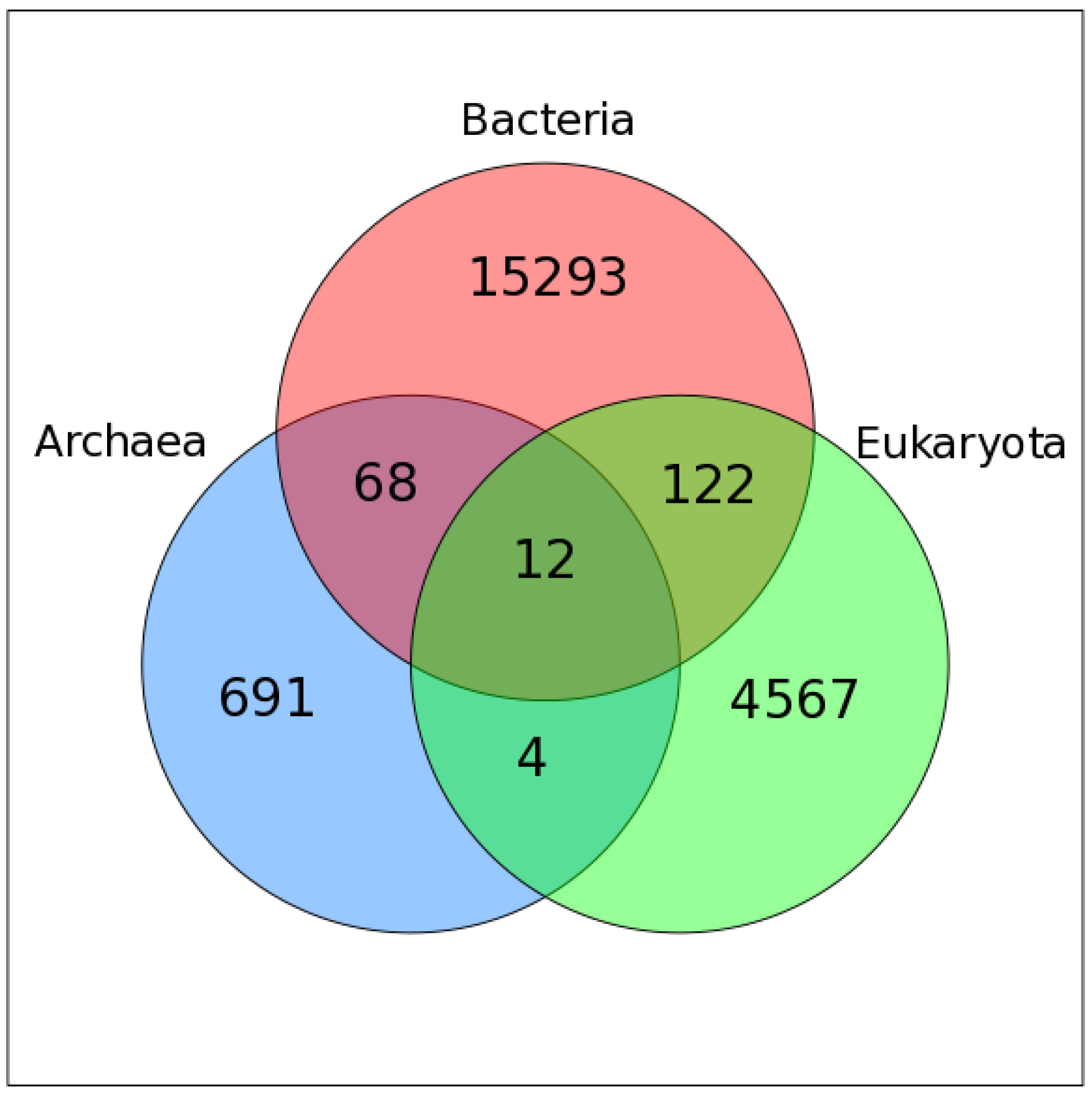
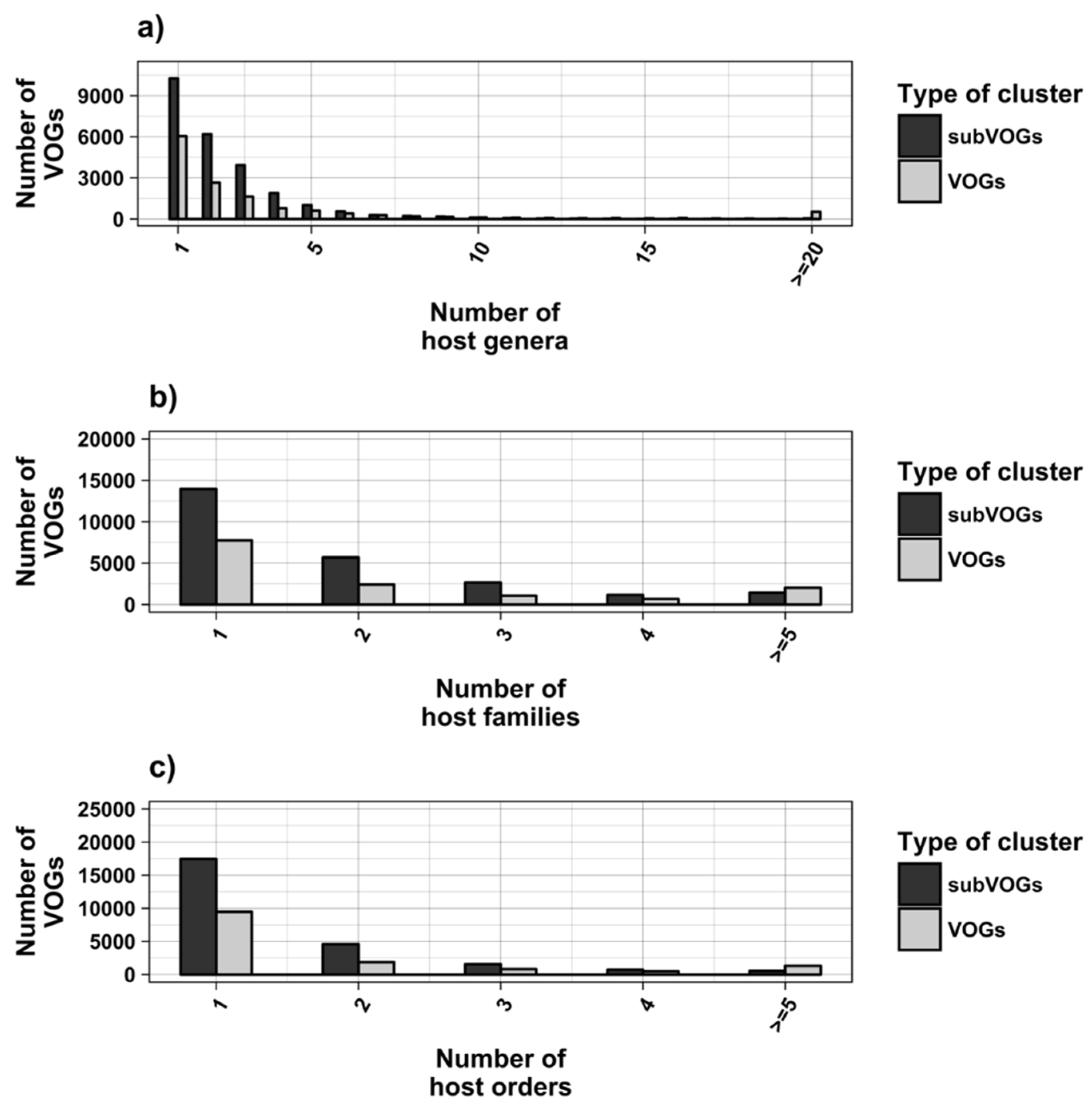
2.2. Mapping VOG Sequences to Specific Hosts
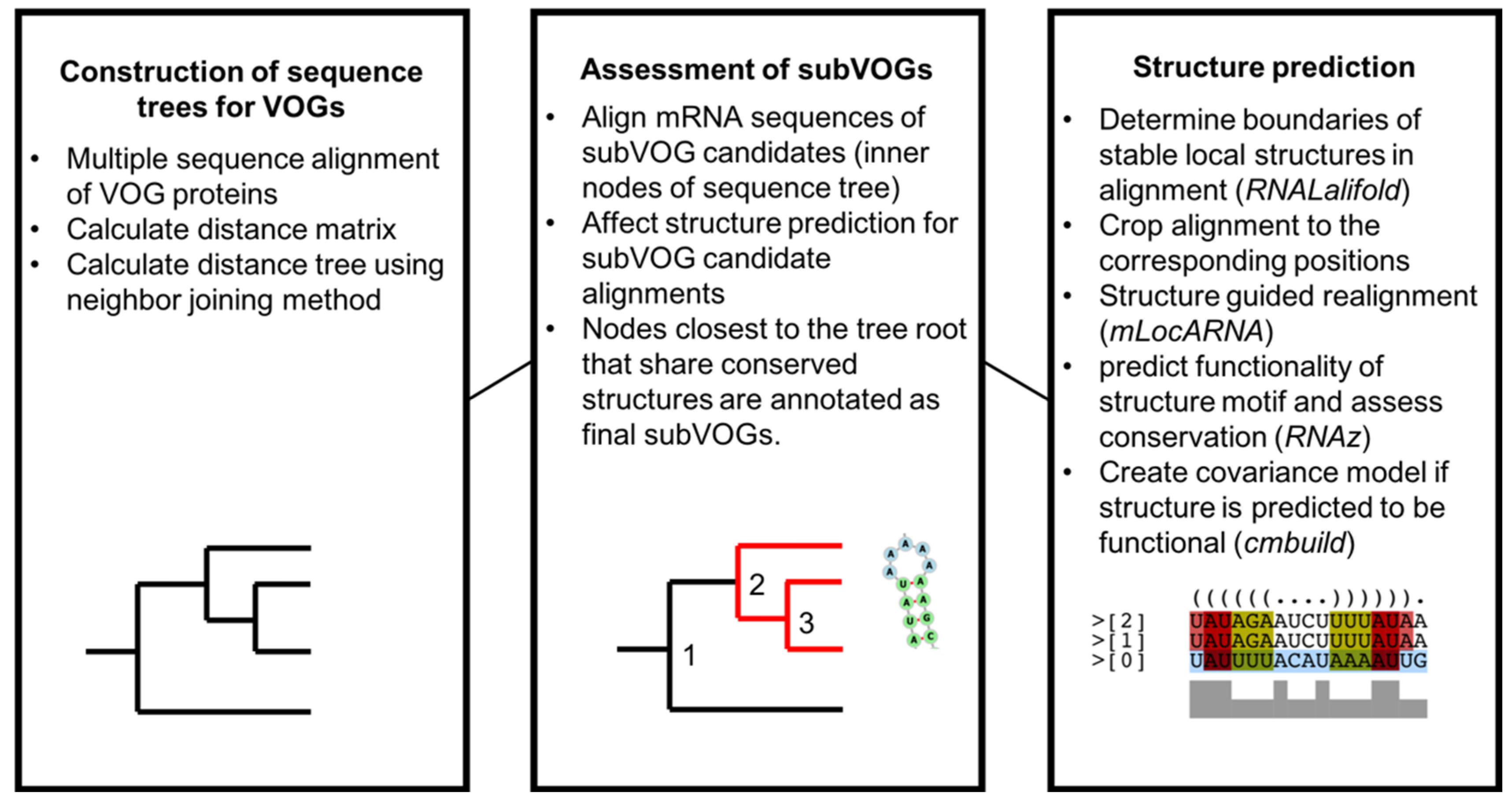
2.3. Distance Trees of VOG Proteins
2.4. Structure Prediction and subVOG Assignment
2.5. mRNA Stability
2.6. mRNA Structures and Protein Function
3. Results
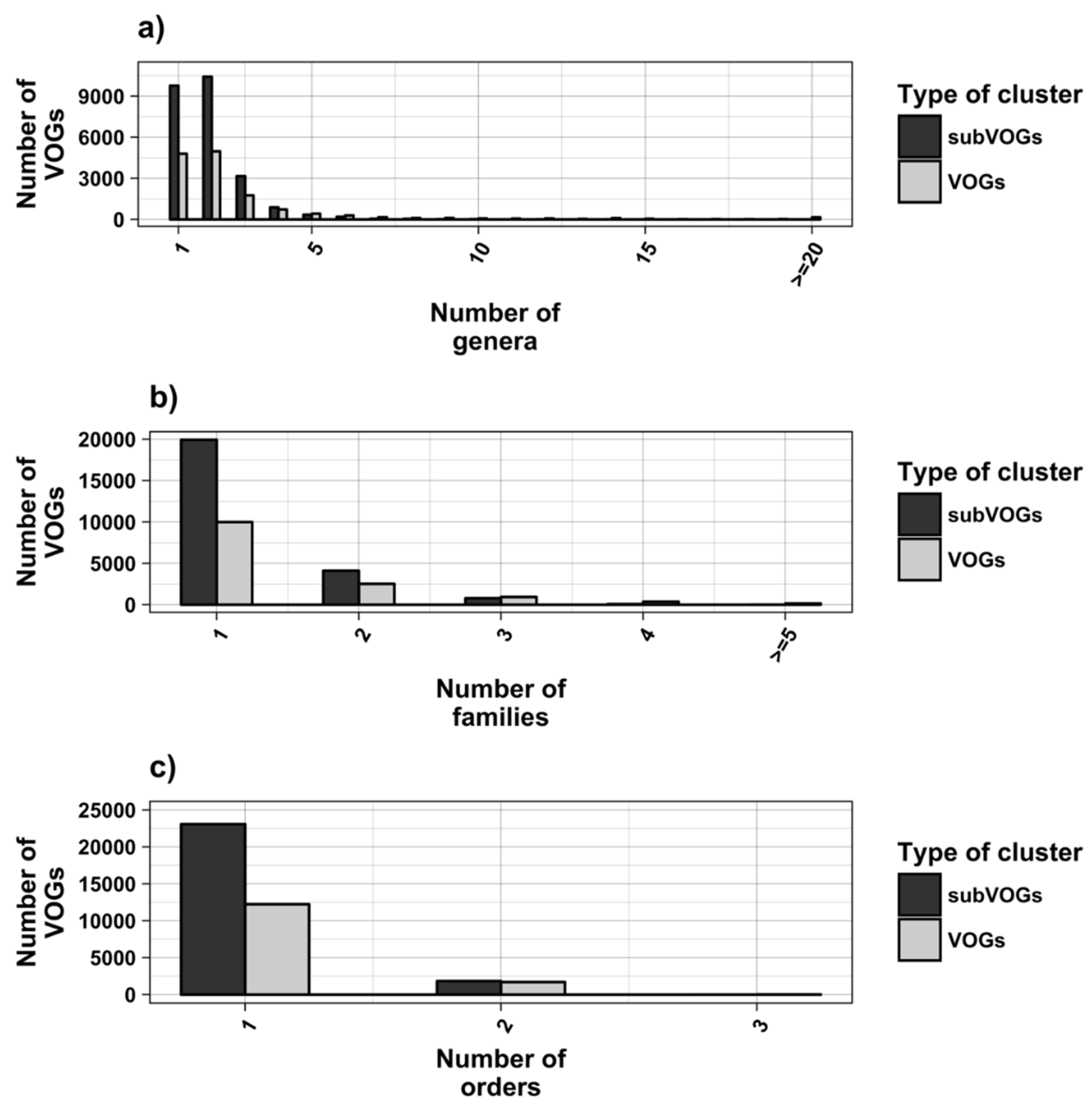
3.1. Overview of the Study
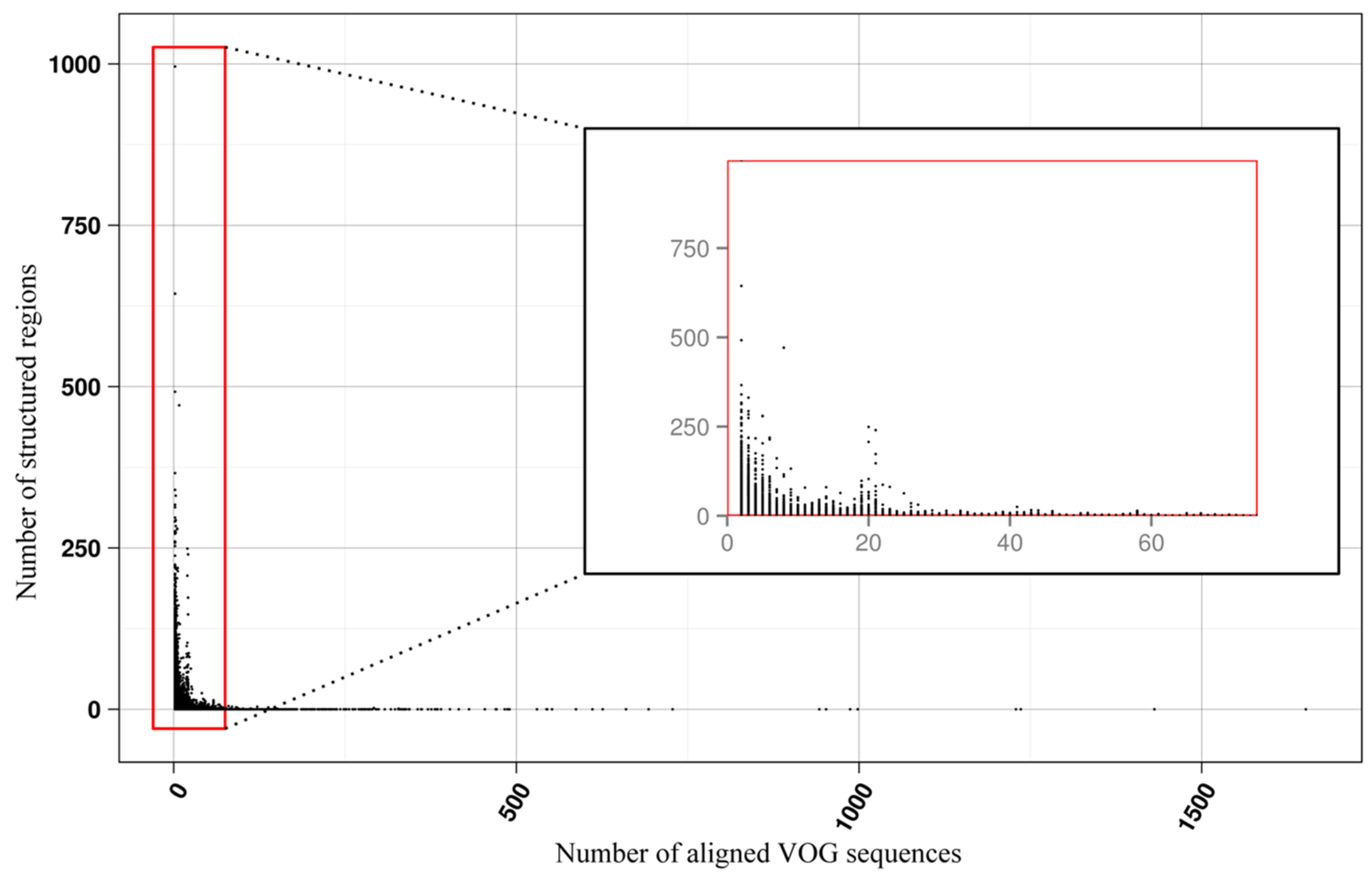
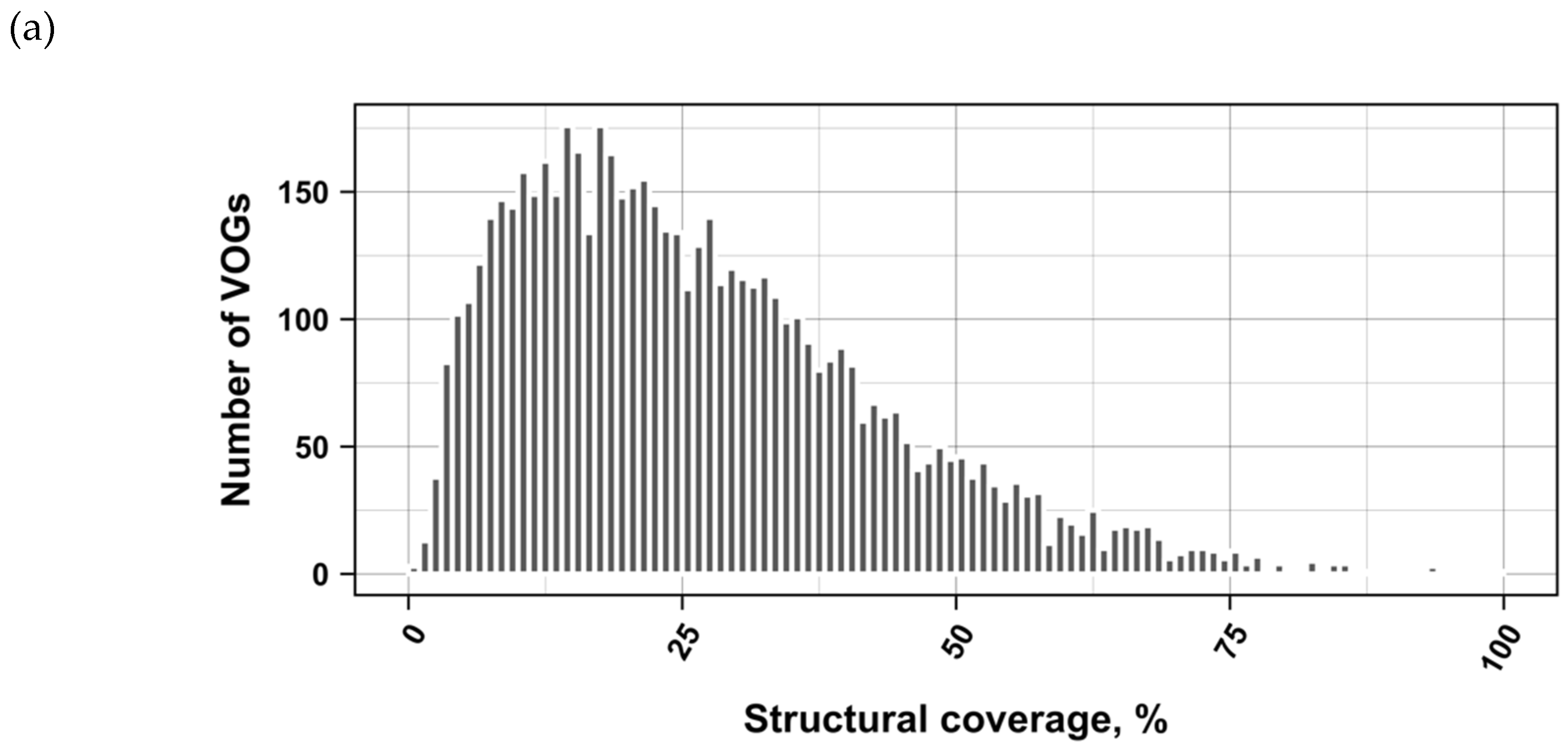
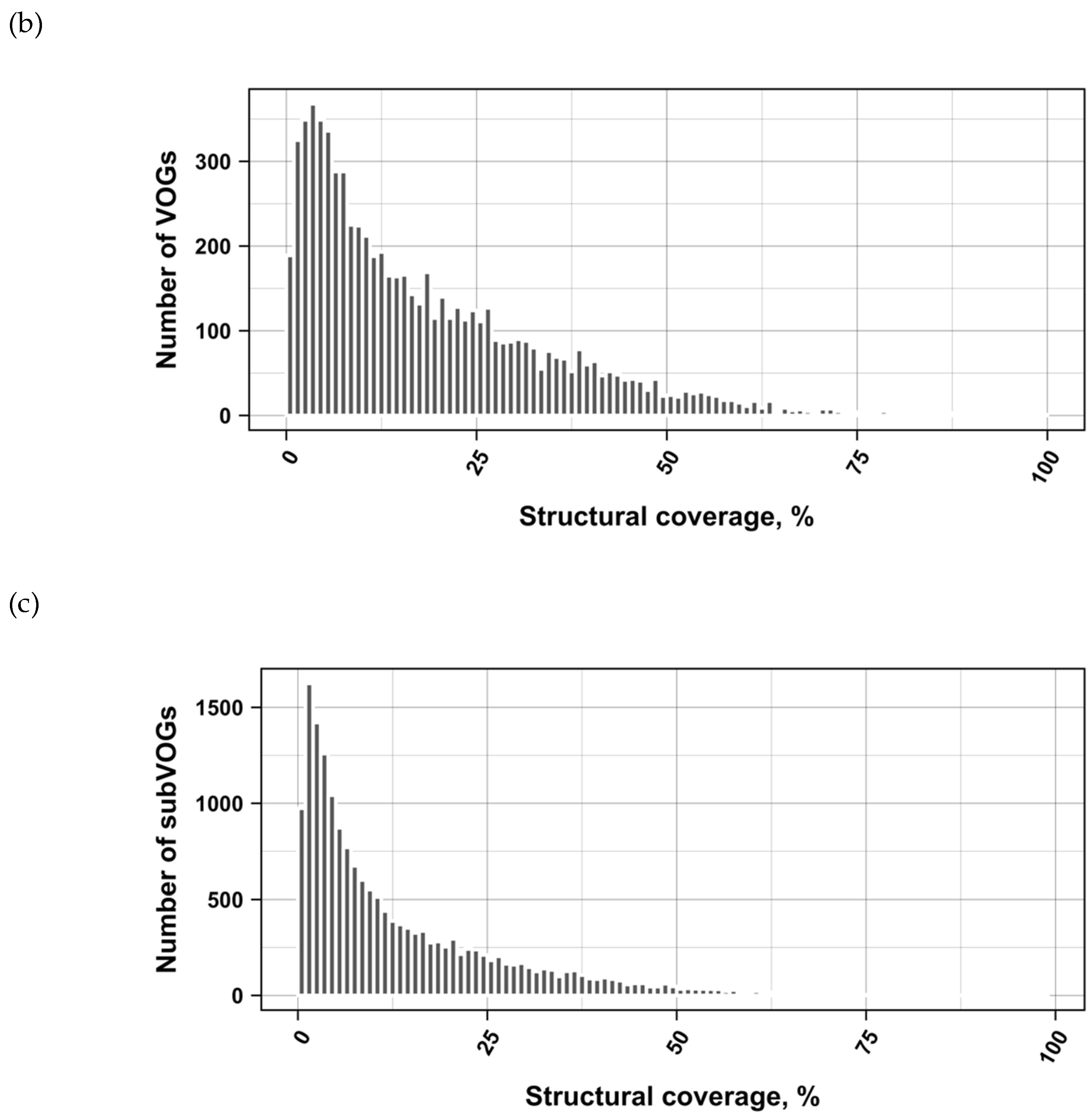
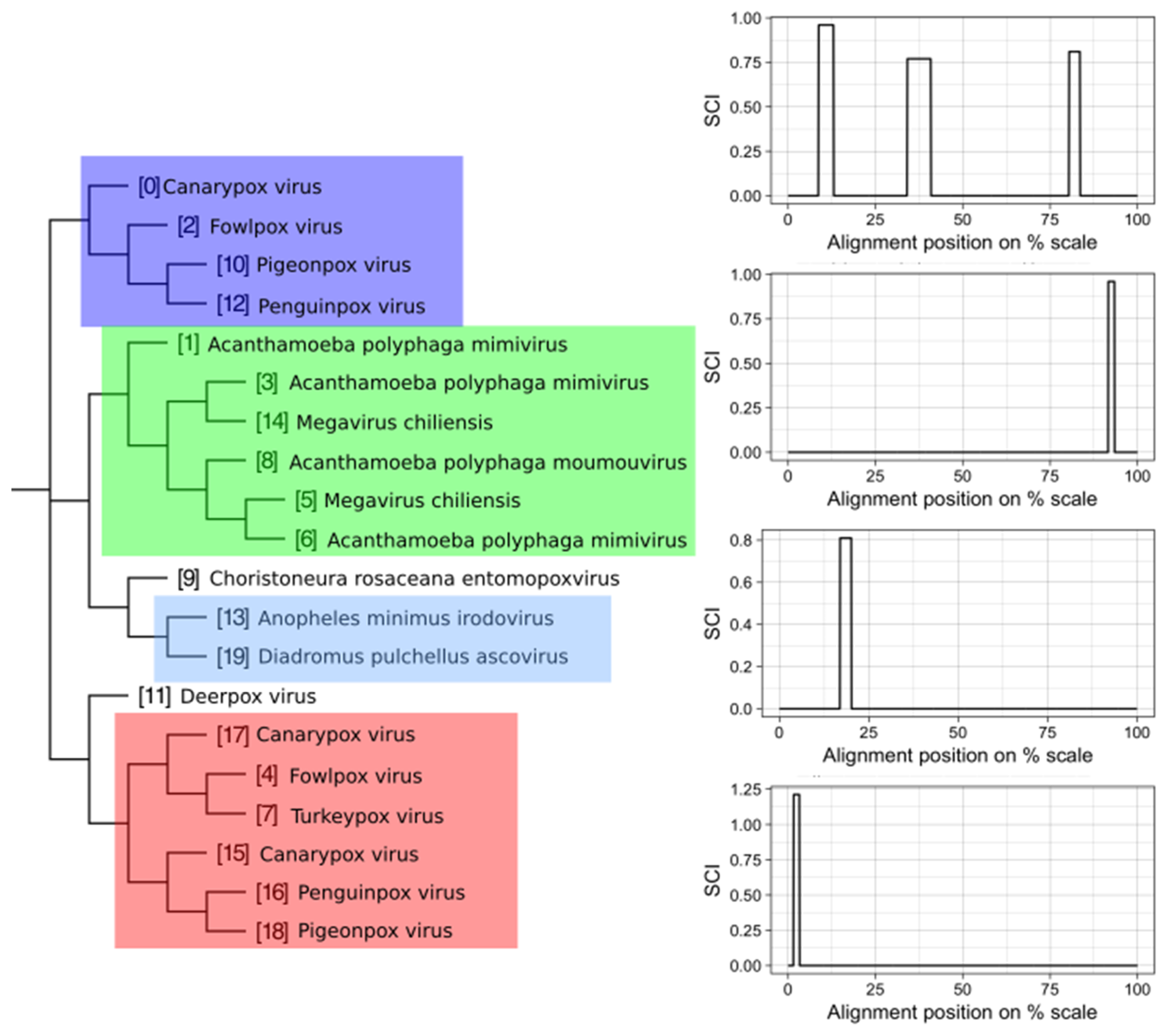
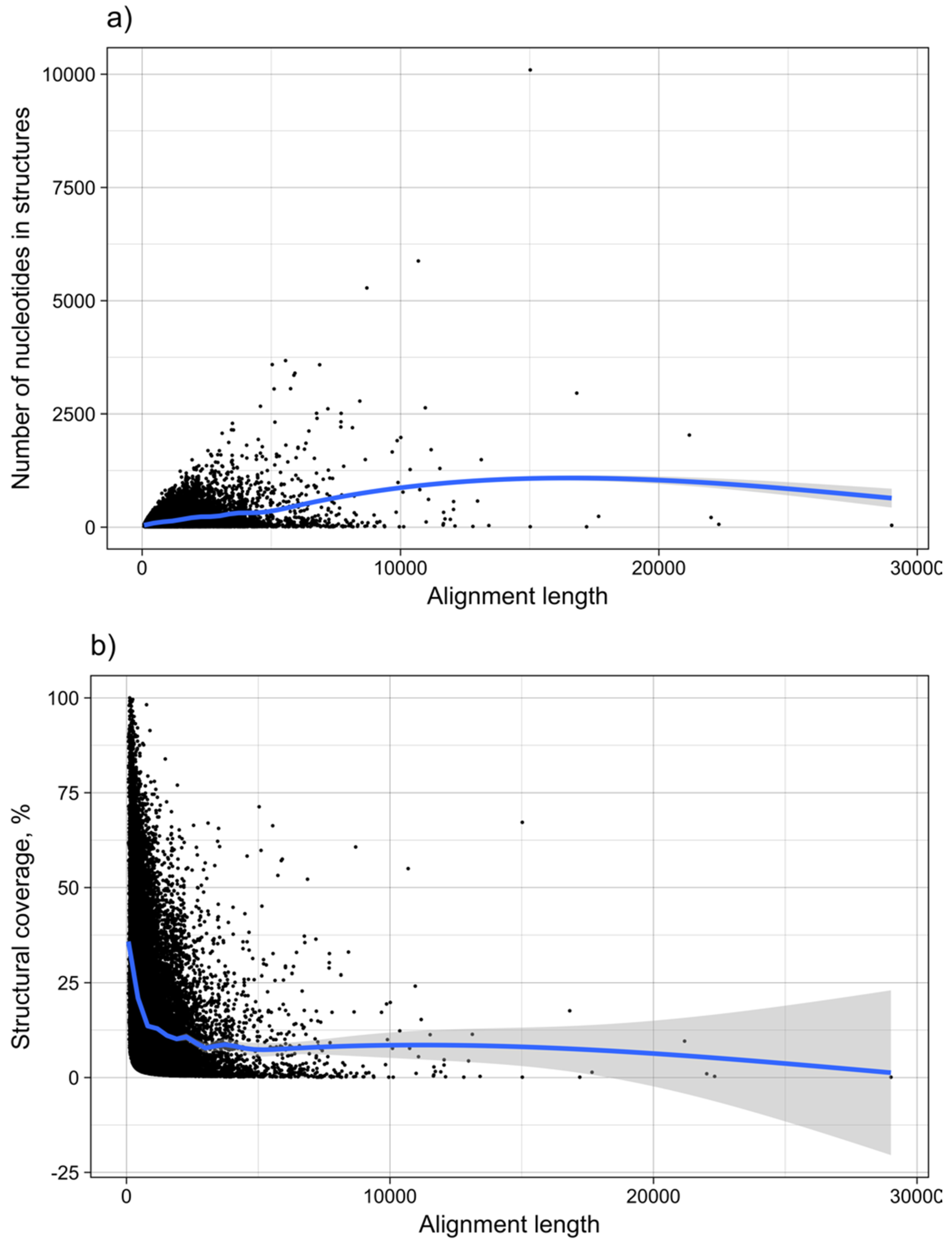
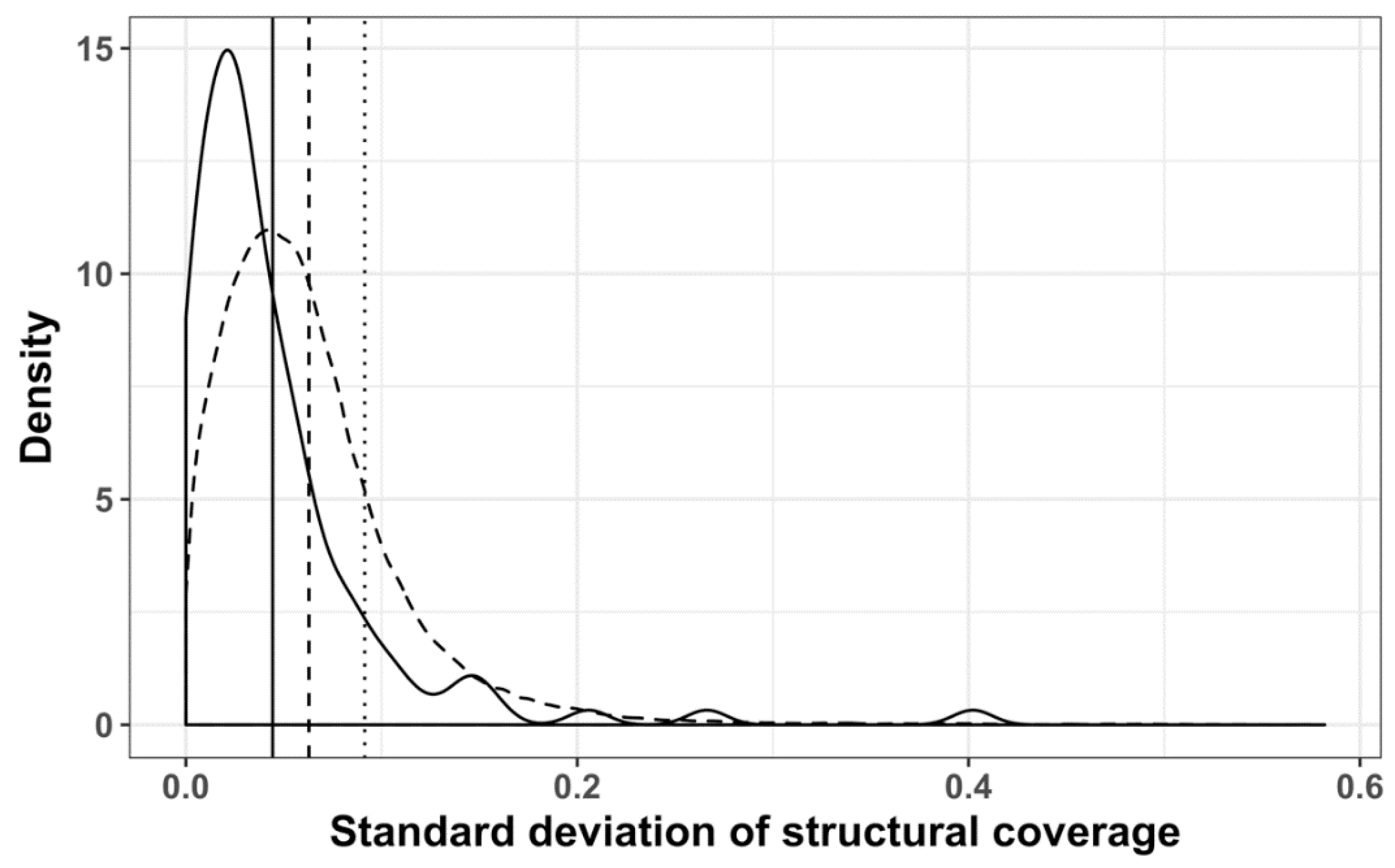
3.2. Structure Conservation in VOGs
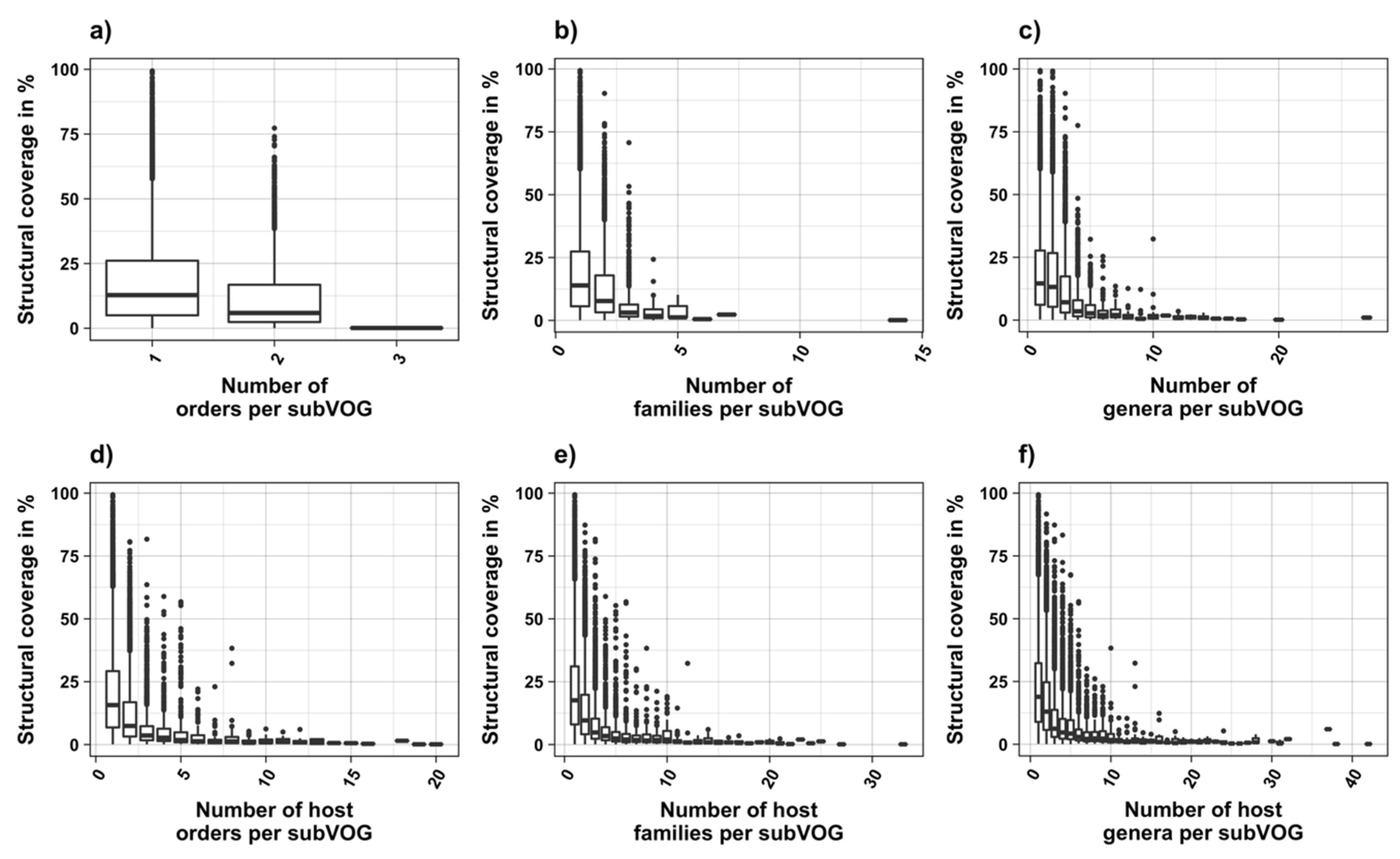
3.3. Structure Conservation in subVOGs
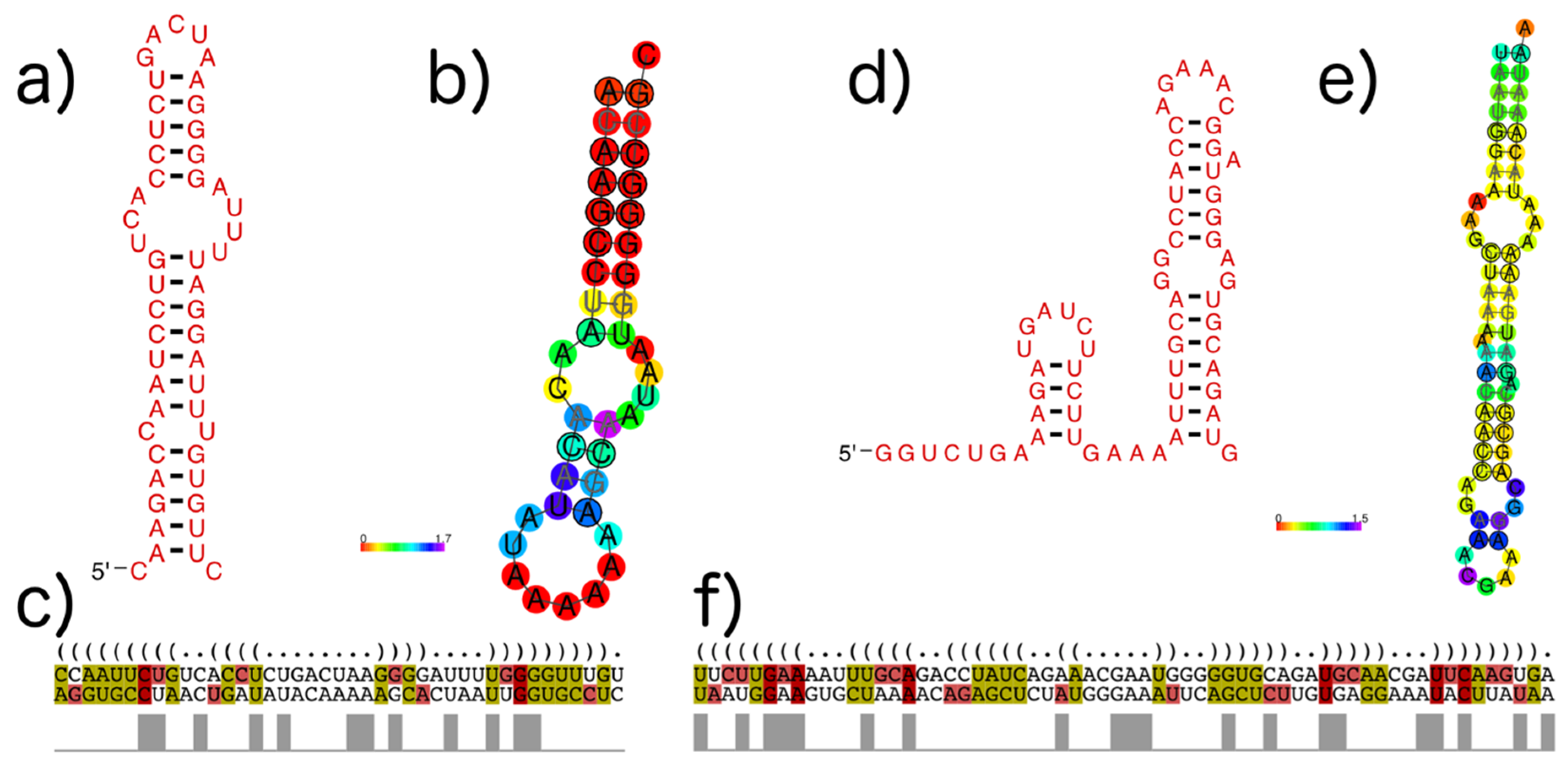
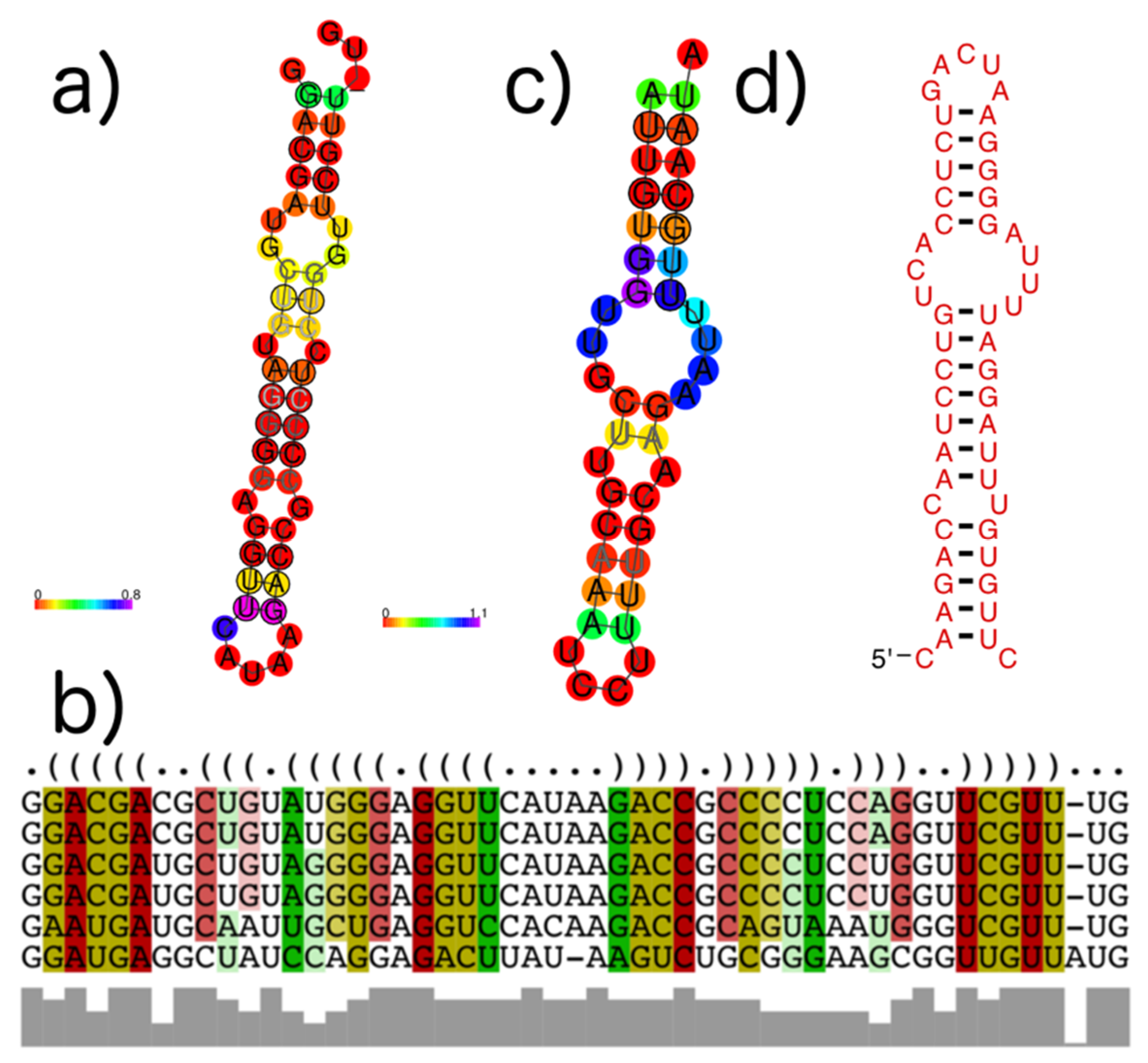
3.4. subVOG Covariance Models
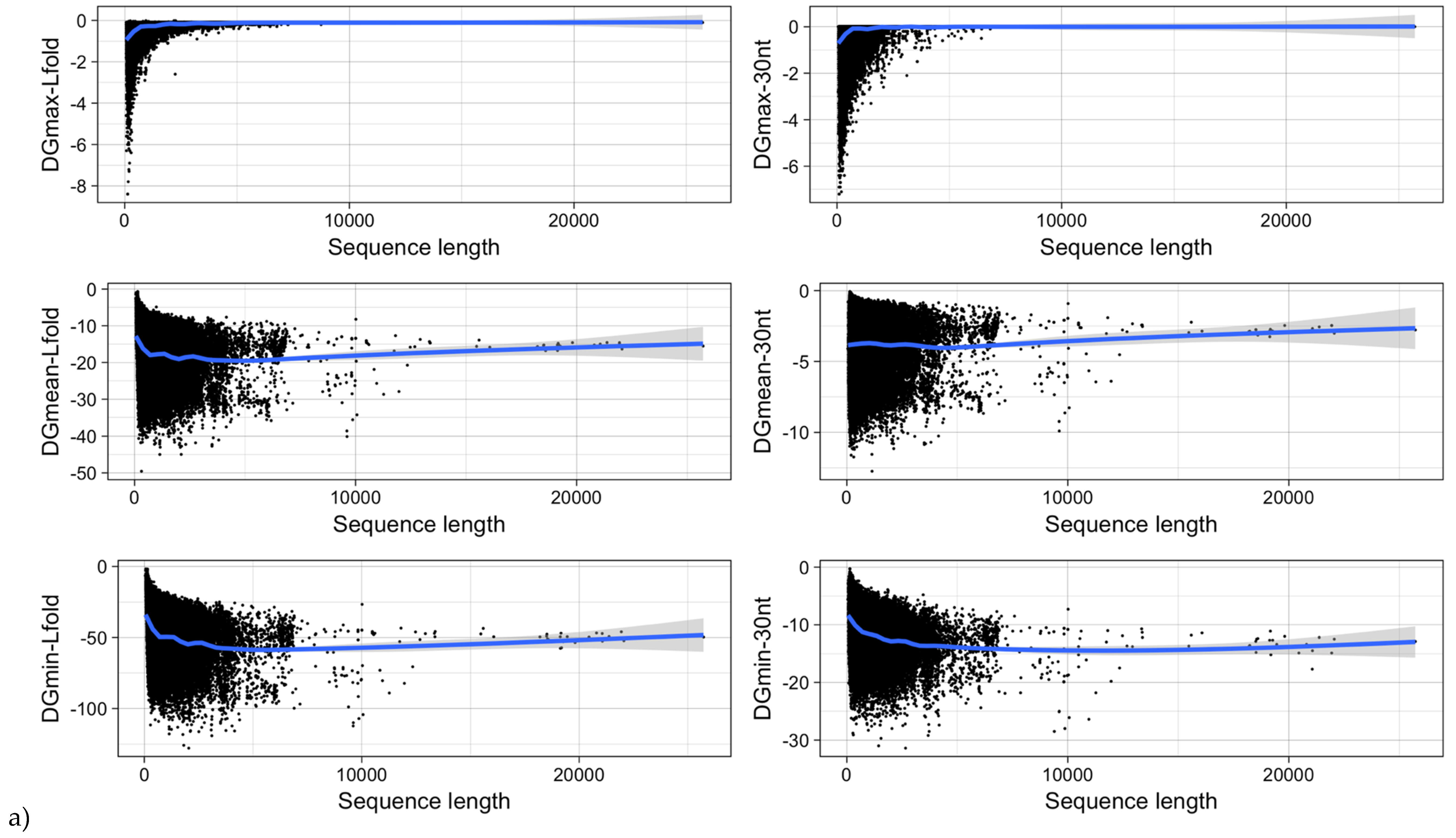
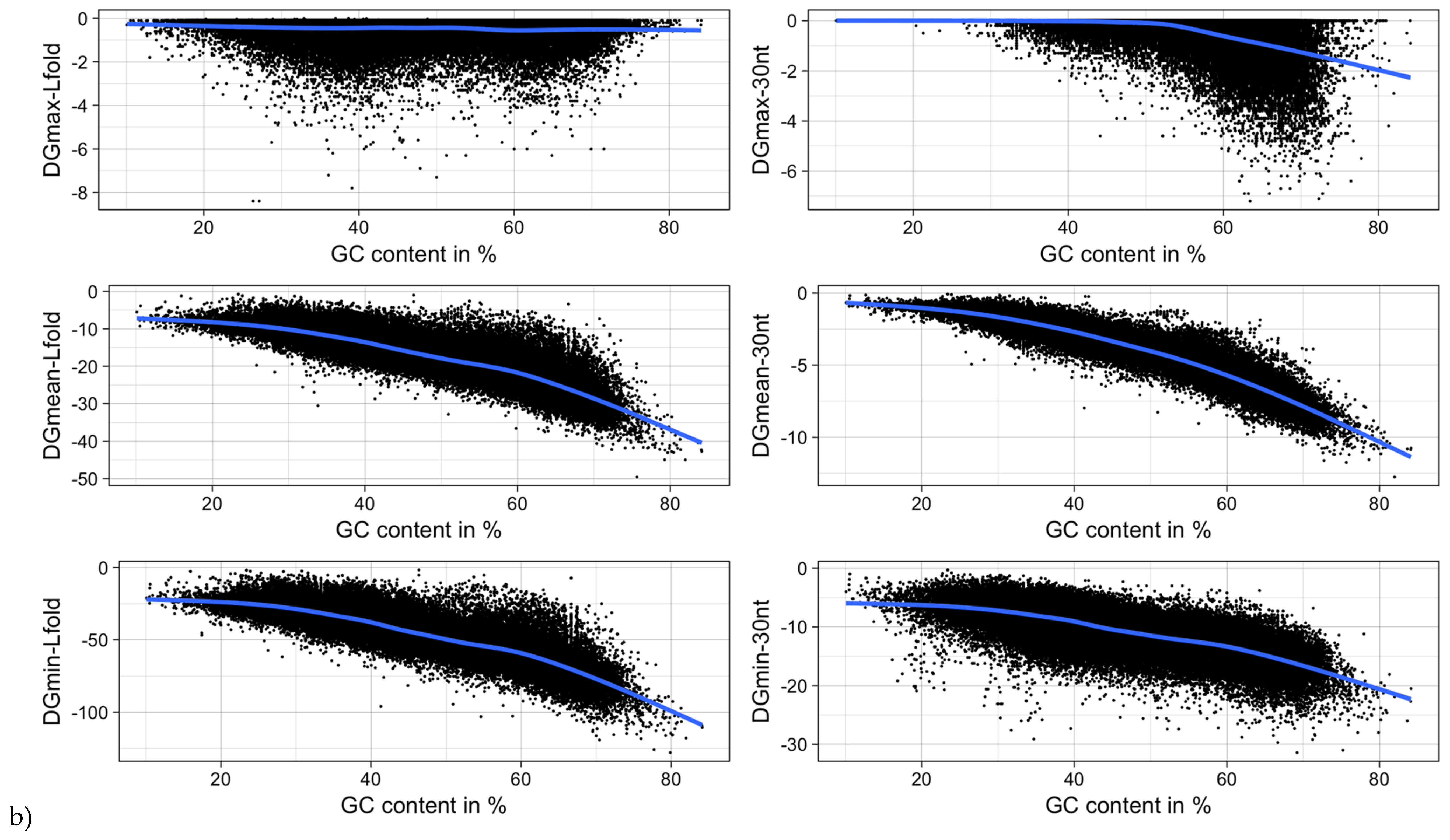
3.5. mRNA Stability and Length
3.6. mRNA Structures and Protein Function
3.7. subVOG Online Resource
- (i)
- General information, i.e., number of mRNAs in the subVOG, the number of proteins and species in the parent VOG, as well as a consensus functional description;
- (ii)
- Information on conserved structures among the subVOG sequences. A plot outlining the SCI for each column of the subVOG MSA gives a brief overview over the structure of the subVOG members. Also provided is a table that shows a list of all structures found, including the corresponding values of SCI, mPID, and the GC content. The consensus structure can also be visualized by Forna, and a covariance model is provided, which can be used to search for similar structures. Additionally, the RNAz results for each individual structured region can be accessed, including structure visualization, dot plots, and the local structure-guided alignments;
