1. Introduction
Studies on the functions of viral genes in human cytomegalovirus (HCMV) replication in vivo and understanding of viral pathogenesis are essential for developing novel drugs and strategies to treat the viral infections, but there are no suitable animal models for HCMV infection at present. Because HCMV proliferates in human cells specifically, grows slowly and has a very long lytic replication cycle in humans, it is quite difficult to study HCMV (genome size 235 kb) gene function and pathogenesis [
1]. However, the murine cytomegalovirus (MCMV) provides an excellent animal model for studying the biology of cytomegaloviruses (CMV) through its specific infection of mice. An MCMV genome of 230 kb is predicted to encode more than 170 open reading frames (ORFs), 78 of which have an extensive homology to those of HCMV [
1,
2,
3]. Moreover, the pathogenesis of MCMV infection in mice is very similar to that of HCMV infection in humans in several aspects such as active infections, establishment of latency, and reactivation after latent infections [
1,
3,
4,
5]. A complete understanding of the biology of MCMV and the function of its genes may provide insights into the pathogenesis of HCMV.
As a structural protein, the HCMV UL80 assembly protein is presumed to function in packaging DNA. A protease is encoded by the N-terminal region of UL80 that cleaves the assembly protein precursor at a site near the C terminus. The MCMV homolog (M80) of the assembly protein of HCMV (UL80) conserves the domain structure and cleavage sites present in HCMV UL80. MCMV is different in that the conserved CD1 and CD2 domains are separated by 100 amino acids, whereas in all other sequenced herpes viruses the two domains are separated by 80 to 84 residues [
2]. The MCMV homolog (M80.5) of HCMV, UL80.5 ORF, is referred to code for protease. The ORFs of MCMV M80 and M80.5 are required for the assembly of proteins and proteases (capsid synthesis) and further virion production. The transcriptions of adjoining MCMV M80 and M80.5 ORFs have different start sites; however, they have the same stop site. Therefore, the overlapping region of MCMV M80 and M80.5 (M80/80.5) is an appropriate target site for virus inhibition.
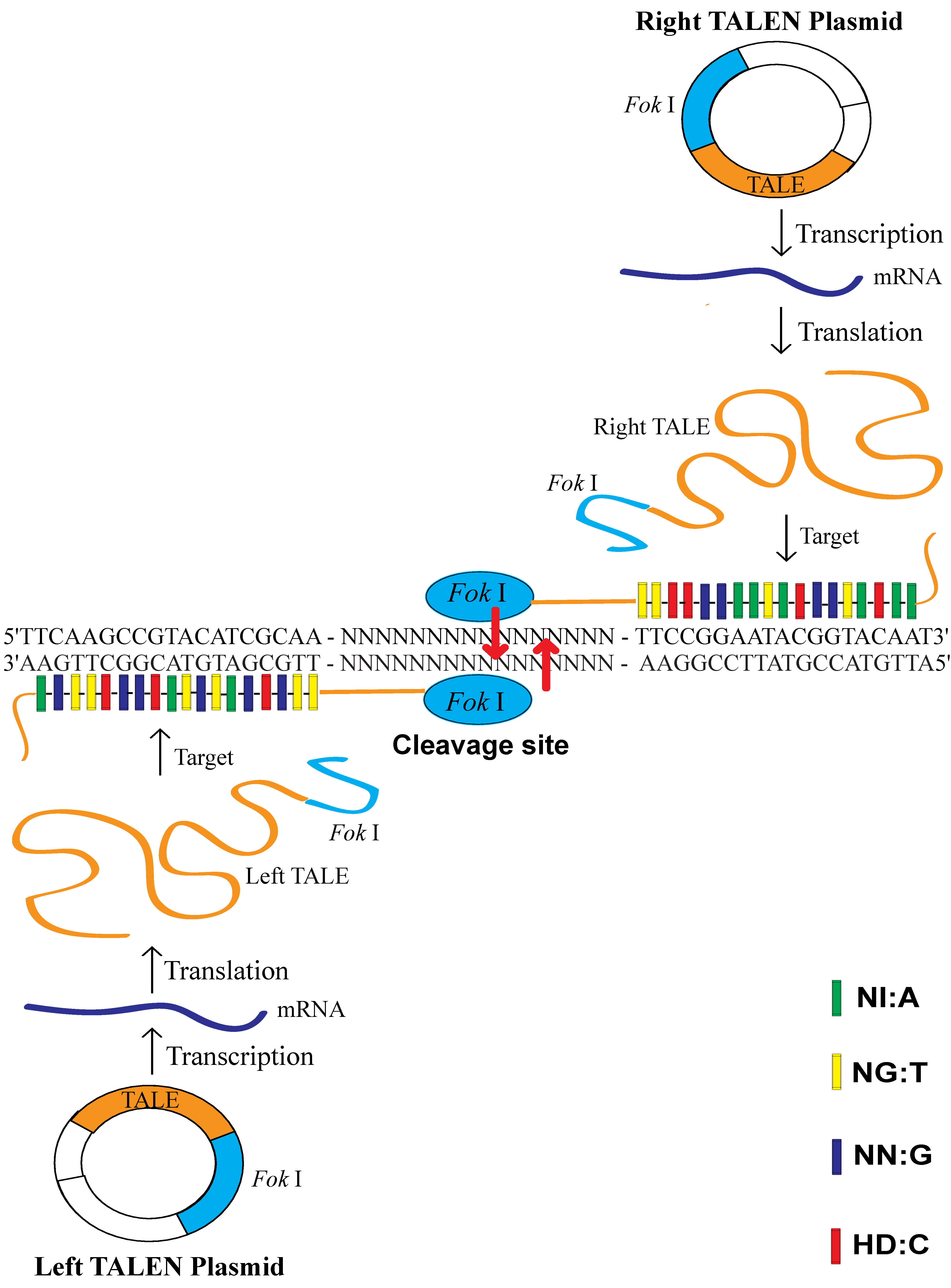
Transcription activator-like effectors (TALEs) are proteins secreted by the plant pathogenic bacteria
Xanthomonas via a type III secretion system where they infect various plant species [
6]. These proteins can bind promoter sequences in the host plant and activate the expression of host genes that aid bacterial infections. TALEs are important virulence factors that act as transcriptional activators in the plant cell nucleus, where they directly bind to DNA via a central domain of tandem repeats [
6]. Each TALE contains a central repetitive region consisting of varying numbers of repeat units (about 17.5 repeats) of 34 amino acids [
6,
7]. The DNA-binding domain contains a highly conserved 34-amino acid sequence, with the exception of the 12th and 13th amino acids [
6]. Only the 12th and 13th amino acids in TALEs are changeable and variable; the other amino acids are constant and stable. These two locations’ RVDs (Repeat Variable Di-residues) are highly variable and show a strong correlation with a specific nucleotide recognition by different frequencies, for example NI recognizes A (55%), NG recognizes T (50%), NN recognizes G (7%) and HD recognizes C (69%) [
