Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Generation of Env Mutant Virus Libraries
2.2. Resistance Profiling
2.3. Analysis of Deep Sequencing Data
2.4. Data Availability and Source Code
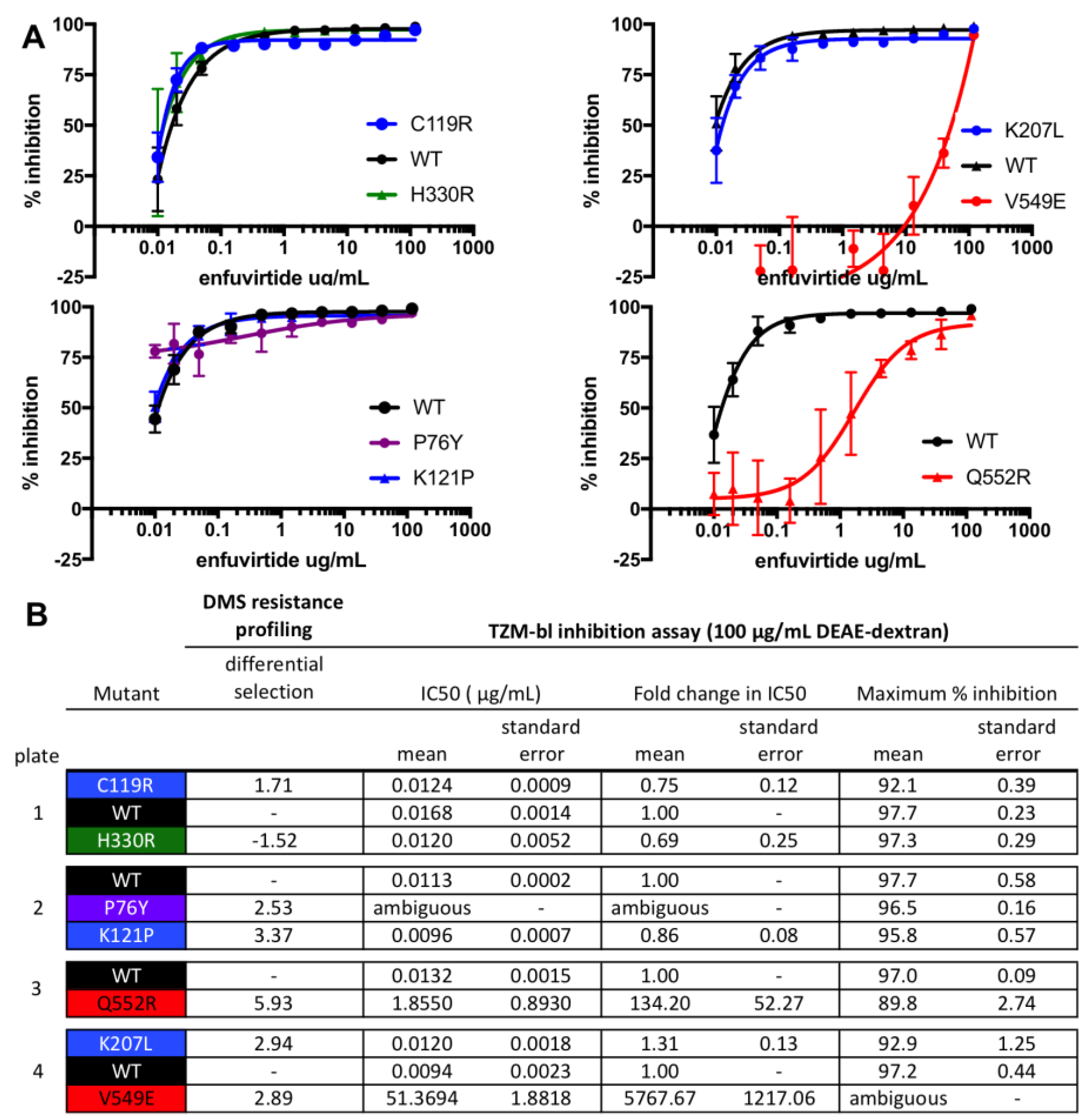
2.5. TZM-BL Inhibition Assays
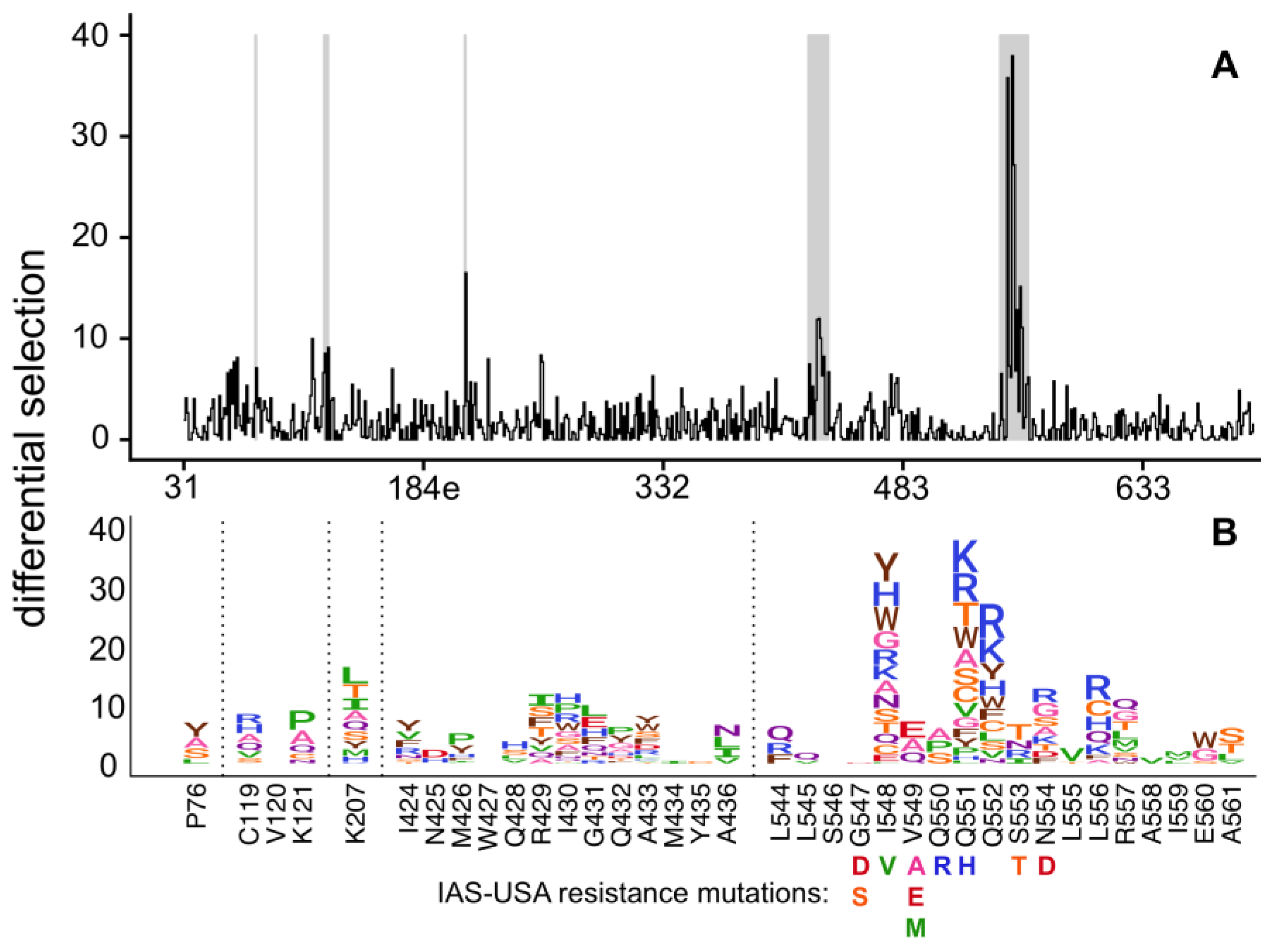
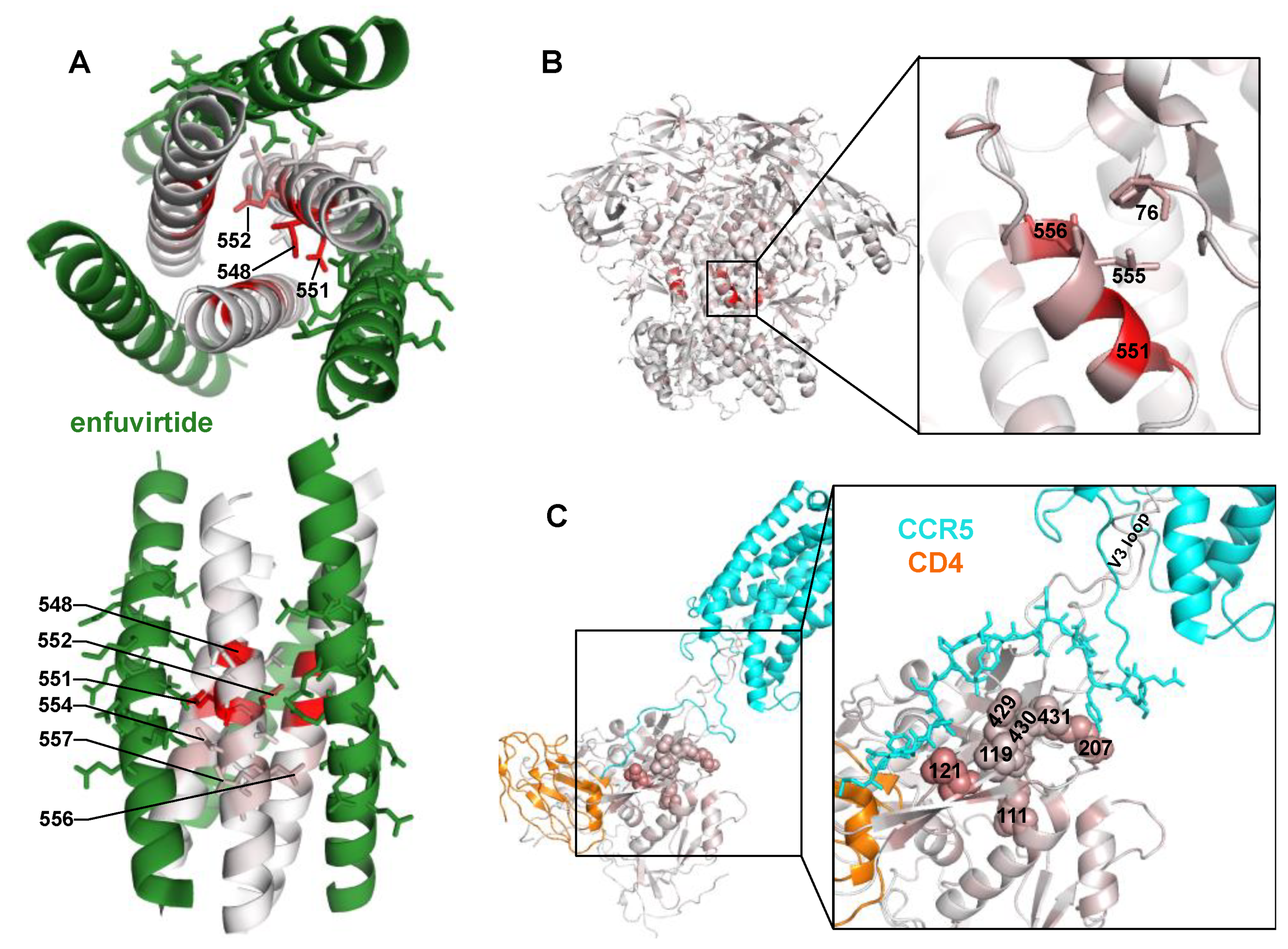
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wild, C.T.; Shugars, D.C.; Greenwell, T.K.; McDanal, C.B.; Matthews, T.J. Peptides corresponding to a predictive alpha-helical domain of human immunodeficiency virus type 1 gp41 are potent inhibitors of virus infection. Proc. Natl. Acad. Sci. USA 1994, 91, 9770–9774. [Google Scholar] [CrossRef]
- Jiang, S.; Lin, K.; Strick, N.; Neurath, A.R. HIV-1 inhibition by a peptide. Nature 1993, 365, 113. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Kim, P.S. HIV entry and its inhibition. Cell 1998, 93, 681–684. [Google Scholar] [CrossRef]
- Zhang, X.; Ding, X.; Zhu, Y.; Chong, H.; Cui, S.; He, J.; Wang, X.; He, Y. Structural and functional characterization of HIV-1 cell fusion inhibitor T20. AIDS 2019, 33, 1–11. [Google Scholar] [CrossRef] [PubMed]
- De Feo, C.J.; Weiss, C.D. Escape from human immunodeficiency virus type 1 (HIV-1) entry inhibitors. Viruses 2012, 4, 3859–3911. [Google Scholar] [CrossRef]
- Keller, P.W.; Morrison, O.; Vassell, R.; Weiss, C.D. HIV-1 gp41 Residues Modulate CD4-Induced Conformational Changes in the Envelope Glycoprotein and Evolution of a Relaxed Conformation of gp120. J. Virol. 2018, 92, e00583-18. [Google Scholar] [CrossRef] [PubMed]
- Reeves, J.D.; Gallo, S.A.; Ahmad, N.; Miamidian, J.L.; Harvey, P.E.; Sharron, M.; Pohlmann, S.; Sfakianos, J.N.; Derdeyn, C.A.; Blumenthal, R.; et al. Sensitivity of HIV-1 to entry inhibitors correlates with envelope/coreceptor affinity, receptor density, and fusion kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 16249–16254. [Google Scholar] [CrossRef] [Green Version]