- (iii)
- (iv)
- The list of subVOG members, including protein names, descriptions, and taxonomy. For each protein, a link to the RefSEQ entry is provided, as well as the amino acid and nucleotide sequences. The leftmost column of the list contains a checkbox for each subVOG member, which can be used to build a subset of members and analyze the RNA structures shared by these.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bevilacqua, P.C.; Blose, J.M. Structures, kinetics, thermodynamics, and biological functions of RNA hairpins. Annu. Rev. Phys. Chem. 2008, 59, 79–103. [Google Scholar] [CrossRef] [PubMed]
- Serganov, A.; Patel, D.J. Ribozymes, riboswitches and beyond: Regulation of gene expression without proteins. Nat. Rev. Genet. 2007, 8, 776–790. [Google Scholar] [CrossRef] [PubMed]
- Gray, N.K.; Hentze, M.W. Regulation of protein synthesis by mRNA structure. Mol. Biol. Rep. 1994, 19, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene 2005, 361, 13–37. [Google Scholar] [CrossRef]
- Katz, L.; Burge, C.B. Widespread selection for local RNA secondary structure in coding regions of bacterial genes. Genome Res. 2003, 13, 2042–2051. [Google Scholar] [CrossRef]
- Mortimer, S.A.; Kidwell, M.A.; Doudna, J.A. Insights into RNA structure and function from genome-wide studies. Nat. Rev. Genet. 2014, 15, 469–479. [Google Scholar] [CrossRef]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef]
- Duan, J.; Wainwright, M.S.; Comeron, J.M.; Saitou, N.; Sanders, A.R.; Gelernter, J.; Gejman, P.V. Synonymous mutations in the human dopamine receptor D2 (DRD2) affect mRNA stability and synthesis of the receptor. Hum. Mol. Genet. 2003, 12, 205–216. [Google Scholar] [CrossRef]