6]. DNA transcribes RNA according to the complementary base pairing rule (A = U, G ≡ C) and RNA translates protein according to the standard genetic code (RNA codon table). After the breaking of the code of the DNA-binding specificity of TALEs [
6,
7], we know that two amino acids can also recognize one nucleotide. The restriction endonuclease
Fok I, naturally found in the bacterium
Flavobacterium okeanokoites, consists of an N-terminal specific DNA-binding domain and a C-terminal nonspecific DNA cleavage domain. The DNA-binding domain recognizes the non-palindromic sequence 5′-GGATG-3′ when the catalytic domain cleaves double-stranded DNA nonspecifically at a fixed distance of 9 and 13 nucleotides downstream of the recognition site.
Fok I exists as an inactive monomer and becomes an active dimer upon binding to its target DNA and in the presence of specific divalent metals [
7]. The DNA cleavage domain of
Fok I functions as a homodimer, requiring two constructs with unique DNA-binding domains for sites in the target genome with proper orientation and spacing. Transcription activator-like effector nucleases (TALENs) are artificial restriction enzymes generated by fusing the specific TALE DNA-binding domain to a nonspecific
Fok I DNA cleavage domain [
8,
9] (
Figure 1).
TALENs were shown to be a valuable tool for precise genome engineering with low toxicity [
10]. Additionally, they have been successfully applied in the genetic engineering of human pluripotent cells [
11,
12], and the generation of knockout animals, such as nematodes (
Caenorhabditis elegans) [
8], rats [
13], and zebrafish [
14,
15]. TALENs can be used to edit genomes by making double-stranded breaks (DSBs), which cells respond to with two repair mechanisms: non-homologous end joining (NHEJ) or homologous recombination (HR). DNA can be introduced into a genome through NHEJ in the presence of exogenous double-stranded DNA fragments. HR can also introduce foreign DNA at the DSB as the transfected double-stranded sequences are used as templates for the repair enzymes. However, TALENs have some potential problems. If TALENs do not specifically target a unique site within the genome of interest, off-target cleavage may occur [
11]. Such off-target cleavage may lead to the production of enough DSB to overcome the repair machinery and consequently result in chromosomal rearrangements and/or cell death [
8].
Previous reports have suggested that TALENs can be engineered to adapt for an antiviral strategy. For example, TALENs were known to be effective in the inactivation of Hepatitis B virus (HBV) replication in cultured cells and in vivo [
16], and in the targeting of the HBV genome [
17]. Also, Epstein–Barr virus (EBV)-encoded nuclear antigen-1 (EBNA1) plays a crucial role in EBV episome replication and persistence. TALEN-mediated targeted disruption of EBNA1 was shown to inhibit the growth of EBV-infected cells, hinting at a possible therapeutic application for EBV-associated disorders [
18]. TALENs may also provide a new strategy for the treatment of CMV infections.