- Khasnis, M.D.; Halkidis, K.; Bhardwaj, A.; Root, M.J. Receptor Activation of HIV-1 Env Leads to Asymmetric Exposure of the gp41 Trimer. PLoS Pathog. 2016, 12, e1006098. [Google Scholar] [CrossRef]
- Zhu, X.; Zhu, Y.; Ye, S.; Wang, Q.; Xu, W.; Su, S.; Sun, Z.; Yu, F.; Liu, Q.; Wang, C.; et al. Improved Pharmacological and Structural Properties of HIV Fusion Inhibitor AP3 over Enfuvirtide: Highlighting Advantages of Artificial Peptide Strategy. Sci. Rep. 2015, 5, 13028. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Xiao, Y.; Song, H.; Liang, Q.; Ju, D.; Chen, X.; Lu, H.; Jing, W.; Jiang, S.; Zhang, L. Design and Evaluation of Sifuvirtide, a Novel HIV-1 Fusion Inhibitor. J. Biol. Chem. 2008, 283, 11126–11134. [Google Scholar] [CrossRef] [Green Version]
- He, Y.; Cheng, J.; Li, J.; Qi, Z.; Lu, H.; Dong, M.; Jiang, S.; Dai, Q. Identification of a Critical Motif for the Human Immunodeficiency Virus Type 1 (HIV-1) gp41 Core Structure: Implications for Designing Novel Anti-HIV Fusion Inhibitors. J. Virol. 2008, 82, 6349–6358. [Google Scholar] [CrossRef]
- Dwyer, J.J.; Wilson, K.L.; Davison, D.K.; Freel, S.A.; Seedorff, J.E.; Wring, S.A.; Tvermoes, N.A.; Matthews, T.J.; Greenberg, M.L.; Delmedico, M.K. Design of helical, oligomeric HIV-1 fusion inhibitor peptides with potent activity against enfuvirtide-resistant virus. Proc. Natl. Acad. Sci. USA 2007, 104, 12772–12777. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Pu, J.; Su, S.; Hua, C.; Su, X.; Wang, Q.; Jiang, S.; Lu, L. Revisiting the mechanism of enfuvirtide and designing an analog with improved fusion inhibitory activity by targeting triple sites in gp41. AIDS 2019, 1. [Google Scholar] [CrossRef]
- Clotet, B.; Cooper, D. Clinical management of treatment-experienced, HIV- infected patients with the fusion inhibitor enfuvirtide: Consensus recommendations. AIDS 2004. [Google Scholar] [CrossRef]