- Ilyinskii, P.O.; Schmidt, T.; Lukashev, D.; Meriin, A.B.; Thoidis, G.; Frishman, D.; Shneider, A.M. Importance of mRNA secondary structural elements for the expression of influenza virus genes. OMICS 2009, 13, 421–430. [Google Scholar] [CrossRef]
- Carlini, D.B.; Chen, Y.; Stephan, W. The relationship between third-codon position nucleotide content, codon bias, mRNA secondary structure and gene expression in the drosophilid alcohol dehydrogenase genes Adh and Adhr. Genetics 2001, 159, 623–633. [Google Scholar] [PubMed]
- Nackley, A.G.; Shabalina, S.A.; Tchivileva, I.E.; Satterfield, K.; Korchynskyi, O.; Makarov, S.S.; Maixner, W.; Diatchenko, L. Human catechol-O-methyltransferase haplotypes modulate protein expression by altering mRNA secondary structure. Science 2006, 314, 1930–1933. [Google Scholar] [CrossRef]
- Gu, W.; Zhou, T.; Wilke, C.O. A universal trend of reduced mRNA stability near the translation-initiation site in prokaryotes and eukaryotes. PLoS Comput. Biol. 2010, 6, e1000664. [Google Scholar] [CrossRef]
- Del Campo, C.; Bartholomäus, A.; Fedyunin, I.; Ignatova, Z. Secondary Structure across the Bacterial Transcriptome Reveals Versatile Roles in mRNA Regulation and Function. PLoS Genet. 2015, 11, e1005613. [Google Scholar] [CrossRef]
- Shabalina, S.A.; Ogurtsov, A.Y.; Spiridonov, N.A. A periodic pattern of mRNA secondary structure created by the genetic code. Nucleic Acids Res. 2006, 34, 2428–2437. [Google Scholar] [CrossRef]
- Simmonds, P.; Tuplin, A.; Evans, D.J. Detection of genome-scale ordered RNA structure (GORS) in genomes of positive-stranded RNA viruses: Implications for virus evolution and host persistence. RNA 2004, 10, 1337–1351. [Google Scholar] [CrossRef]