Latency is a specific phase in viral life cycles, in which viral particles stop producing after infection, but the viral genome has not been completely removed. Proviral latency and episomal latency are two known viral latency models. CMV belongs to the episomal latency model which is essentially quiescent in myeloid progenitor cells, and is reactivated by differentiation, inflammation, immunosuppression or critical diseases [
19]. CMV latency has been defined as the absence of infectious viruses, despite the presence of viral DNA. Although the molecular mechanisms by which latency is established and maintained have not been clear, transcriptional control of viral gene expression is very important in controlling viral latency and reactivation. Viral replication is initiated by the expression of
ie (immediately early) genes. Studies with CMV have suggested that latency is established through the repression of
ie-1 gene expression.
Ie-1 proteins, the first proteins expressed by the virus during productive infection, are transcriptionally regulatory proteins that are required for the induction of early and late gene expression, viral DNA synthesis, and virion production [
20,
21,
22]. Since MCMV
ie-1 proteins are required for viruses to start replication from latency, the
ie-1 gene is one of the key targets for latency in animal studies. In this study, our goal is to develop an effective strategy to inhibit the growth of MCMV in cell culture and animals specifically, particularly for the removal of latent viruses.
2. Materials and Methods
2.1. Ethics Statement
For all experiments on live mice, we confirm that all methods were carried out in accordance with relevant guidelines and regulations. The protocol for all animal experiments was approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California at Berkeley, USA (Protocol #R240 and #R276). All efforts were made to minimize suffering.
2.2. Viruses and Cell Culture
The MCMV Smith strain and mouse embryonic fibroblast NIH3T3 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). The MCMV was grown in NIH3T3 cells (ATCC) NIH3T3 cells were cultured in 500 mL Dulbecco’s modified Eagle’s medium (DMEM) (ThermFischer Scientific, Waltham, MA, USA) supplemented with 10% Nu-Serum (Coring, Union City, CA, USA), 1% Pen-Strep (100 U/mL of penicillin and 100 µg/mL of streptomycin), 1× MEM essential amino acids (EAA), 1× MEM nonessential amino acids (NEAA) and 12 mL sodium bicarbonate (ThermoFischer Scientific, USA).
2.3. Mice
The three-week-old immunocompetent Balb/c mice were purchased from the Jackson Laboratory, USA and used at four weeks of age.
2.4. Primers and Probes
MCMV M80/80.5 forward primers (5′-CTTGCCTCAGGTGCCCTCTTATTACGGAAT-3′) and reverse primers (5′- ATAAATCACACGTTCACTCCGTTAGTCCGG -3′) were both synthesized by Life technologies, Camarillo, CA, USA.
MCMV ie-1 forward primers (5′-TCAGCCATCAACTCTGCTACCAAC-3′) and reverse primers (5′-ATCTGAAACAGCCGTATATCATCTTG-3′) were synthesized by Life technologies, USA. TaqMan probes (5′-TTCTCTGTCAGCTAGCCAATGATATCTTCGAGC-3′) were synthesized by Genscript, Nanjing, Jiangsu, China. The probe was labeled at the 5′ end with the reporter dye FAM and at the 3′ end with the quencher dye TAMRA.
2.5. Transcription Activator-Like Effector Nuclease (TALEN) Plasmids
Three specific pairs (left and right) of TALEN plasmids for each targeting site MCMV1-2, 3-4, 5-6, and two nonspecific pairs W1FS-W7R1, KSHV1-2 were constructed by others. They all contain a CMV promoter, a
Fok I gene (cleavage domain) and a TALE DNA sequence using pTAL4 Leu (8467 bp, Addgene, Watertown, MA, USA) as the backbone vector. All ten TALEN plasmids are listed in
Table 1.
2.6. Determination of the Murine Cytomegalovirus (MCMV) Growth Curve in Host Cells
NIH3T3 cells (1.00 × 105 cells/well) were plated in a 12-well format containing 1 mL of growth medium and infected with MCMV (multiplicity of infection, MOI = 0.05) 1 day later (initial titer: 5.00 × 103 pfu/mL). In the preliminary test, the viral titers were 4.20 × 103, 1.40 × 105, 5.10 × 105, 2.70 × 105 pfu/mL at 1, 3, 5 and 7 days post infection, respectively. We found that the viral titers were increasing for 1 to 5 days post infection, but gradually decreasing after 5 days. The results demonstrated that MCMV reached the highest titer at the 5th day post infection.