- Miller, M.D.; Hazuda, D.J. HIV resistance to the fusion inhibitor enfuvirtide: Mechanisms and clinical implications. Drug Resist. Updat. 2004, 7, 89–95. [Google Scholar] [CrossRef]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef]
- Melby, T.; Sista, P.; DeMasi, R.; Kirkland, T.; Roberts, N.; Salgo, M.; Heilek-Snyder, G.; Cammack, N.; Matthews, T.J.; Greenberg, M.L. Characterization of envelope glycoprotein gp41 genotype and phenotypic susceptibility to enfuvirtide at baseline and on treatment in the phase III clinical trials TORO-1 and TORO-2. AIDS Res. Hum. Retrovir. 2006, 22, 375–385. [Google Scholar] [CrossRef]
- Su, C.; Melby, T.; DeMasi, R.; Ravindran, P.; Heilek-Snyder, G. Genotypic changes in human immunodeficiency virus type 1 envelope glycoproteins on treatment with the fusion inhibitor enfuvirtide and their influence on changes in drug susceptibility in vitro. J. Clin. Virol. 2006, 36, 249–257. [Google Scholar] [CrossRef]
- Poveda, E.; Rodés, B.; Lebel-Binay, S.; Faudon, J.L.; Jimenez, V.; Soriano, V. Dynamics of enfuvirtide resistance in HIV-infected patients during and after long-term enfuvirtide salvage therapy. J. Clin. Virol. 2005, 34, 295–301. [Google Scholar] [CrossRef]
- Greenberg, M.L.; Cammack, N. Resistance to enfuvirtide, the first HIV fusion inhibitor. J. Antimicrob. Chemother. 2004, 54, 333–340. [Google Scholar] [CrossRef] [Green Version]
- Marcelin, A.G.; Reynes, J.; Yerly, S.; Ktorza, N.; Segondy, M.; Piot, J.C.; Delfraissy, J.F.; Kaiser, L.; Perrin, L.; Katlama, C.; et al. Characterization of genotypic determinants in HR-1 and HR-2 gp41 domains in individuals with persistent HIV viraemia under T-20. AIDS 2004. [Google Scholar] [CrossRef]
- Sista, P.R.; Melby, T.; Davison, D.; Jin, L.; Mosier, S.; Mink, M.; Nelson, E.L.; DeMasi, R.; Cammack, N.; Salgo, M.P.; et al. Characterization of determinants of genotypic and phenotypic resistance to enfuvirtide in baseline and on-treatment HIV-1 isolates. AIDS 2004, 18, 