- Meyer, I.M.; Miklós, I. Statistical evidence for conserved, local secondary structure in the coding regions of eukaryotic mRNAs and pre-mRNAs. Nucleic Acids Res. 2005, 33, 6338–6348. [Google Scholar] [CrossRef]
- Olivier, C.; Poirier, G.; Gendron, P.; Boisgontier, A.; Major, F.; Chartrand, P. Identification of a conserved RNA motif essential for She2p recognition and mRNA localization to the yeast bud. Mol. Cell. Biol. 2005, 25, 4752–4766. [Google Scholar] [CrossRef]
- Fricke, M.; Gerst, R.; Ibrahim, B.; Niepmann, M.; Marz, M. Global importance of RNA secondary structures in protein-coding sequences. Bioinformatics 2019, 35, 579–583. [Google Scholar] [CrossRef]
- Thurner, C.; Witwer, C.; Hofacker, I.L.; Stadler, P.F. Conserved RNA secondary structures in Flaviviridae genomes. J. Gen. Virol. 2004, 85, 1113–1124. [Google Scholar] [CrossRef]
- Fricke, M.; Dünnes, N.; Zayas, M.; Bartenschlager, R.; Niepmann, M.; Marz, M. Conserved RNA secondary structures and long-range interactions in hepatitis C viruses. RNA 2015, 21, 1219–1232. [Google Scholar] [CrossRef]
- Pirakitikulr, N.; Kohlway, A.; Lindenbach, B.D.; Pyle, A.M. The Coding Region of the HCV Genome Contains a Network of Regulatory RNA Structures. Mol. Cell 2016, 62, 111–120. [Google Scholar] [CrossRef]
- Simmonds, P.; Smith, D.B. Structural constraints on RNA virus evolution. J. Virol. 1999, 73, 5787–5794. [Google Scholar]
- Moss, W.N.; Priore, S.F.; Turner, D.H. Identification of potential conserved RNA secondary structure throughout influenza A coding regions. RNA 2011, 17, 991–1011. [Google Scholar] [CrossRef]
- Clyde, K.; Harris, E. RNA secondary structure in the coding region of dengue virus type 2 directs translation start codon selection and is required for viral replication. J. Virol. 2006, 80, 2170–2182. [Google Scholar] [CrossRef]