2.7. Determination of Transfection Efficiency
Lipofectamine™ 2000 transfection reagent was commercially obtained from Life Technologies, USA. The other transfection reagent NKS11, a new lipoid, was synthesized by others. In a safety test, the NKS11 formulation was proved to be nontoxic to Balb/c mice by weight measurement and health observation.
One day before transfection, adherent NIH3T3 cells (1.00 × 105 cells/well) in 1 mL growth medium without antibiotics were plated in a 12-well format so that cells would be 90–95% confluent at the time of transfection. GFP (green fluorescent protein) plasmids (1.6 µg/well, pRK-9-Flag-EGFP, 5520 bp, Addgene, USA) were transfected into the cells to determine the transfection efficiency. The medium was changed after 4–6 h. The growth medium was removed and the cells were washed with phosphate buffered saline (PBS), followed by trypsinizing the cell pellet for 5 min using trypsin (100 µL/well) at 1, 2, and 3 days post transfection, respectively. The cell pellet was resuspended with 1 ml growth medium and cell number was counted using a hemocytometer under the fluorescence microscope.
By the data we obtained, the transfection efficiency increased during the period 1, 2, and 3 days post transfection, and then saturated at the 3rd day. The percentages were about 14.3%, 21.4%, and 21.4% using lipofectamine. The results revealed that the highest transfection efficiency for plasmids in NIH 3T3 cells was about 20–25% and it took about 2–3 days for the plasmids to transfect into cells completely. NKS 11 showed almost the same efficiency for transfection in NIH 3T3 cells.
2.8. Cell Count
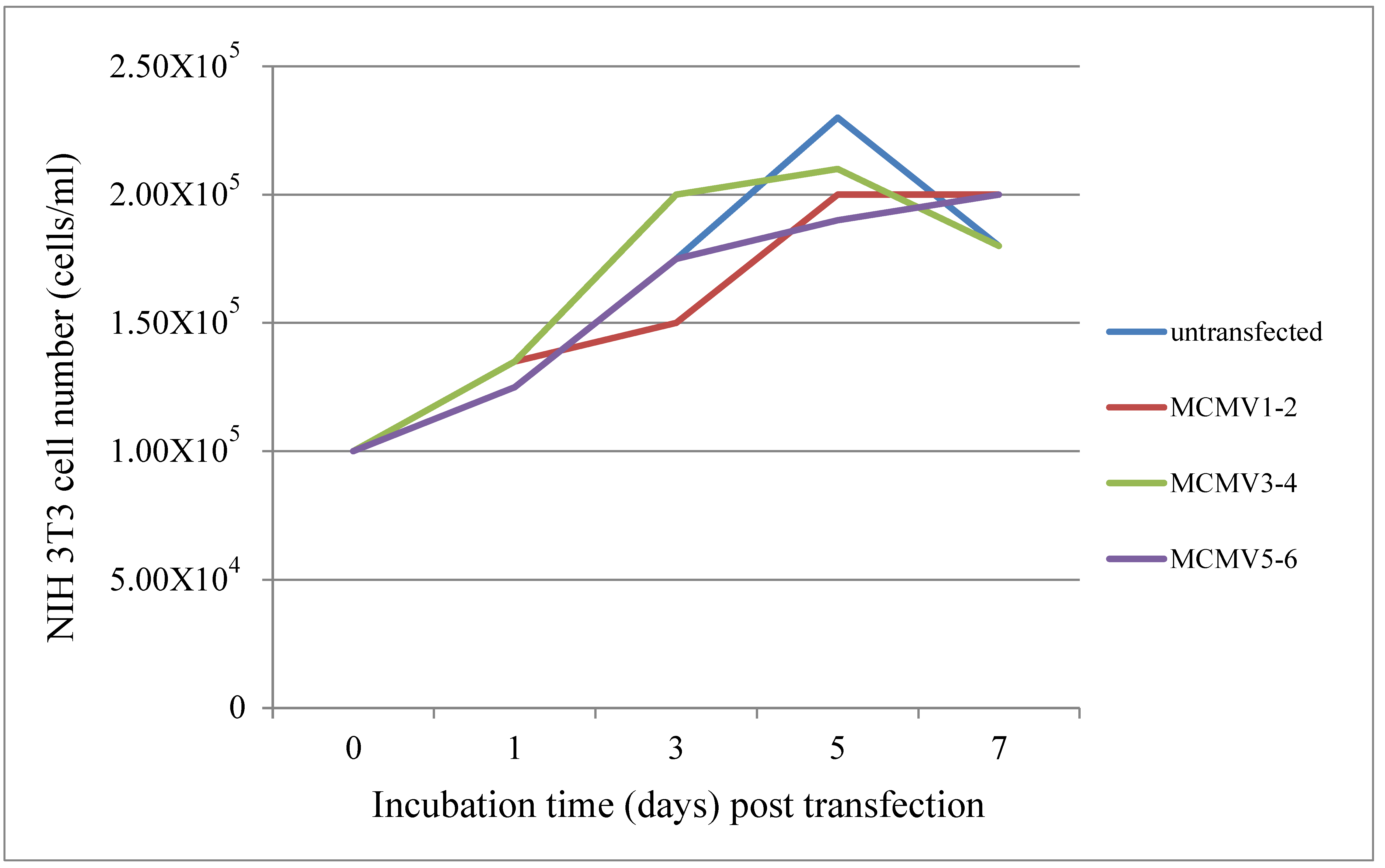
One day before transfection, NIH3T3 cells (1.00 × 105 cells/well) were plated in a 12-well format containing 1 mL of growth medium without antibiotics so that adherent cells would be 90–95% confluent at the time of transfection. TALEN plasmids MCMV 1–2, 3–4 or 5–6 were transfected for each well (1.6 µg/well, 0.8 µg for each plasmid), or none were transfected as a negative control using lipofectamine. GFP plasmids (1.6 µg/well) were transfected into the cells as a positive control. The medium was changed after 4–6 h. The growth medium was removed and the cells were washed with PBS. The cell pellet was harvested using trypsin (100 µL/well) at 1, 3, 5 and 7 days post transfection, respectively. The cell pellet was resuspended with 1 mL growth medium. We counted the cell number using a hemocytometer under a light microscope.