1787–1794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mink, M.; Mosier, S.M.; Janumpalli, S.; Davison, D.; Jin, L.; Melby, T.; Sista, P.; Erickson, J.; Lambert, D.; Stanfield-Oakley, S.A.; et al. Impact of Human Immunodeficiency Virus Type 1 gp41 Amino Acid Substitutions Selected during Enfuvirtide Treatment on gp41 Binding and Antiviral Potency of Enfuvirtide In Vitro. J. Virol. 2005, 79, 12447–12454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rimsky, L.T.; Shugars, D.C.; Matthews, T.J. Determinants of Human Immunodeficiency Virus Type 1 Resistance to gp41-Derived Inhibitory Peptides. J. Virol. 1998, 72, 986–993. [Google Scholar]
- Xu, L.; Ratcliffe, D.; Workman, J.; Joall, A.; Myers, R.; Smit, E.; Cane, P.A.; Greenberg, M.L.; Pillay, D.; Pozniak, A.; et al. Emergence and Evolution of Enfuvirtide Resistance following Long-Term Therapy Involves Heptad Repeat 2 Mutations within Emergence and Evolution of Enfuvirtide Resistance following Long-Term Therapy Involves Heptad Repeat 2 Mutations within gp41. Antimicrob. Agents Chemother. 2005, 49, 1113–1119. [Google Scholar] [CrossRef] [PubMed]
- Derdeyn, C.A.; Decker, J.M.; Sfakianos, J.N.; Wu, X.; O’Brien, W.A.; Ratner, L.; Kappes, J.C.; Shaw, G.M.; Hunter, E. Sensitivity of Human Immunodeficiency Virus Type 1 to the Fusion Inhibitor T-20 Is Modulated by Coreceptor Specificity Defined by the V3 Loop of gp120. J. Virol. 2000, 74, 8358–8367. [Google Scholar] [CrossRef] [Green Version]
- Derdeyn, C.A.; Decker, J.M.; Sfakianos, J.N.; Zhang, Z.; O’Brien, W.A.; Ratner, L.; Shaw, G.M.; Hunter, E. Sensitivity of human immunodeficiency virus type 1 to fusion inhibitors targeted to the gp41 first heptad repeat involves distinct regions of gp41 and is consistently modulated by gp120 interactions with the coreceptor. J. Virol. 2001, 75, 8605–8614. [Google Scholar] [CrossRef]