- Goz, E.; Tuller, T. Widespread signatures of local mRNA folding structure selection in four Dengue virus serotypes. BMC Genomics. 2015, 16 Suppl. 10, S4. [Google Scholar] [CrossRef]
- Goz, E.; Tuller, T. Evidence of a Direct Evolutionary Selection for Strong Folding and Mutational Robustness Within HIV Coding Regions. J. Comput. Biol. 2016, 23, 641–650. [Google Scholar] [CrossRef]
- Díez, J.; Jungfleisch, J. Translation control: Learning from viruses, again. RNA Biol. 2017, 14, 835–837. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Brown, G.R.; Maglott, D.R. NCBI Reference Sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic Acids Res. 2012, 40, D130–D135. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Höner Zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef]
- Gruber, A.R.; Findeiß, S.; Washietl, S.; Hofacker, I.L.; Stadler, P.F. RNAz 2.0: Improved noncoding RNA detection. Pac. Symp Biocomput 2010, 69–79. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Hulo, C.; de Castro, E.; Masson, P.; Bougueleret, L.; Bairoch, A.; Xenarios, I.; Le Mercier, P. ViralZone: A knowledge resource to understand virus diversity. Nucleic Acids Res. 2011, 39, D576–D582. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal omega. Curr. Protoc. Bioinformatics 2014, 48, 3.13.1–3.13.16. [Google Scholar]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009, 23, 205–211. [Google Scholar]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2011, 9, 173–175. [Google Scholar] [CrossRef]
- Enright, A.J.; Van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef]
- UniProt Consortium UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking Virus Genomes with Host Taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef]