2.9. Cell Viability Assay
The viable cells were assayed using the MTT Cell Growth Assay Kit (Sigma-Alderich, Temecula, CA, USA). The trypsinized cells were appropriately diluted to adjust the cell number range of 1000–50,000 cells/well with growth medium. An amount of 0.2 mL of cell dilution was plated into each well in a 96-well format. The cells were incubated at 37 ℃ overnight. The medium was changed to make the final volume of each well 0.1 mL. We added 0.01 mL AB Solution (MTT) to each well. We incubated the cells at 37 °C for 4 h for cleavage of MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide, Thiazolyl Blue Tetrazolium Bromide). We added 0.1 mL isopropanol with 0.04 N HCl to each well. The absorbance was measured on an ELISA plate reader (Bio-rad, Hercules, CA, USA) with a test wavelength of 570 nm and a reference wavelength of 630 nm within 1 h.
2.10. TALEN Plasmid Transfection and MCMV Infection
On the 1st day, NIH3T3 cells (1.00 × 105 cells/well) in 1 mL growth medium without antibiotics were plated in a 12-well format so that adherent cells would be 90–95% confluent at the time of transfection. On the 2nd day, one pair of TALEN plasmids for each well (1.6 µg/well, 0.8 µg for each plasmid) was transfected, or none were transfected as a negative control, into the cells. GFP plasmids (1.6 µg/well) were transfected into the cells as a positive control. The medium was changed after 4–6 h. On the 3rd day, the cells were infected with MCMV (MOI = 0.05). The steps taken on the 2nd and 3rd day would be reversed if MCMV infection was prior to TALEN plasmid transfection. The medium was changed after 1 h. The cells were incubated at 37 °C in a CO2 incubator for 5–7 days and we changed the medium every three days.
2.11. Virus Titration Assay
NIH3T3 cells grown to 60–70% confluent in 12-well format were prepared for virus titration. At 1, 3, 5, and 7 days post infection, the infected cells together with the medium were harvested and followed by 10 folds of serial dilution. After 1 h of incubation with the dilution at 37 °C in a CO2 incubator, the prepared cells were overlaid with 2 mL fresh complete medium containing 1% low melting agarose and cultured for 4 to 5 days before the plaques were counted under a light microscope. The viral titer (pfu/mL) was determined by plaque assays. The values of the viral titers were the average of triplicate experiments.
2.12. Harvest of the Total DNA and Amplification of the Target DNA Sequence
The total DNA was harvested from the cell culture including the cell pellet and supernatant using the Blood and Tissue DNeasy kit (Qiagen, Germantown, MD, USA) and quantified by UV260 with a spectrophotometer. We amplified the specific product using MCMVM80/80.5 primers and the total DNA as a template by a polymerase chain reaction (PCR), under the conditions of 300 nM for each primer and 1× Hotstar Taq master mix (Qiagen, USA) in a 50 µL mixture. The thermal cycling conditions were 95 °C for 15 min followed by 35 cycles of 94 °C for 30 s, 60 °C for 30 s, 72 °C for 1 min, and 72 °C for 10 min.