- Reeves, J.D.; Miamidian, J.L.; Biscone, M.J.; Lee, F.; Ahmad, N.; Pierson, T.C.; Doms, R.W. Impact of Mutations in the Coreceptor Binding Site on Human Immunodeficiency Virus Type 1 Fusion, Infection, and Entry Inhibitor Sensitivity. J. Virol. 2004, 78, 5476–5485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roman, F.; Gonzalez, D.; Lambert, C.; Deroo, S.; Fischer, A.; Baurith, T.; Staub, T.; Boulmé, R.; Arendt, V.; Schneider, F.; et al. Uncommon mutations at residue positions critical for enfuvirtide (T-20) resistance in enfuvirtide-naive patients infected with subtype B and non-B HIV-1 strains. J. Acquir. Immune Defic. Syndr. 2003, 33, 134–139. [Google Scholar] [CrossRef]
- Yu, X.; Yuan, L.; Huang, Y.; Xu, W.; Fang, Z.; Liu, S.; Shao, Y.; Jiang, S.; Ma, L. Susceptibility of HIV-1 subtypes B′, CRF07_BC and CRF01_AE that are predominantly circulating in China to HIV-1 entry inhibitors. PLoS ONE 2011, 6, 1–8. [Google Scholar] [CrossRef]
- Cilliers, T.; Patience, T.; Pillay, C.; Papathanasopoulos, M.; Morris, L. Sensitivity of HIV Type 1 Subtype C Isolates to the Entry Inhibitor T-20. AIDS Res. Hum. Retrovir. 2004. [Google Scholar] [CrossRef]
- Taylor, B.S.; Sobieszczyk, M.E.; McCutchan, F.E.; Hammer, S.M. The Challenge of HIV-1 Subtype Diversity. N. Engl. J. Med. 2008, 358, 1590–1602. [Google Scholar] [CrossRef] [Green Version]
- D’Arrigo, R.; Ciccozzi, M.; Gori, C.; Montieri, S.; Aquaro, S.; Bellagamba, R.; Boumis, E.; Di Perri, G.; Pizzi, D.; Antinori, A.; et al. gp41 Sequence Variability in HIV Type 1 Non-B Subtypes Infected Patients Undergoing Enfuvirtide Pressure. AIDS Res. Hum. Retrovir. 2007, 23, 1296–1302. [Google Scholar] [CrossRef]
- Haddox, H.K.; Dingens, A.S.; Hilton, S.K.; Overbaugh, J.; Bloom, J.D. Mapping mutational effects along the evolutionary landscape of HIV envelope. Elife 2018, 7, e34420. [Google Scholar] [CrossRef]
- Haddox, H.K.; Dingens, A.S.; Bloom, J.D. Experimental Estimation of the Effects of All Amino-Acid Mutations to HIV’s Envelope Protein on Viral Replication in Cell Culture. PLoS Pathog. 2016, 12, e1006114. [Google Scholar] [CrossRef]