- Kazlauskas, D.; Krupovic, M.; Venclovas, Č. The logic of DNA replication in double-stranded DNA viruses: Insights from global analysis of viral genomes. Nucleic Acids Res. 2016, 44, 4551–4564. [Google Scholar] [CrossRef]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Stajich, J.E.; Block, D.; Boulez, K.; Brenner, S.E.; Chervitz, S.A.; Dagdigian, C.; Fuellen, G.; Gilbert, J.G.R.; Korf, I.; Lapp, H.; et al. The Bioperl toolkit: Perl modules for the life sciences. Genome Res. 2002, 12, 1611–1618. [Google Scholar] [CrossRef]
- Will, S.; Joshi, T.; Hofacker, I.L.; Stadler, P.F.; Backofen, R. LocARNA-P: Accurate boundary prediction and improved detection of structural RNAs. RNA 2012, 18, 900–914. [Google Scholar] [CrossRef]
- Cui, X.; Lu, Z.; Wang, S.; Jing-Yan Wang, J.; Gao, X. CMsearch: Simultaneous exploration of protein sequence space and structure space improves not only protein homology detection but also protein structure prediction. Bioinformatics 2016, 32, i332–i340. [Google Scholar] [CrossRef]
- Tuller, T.; Veksler-Lublinsky, I.; Gazit, N.; Kupiec, M.; Ruppin, E.; Ziv-Ukelson, M. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011, 12, R110. [Google Scholar] [CrossRef]
- Faure, G.; Ogurtsov, A.Y.; Shabalina, S.A.; Koonin, E.V. Role of mRNA structure in the control of protein folding. Nucleic Acids Res. 2016, 44, 10898–10911. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Vandivier, L.; Li, F.; Zheng, Q.; Willmann, M.; Chen, Y.; Gregory, B. Arabidopsis mRNA secondary structure correlates with protein function and domains. Plant. Signal. Behav. 2013, 8, e24301. [Google Scholar] [CrossRef]
- Binns, D.; Dimmer, E.; Huntley, R.; Barrell, D.; O’Donovan, C.; Apweiler, R. QuickGO: A web-based tool for Gene Ontology searching. Bioinformatics 2009, 25, 3045–3046. [Google Scholar] [CrossRef]