2.13. Surveyor Nuclease Mutation Detection Assay
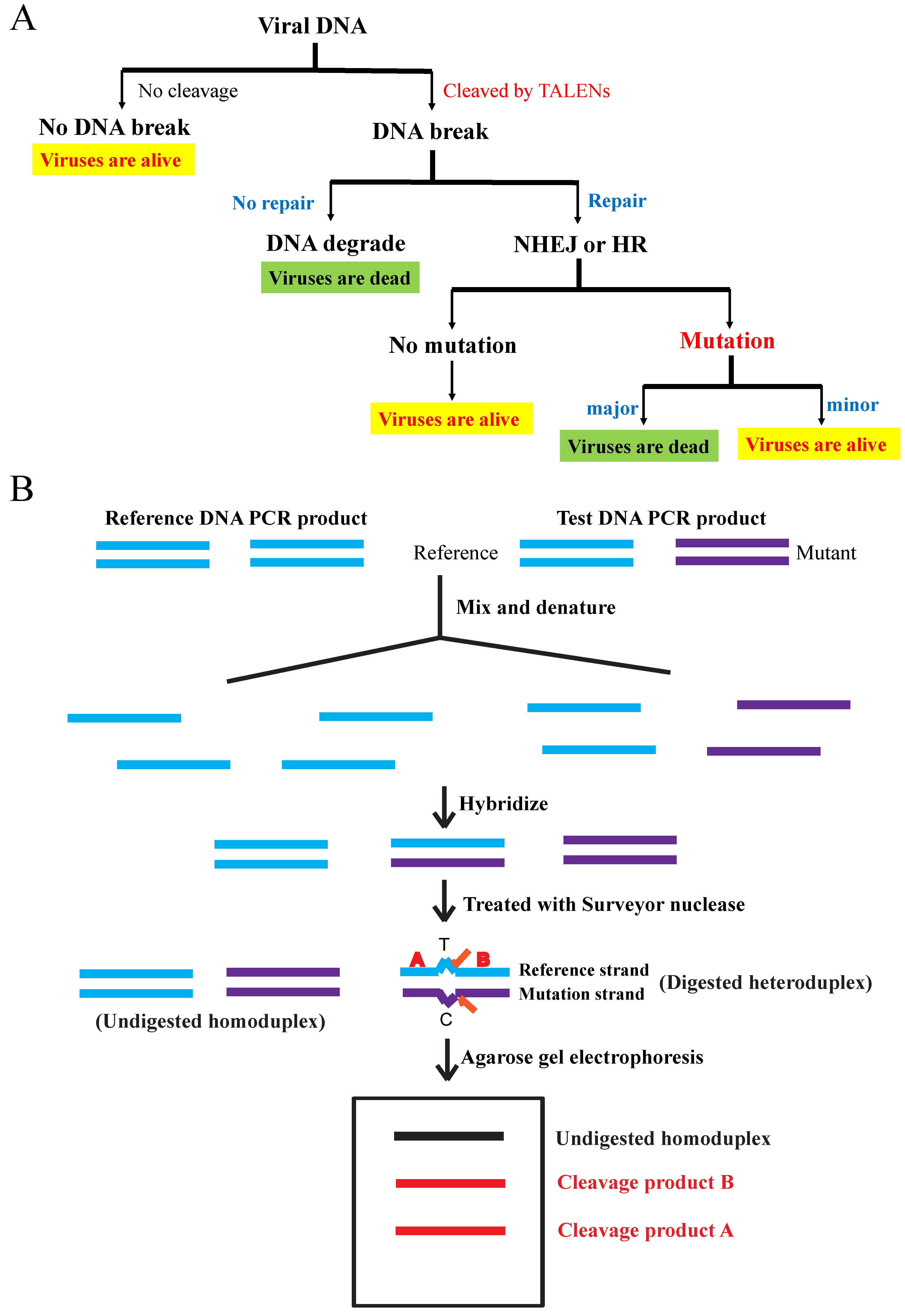
A Surveyor nuclease mutation detection kit (Surveyor nuclease and G + C control included) was obtained from IDT Integrated Technologies, USA [
23].
We amplified wild-type (reference) and mutant (test) total DNA by PCR using MCMV M80/80.5 primers. We mixed equal amounts of reference and test PCR products. We incubated the mixture at 95 °C for 5 min in a beaker filled with 800 mL of water. We then allowed the mixture to denature in order to rehybridize, by heating and cooling it to form heteroduplexes and homoduplexes (finally leaving the water at <30 °C). We treated the annealed mixture with the Surveyor nuclease and incubated at 42 °C for 1 h. The reference PCR product was treated alone as a negative control. DNA fragments were separated by 2% agarose gel electrophoresis [
23].
The cleavage efficiency of PCR products was calculated by scanning the signal strength of DNA bands on the UV illuminator. They were indicated by the percentage (%) of extra bands divided by the total bands (major bands + extra bands) in signal strength.