- Dingens, A.S.; Haddox, H.K.; Overbaugh, J.; Bloom, J.D. Comprehensive Mapping of HIV-1 Escape from a Broadly Neutralizing Antibody. Cell Host Microbe 2017, 21, 777–787.e4. [Google Scholar] [CrossRef]
- Dingens, A.S.; Arenz, D.; Weight, H.; Overbaugh, J.; Bloom, J.D. An Antigenic Atlas of HIV-1 Escape from Broadly Neutralizing Antibodies Distinguishes Functional and Structural Epitopes. Immunity 2019, 50, 520–532.e3. [Google Scholar] [CrossRef]
- Dingens, A.S.; Acharya, P.; Haddox, H.K.; Rawi, R.; Xu, K.; Chuang, G.-Y.; Wei, H.; Zhang, B.; Mascola, J.R.; Carragher, B.; et al. Complete functional mapping of infection- and vaccine-elicited antibodies against the fusion peptide of HIV. PLoS Pathog. 2018, 14, e1007159. [Google Scholar] [CrossRef]
- Bloom, J.D. Software for the analysis and visualization of deep mutational scanning data. BMC Bioinform. 2015, 16, 1–13. [Google Scholar] [CrossRef]
- Doud, M.B.; Hensley, S.E.; Bloom, J.D. Complete mapping of viral escape from neutralizing antibodies. PLoS Pathog. 2017, 13, e1006271. [Google Scholar] [CrossRef]
- Wensing, A.M.; Calvez, V.; Günthard, H.F.; Johnson, V.A.; Paredes, R.; Pillay, D.; Shafer, R.W.; Richman, D.D. 2017 Update of the Drug Resistance Mutations in HIV-1. Top. Antivir. Med. 2016, 24, 132–133. [Google Scholar]
- Shaik, M.; Peng, H.; Lu, J.; Rits-volloch, S.; Xu, C.; Liao, M.; Chen, B. Structural basis of coreceptor recognition by HIV-1 envelope spike. Nature 2018, 4. [Google Scholar] [CrossRef] [PubMed]
- Sampah, M.E.S.; Shen, L.; Jilek, B.L.; Siliciano, R.F. Dose-response curve slope is a missing dimension in the analysis of HIV-1 drug resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 7613–7618. [Google Scholar] [CrossRef]
- Herschhorn, A.; Gu, C.; Moraca, F.; Ma, X.; Farrell, M.; Smith, A.B.; Pancera, M.; Kwong, P.D.; Schön, A.; Freire, E.; et al. The β20–β21 of gp120 is a regulatory switch for HIV-1 Env conformational transitions. Nat. Commun. 2017, 8, 1049. [Google Scholar] [CrossRef]