- Giglio, M.; Tauber, R.; Nadendla, S.; Munro, J.; Olley, D.; Ball, S.; Mitraka, E.; Schriml, L.M.; Gaudet, P.; Hobbs, E.T.; et al. ECO, the Evidence & Conclusion Ontology: Community standard for evidence information. Nucleic Acids Res. 2019, 47, D1186–D1194. [Google Scholar]
- Supek, F.; Bošnjak, M.; Škunca, N.; Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 2011, 6, e21800. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Nogales, A.; Baker, S.F.; Martinez-Sobrido, L.; Turner, D.H. Mutations Designed by Ensemble Defect to Misfold Conserved RNA Structures of Influenza A Segments 7 and 8 Affect Splicing and Attenuate Viral Replication in Cell Culture. PLoS ONE 2016, 11, e0156906. [Google Scholar] [CrossRef]
- Kutchko, K.M.; Madden, E.A.; Morrison, C.; Plante, K.S.; Sanders, W.; Vincent, H.A.; Cruz Cisneros, M.C.; Long, K.M.; Moorman, N.J.; Heise, M.T.; et al. Structural divergence creates new functional features in alphavirus genomes. Nucleic Acids Res. 2018, 46, 3657–3670. [Google Scholar] [CrossRef]
- Weinberg, Z.; Breaker, R.R. R2R--software to speed the depiction of aesthetic consensus RNA secondary structures. BMC Bioinformatics 2011, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Li, P.; Gutenkunst, R.N. Systematic Effects Of mRNA Secondary Structure On Gene Expression And Molecular Function In Budding Yeast. BioRxiv 2017. [Google Scholar] [CrossRef]
- Li, F.; Zheng, Q.; Vandivier, L.E.; Willmann, M.R.; Chen, Y.; Gregory, B.D. Regulatory impact of RNA secondary structure across the Arabidopsis transcriptome. Plant. Cell 2012, 24, 4346–4359. [Google Scholar] [CrossRef]
- Zur, H.; Tuller, T. Strong association between mRNA folding strength and protein abundance in S. cerevisiae. EMBO Rep. 2012, 13, 272–277. [Google Scholar] [CrossRef]