2.14. Latency Establishment, Treatment and Reactivation
In total, 27 Balb/c mice were infected with Smith strain MCMV (1 × 10
5 pfu/mouse) intraperitoneally (IP), but 3 Balb/c mice were not infected with MCMV. In total, 30 mice were housed to establish their latency for 4–5 months [
3,
22,
24]. We sacrificed 3 mice infected with MCMV to harvest their organs (livers, lungs, spleens, kidneys and salivary glands), in order to test whether MCMV latency was established. The remaining 24 infected Balb/c mice were divided into five experimental groups (3–5 mice/group). They were untreated or treated with TALEN plasmids by tail vein injection 8 times (once/ 5–6 days). The treatment formulation formerly confirmed to be safe for mice was as follows: for total 200 µL injection, 6 µg TALEN plasmids (0.5 µg/µL), 30 µg NKS11 (10 µg/µL), 3.125 mM Sodium Acetate (25 mM, pH5.5) and PBS in each mouse. After treatment, all 24 mice were injected with an immunosuppressive agent cyclophosphamide (Sigma-Alderich, USA) at 150 mg/kg body weight twice (1 dose/5–6 days) to reactivate latency by tail vein injection [
25,
26,
27]. Five days later, all mice were sacrificed and their organs harvested. We sonicated the organs to harvest the homogenate and total DNA.
2.15. Plaque Assay of MCMV in Mouse Organs
The organ homogenates were prepared by sonication and stored in 10% skim milk at −80 °C. The concentrations of all homogenates were adjusted to 10% (100 mg/mL). The presence of infectious viruses in the livers, lungs, spleens, kidneys, and salivary glands were determined by titrating organ homogenates. Plaque assays were performed by virus titration assay. The values given were calculated as PFUs per mg of tissue.
2.16. Quantitative Real-Time Polymerase Chain Reaction (qPCR) Analysis for DNA Copy Number
The total DNAs were harvested in 200 µL organ homogenates (100 mg/mL) in mice tissue using a Blood and Tissue DNeasy kit. Total DNA was dissolved in 50 µL Buffer AE (Qiagen, USA).
For the generation of a standard curve, the total DNA products were amplified by PCR using MCMV ie-1 forward and reverse primers and the total MCMV DNA as a template, under the conditions of 500 nM for each primer and 1X Hotstar Taq polymerase master mix (Qiagen, USA) in a 50 µL mixture. The thermal cycling conditions were 95 °C for 15 min followed by 43 cycles of 94 °C for 30 s, 60 °C for 30 s, 72 °C for 1 min, and 72 °C for 10 min. The MCMV ie-1 DNA PCR product (100 bp, nucleotide no.:181091-181190 in MCMV genome) was isolated to create the DNA dilution standard using the QIAquick Gel Kit Protocol. For absolute quantification of the MCMV DNA copy number in the organs, a standard curve was generated by serial dilutions of MCMV ie-1 DNA PCR products, such that 1 µL of the standard curve template contained 5 × 101, 5 × 102, 5 × 103, 5 × 104, 5 × 105, 5 × 106 for ie-1 DNA copies. The standard curve was obtained by plotting the average threshold cycle (Ct) values against the logarithm of the target template molecules eluted from the MCMV ie-1 DNA PCR products, followed by a sum of least squares regression analysis. Results were expressed as DNA copies/ mg of tissue. Since DNA yield per mg of tissue differs from tissue to tissue, DNA was extracted from 20 mg of each tissue and all extractions were done in triplicate and the average was used to determine the DNA yield/mg of each tissue type.
All qPCRs were performed with the TaqMan gene expression master mix (2× Hotstar Taq polymerase, Qiagen, USA) using the standard curve assay. Each sample was analyzed in triplicate at a 20 µL volume. For the
ie-1 DNA copy number assay, reaction mixtures contained 150 nM of each MCMV
ie-1 primer and 100 nM of the TaqMan probe. The amplification conditions were 50 °C for 2 min, 95 °C for 10 min, followed by 43 cycles of 95 °C for 15 s and 60 °C for 1 min. Values were calculated as copies per mg of tissue [
28].
2.17. Statistical Analysis
The data were analyzed statistically using Microsoft Excel. In all cases, the values were the average of triplicate experiments and indicated as Mean ± SD (standard deviation).
Significant differences between the 2 groups in MCMV DNA copies in each organ of mice were determined using a two-tail Student’s t test (type 3). Data were calculated in triplicate and expressed as Mean ± SEM (standard error of the mean). To determine real-time PCR techniques and their relative sensitivity, all real-time PCR data were calculated to DNA copies/mg of tissue. In all cases, a p value of ≤0.05 was considered statistically significant.
4. Discussion
The transfection efficiency of plasmids in NIH3T3 cells was about 20–25% in cell culture. We realized that an elevated level of transfection efficiency could increase the efficacy in inhibiting virus growth in cell culture. Despite this, we did not sort the transfected cells using flow cytometry for the studies in cell culture, because it was not feasible for us to do so in mice. To establish a more similar animal model, we used all the cells including untransfected and transfected cells for our ex vivo studies. To enhance the efficacy, multiple round injections for the formulation transfection during the treatment period are required in animal studies.