- Alam, S.M.; Paleos, C.A.; Liao, H.-X.; Scearce, R.; Robinson, J.; Haynes, B.F.; Alam, S.M.; Paleos, C.A.; Liao, H.X.; Scearce, R.; et al. An inducible HIV type 1 gp41 HR-2 peptide-binding site on HIV type 1 envelope gp120. AIDS Res. Hum. Retrovir. 2004, 20, 836–845. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lu, H.; Niu, J.; Xu, Y.; Wu, S.; Jiang, S. Different from the HIV fusion inhibitor C34, the anti-HIV drug fuzeon (T-20) inhibits HIV-1 entry by targeting multiple sites in gp41 and gp120. J. Biol. Chem. 2005, 280, 11259–11273. [Google Scholar] [CrossRef]
- McCoy, L.E.; Falkowska, E.; Doores, K.J.; Le, K.; Sok, D.; van Gils, M.J.; Euler, Z.; Burger, J.A.; Seaman, M.S.; Sanders, R.W.; et al. Incomplete Neutralization and Deviation from Sigmoidal Neutralization Curves for HIV Broadly Neutralizing Monoclonal Antibodies. PLoS Pathog. 2015, 11, 1–19. [Google Scholar] [CrossRef]
- Melby, T.; DeSpirito, M.; DeMasi, R.; Heilek-Snyder, G.; Greenberg, M.L.; Graham, N. HIV-1 Coreceptor Use in Triple-Class Treatment–Experienced Patients: Baseline Prevalence, Correlates, and Relationship to Enfuvirtide Response. J. Infect. Dis. 2006, 194, 238–246. [Google Scholar] [CrossRef]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dingens, A.S.; Arenz, D.; Overbaugh, J.; Bloom, J.D. Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide. Viruses 2019, 11, 439. https://doi.org/10.3390/v11050439
Dingens AS, Arenz D, Overbaugh J, Bloom JD. Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide. Viruses. 2019; 11(5):439. https://doi.org/10.3390/v11050439
Chicago/Turabian StyleDingens, Adam S., Dana Arenz, Julie Overbaugh, and Jesse D. Bloom. 2019. "Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide" Viruses 11, no. 5: 439. https://doi.org/10.3390/v11050439
APA StyleDingens, A. S., Arenz, D., Overbaugh, J., & Bloom, J. D. (2019). Massively Parallel Profiling of HIV-1 Resistance to the Fusion Inhibitor Enfuvirtide. Viruses, 11(5), 439. https://doi.org/10.3390/v11050439