- Federhen, S. The NCBI Taxonomy database. Nucleic Acids Res. 2012, 40, D136–D143. [Google Scholar] [CrossRef]
- Yachdav, G.; Wilzbach, S.; Rauscher, B.; Sheridan, R.; Sillitoe, I.; Procter, J.; Lewis, S.E.; Rost, B.; Goldberg, T. MSAViewer: Interactive JavaScript visualization of multiple sequence alignments. Bioinformatics 2016, 32, 3501–3503. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.A.; Clamp, M.; Barton, G.J. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of ΔG | Pearson Correlation Coefficient | |
|---|---|---|
| ΔG vs. Sequence Length | ΔG vs. GC-Content | |
| ΔGmin | −0.27 (<2.2−16) | −0.73 (<2.2−16) |
| ΔGmean | 0.004 (0.1655) | −0.94 (<2.2−16) |
| ΔGmax | 0.17 (<2.2−16) | −0.50 (<2.2−16) |
| ΔGmin (RNALfold) | −0.24 (<2.2−16) | −0.86 (<2.2−16) |
| ΔGmean (RNALfold) | −0.16 (<2.2−16) | −0.86 (<2.2−16) |
| ΔGmax (RNALfold) | 0.29 (<2.2−16) | −0.07 (<2.2−16) |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiening, M.; Ochsenreiter, R.; Hellinger, H.-J.; Rattei, T.; Hofacker, I.; Frishman, D. Conserved Secondary Structures in Viral mRNAs. Viruses 2019, 11, 401. https://doi.org/10.3390/v11050401
Kiening M, Ochsenreiter R, Hellinger H-J, Rattei T, Hofacker I, Frishman D. Conserved Secondary Structures in Viral mRNAs. Viruses. 2019; 11(5):401. https://doi.org/10.3390/v11050401
Chicago/Turabian StyleKiening, Michael, Roman Ochsenreiter, Hans-Jörg Hellinger, Thomas Rattei, Ivo Hofacker, and Dmitrij Frishman. 2019. "Conserved Secondary Structures in Viral mRNAs" Viruses 11, no. 5: 401. https://doi.org/10.3390/v11050401
APA StyleKiening, M., Ochsenreiter, R., Hellinger, H.-J., Rattei, T., Hofacker, I., & Frishman, D. (2019). Conserved Secondary Structures in Viral mRNAs. Viruses, 11(5), 401. https://doi.org/10.3390/v11050401