Cultured NIH3T3 cells are not specific for latency studies. If the viral infection is prior to the plasmid transfection, NIH3T3 cells are vulnerable to viruses because they do not have the same immune system as animals. In this case, TALENs can hardly protect the host cells from the viral infection, because the viral titer increases rapidly to about 105 pfu/mL within 1–3 days. However, if the plasmid transfection is prior to the viral infection, TALEN plasmid copy number might have increased and induced innate immune responses of host cells to secret cytokine or other factors to fight against invading viruses. Therefore, TALENs could inhibit virus growth by about 50–75% when the viral titer was 103–104 pfu/mL, and about 25% when the viral titer was about 105 pfu/mL.
In cell culture, the results for specific target and cleavage efficiency of TALEN plasmids reveal that some of the plasmids work well on MCMV M80/80.5 target sites. Specific TALEN plasmids also demonstrated their effects on the inhibition of virus growth, ranging from 25–75% depending on the viral titer. Although the decreasing level of viral titers was varied under different conditions, they had the same trend for the inhibition of virus growth. We also found that the higher the viral titer, the lower the effect of the TALEN plasmids on virus growth. Our findings indicated that the inhibition effect on MCMV is about 20–25% when the viral titer reaches the highest level (105). The reasons might be that the amino acid-nucleotide recognition frequency is not absolute (e.g., NI-A: 55%, NG-T: 50%, NN-G: 7%, HD-C: 69%) and the transfection efficiency is about 20–25% in cell culture.
The specificity, efficiency and biosafety of delivery tools are critical for drug delivery in animal studies and human clinical trials [
29]. Lipofectamine, one of the most common transfection reagents used in cell culture, is known to be toxic to animals. Our results have shown that the new transfection reagent NKS11 can work almost as well as lipofectamine for inhibiting virus lytic replication in cell culture, when TALEN plasmid transfection is prior to MCMV infection. Fortunately, NKS11 also proved to be nontoxic to Balb/c mice in the preliminary tests.
The viral
ie-1 DNA copies increased once Balb/c mice were infected with MCMV. An absence of plaques meant that there were no viruses during the lytic cycle. In latency, plaques were not detectable and the
ie-1 DNA copy number was low but detectable. The
ie-1 DNA copies increased once latency was reactivated. In
Table 4, the viral load significantly increased after reactivation in all five organs of mice if there was no treatment (Group 1), compared with the treatment groups (Group 2–5). After treatment, the latent MCMV was removed by TALENs, so the viral load was undetected or significantly decreased even though latency was reactivated using an immunosuppressive agent (Group 2–5). Additionally, if the TALENs are only targeting the reactivating viruses and not the latent pools, it is impossible for the specific treatment group (Group 2) to be all ND (not detected) in the
ie-1 DNA copy number assay for all five organs of mice. Thus, we consider that it is possible to remove latent viruses using this strategy.
In animal studies, the less specific and nonspecific treatment groups (Group 3, 4, 5) also worked well in reducing viral load, although they were less efficient than the specific treatment group (Group 2). This might be explained as follows. Firstly, during the multiple-round injection for TALEN plasmid treatment, recognition of foreign DNA in intracellular compartments or in the cytoplasm of host cells sends a signal of pathogenic invasion. In response, the innate immune DNA-sensing pathways start an antimicrobial type I interferon (IFN) (mainly IFNα and β) secretion to act against viruses. Acting in paracrine or autocrine models, IFNs stimulate intracellular and intercellular networks for regulating innate and acquired immunity in mice that are resistant to the viral infections [
30,
31,
32,
33,
34,
35]. However, there are no data that categorically show that the latent virus has been eradicated and that this is an IFN effect in our studies. Secondly, there are three possible mechanisms for TALEN plasmids to work on DNA, namely cleavage of target DNA, induction of DNA mutation, and inhibition of DNA transcription and translation [
17]. Thirdly, treatment is very complex in animals; it is also influenced by other factors such as the individual diversity of mice and the efficiency of tail vein injection.
Currently, it is impossible to eradicate latent CMV viruses in animals, although there are some effective drugs (e.g., Ganciclovir, Valganciclovir, and Foscarnet) for the treatment of active infections. Our data indicated that TALEN plasmids which could specifically target and cleave MCMV M80/80.5 ORF would effectively reduce the viral load in Balb/c mice, so that they resulted in the implicative decrease of ie-1 DNA copies level. The viral latent infection in humans is a major barrier for effective treatment and also a long-term risk to the host. Although the mechanisms for inhibiting MCMV are still poorly understood, our studies demonstrate that the removal of latent MCMV in animals is possible using TALENs.











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}