Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Suppresses Inflammation and Bacterial Clearance during Influenza-Bacterial Super-Infection

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Experimental Infections and Animal Treatments
2.3. Measurement of Lung Inflammation
2.4. Flow Cytometry
2.5. Statistical Analysis
2.6. Human Data Source
2.7. Definition of Variables
2.8. Human Data Statistical Analysis
3. Results
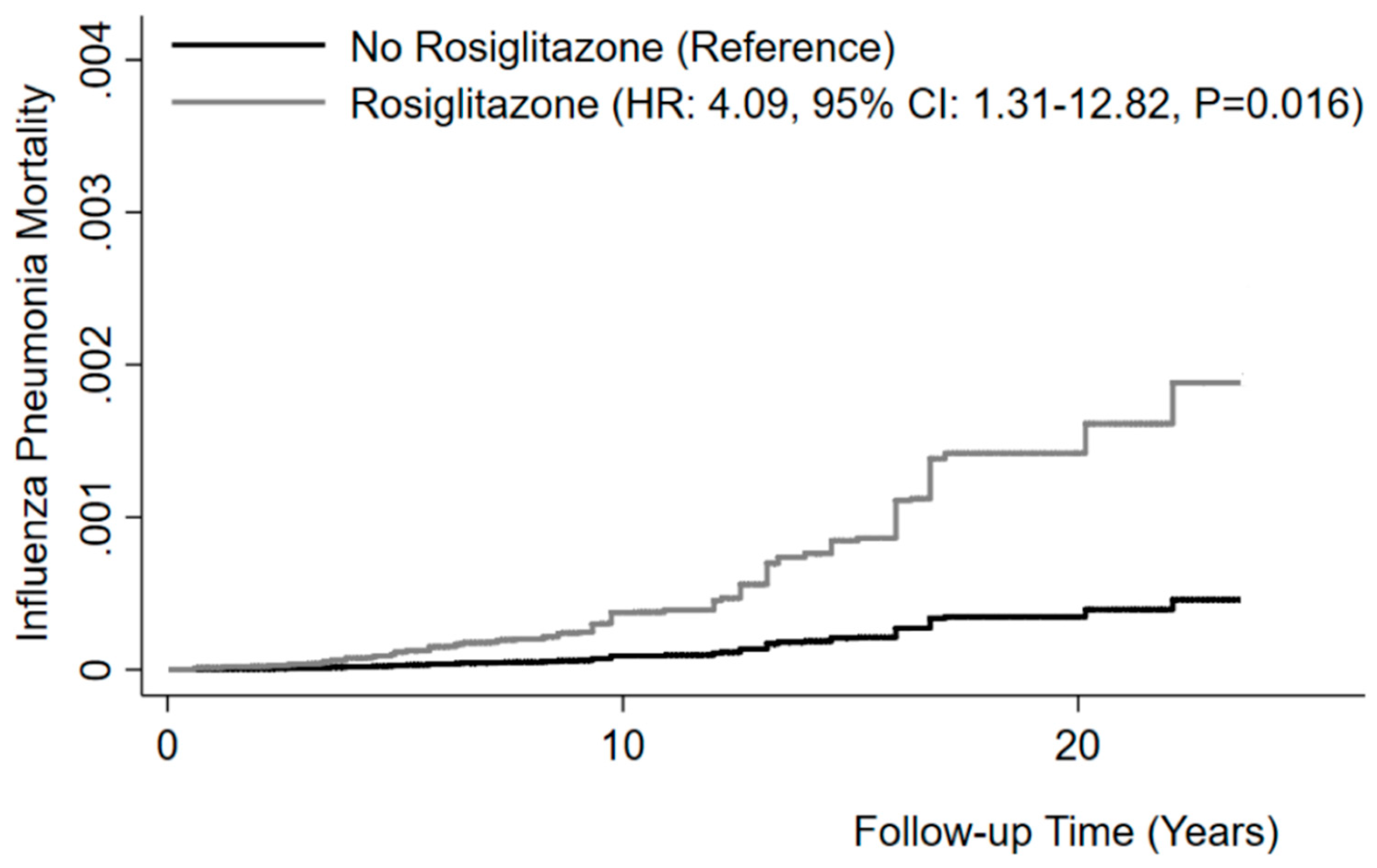
3.1. Influenza/Pneumonia Mortality Is Increased in Diabetic Patients in Response to Rosiglitazone Treatment
3.2. PPARγ Expression Is Suppressed in Response to Influenza Infection
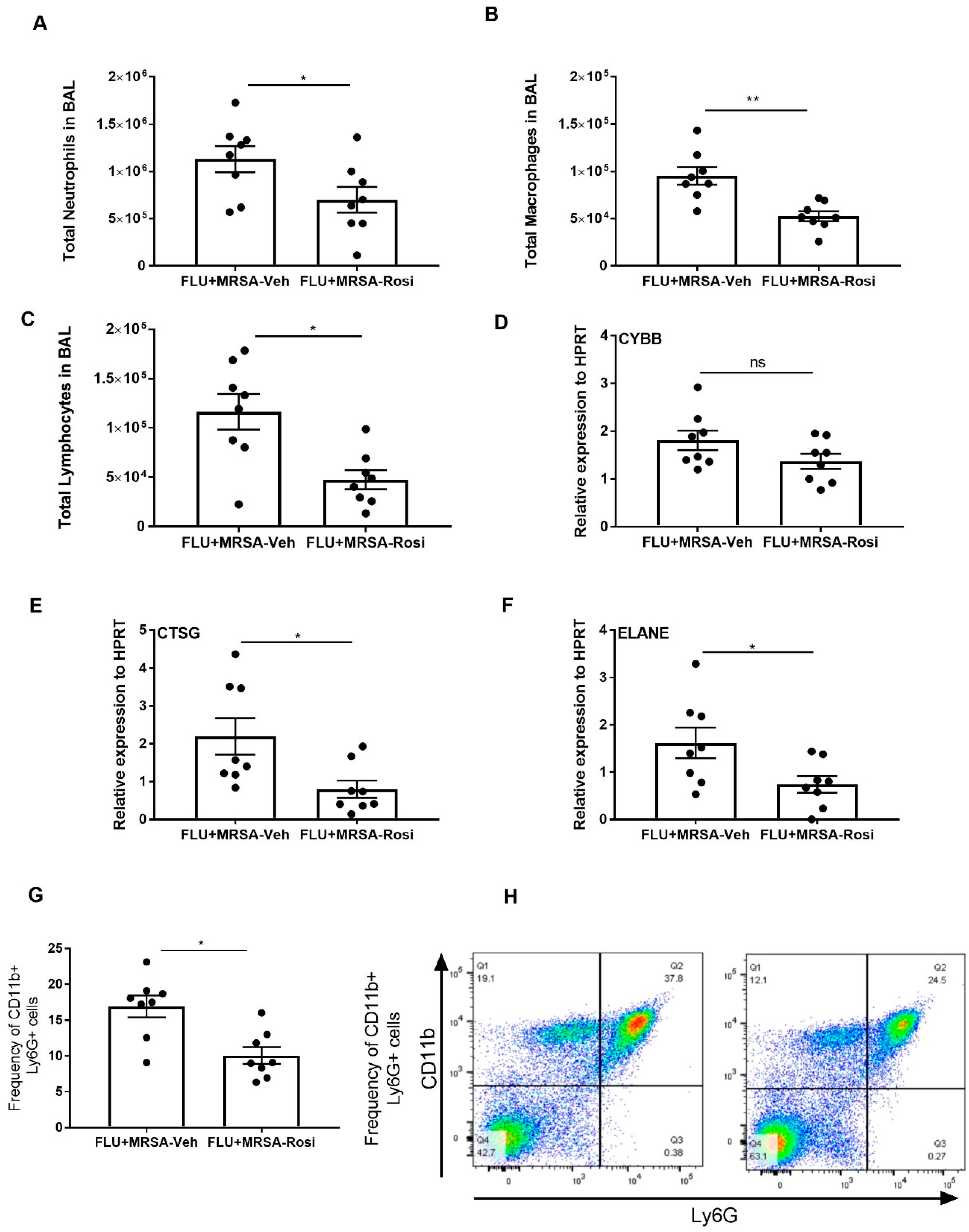
3.3. Rosiglitazone Treatment Suppresses Influenza Viral Burden and Reduces Inflammatory Cells in BAL during Influenza Infection
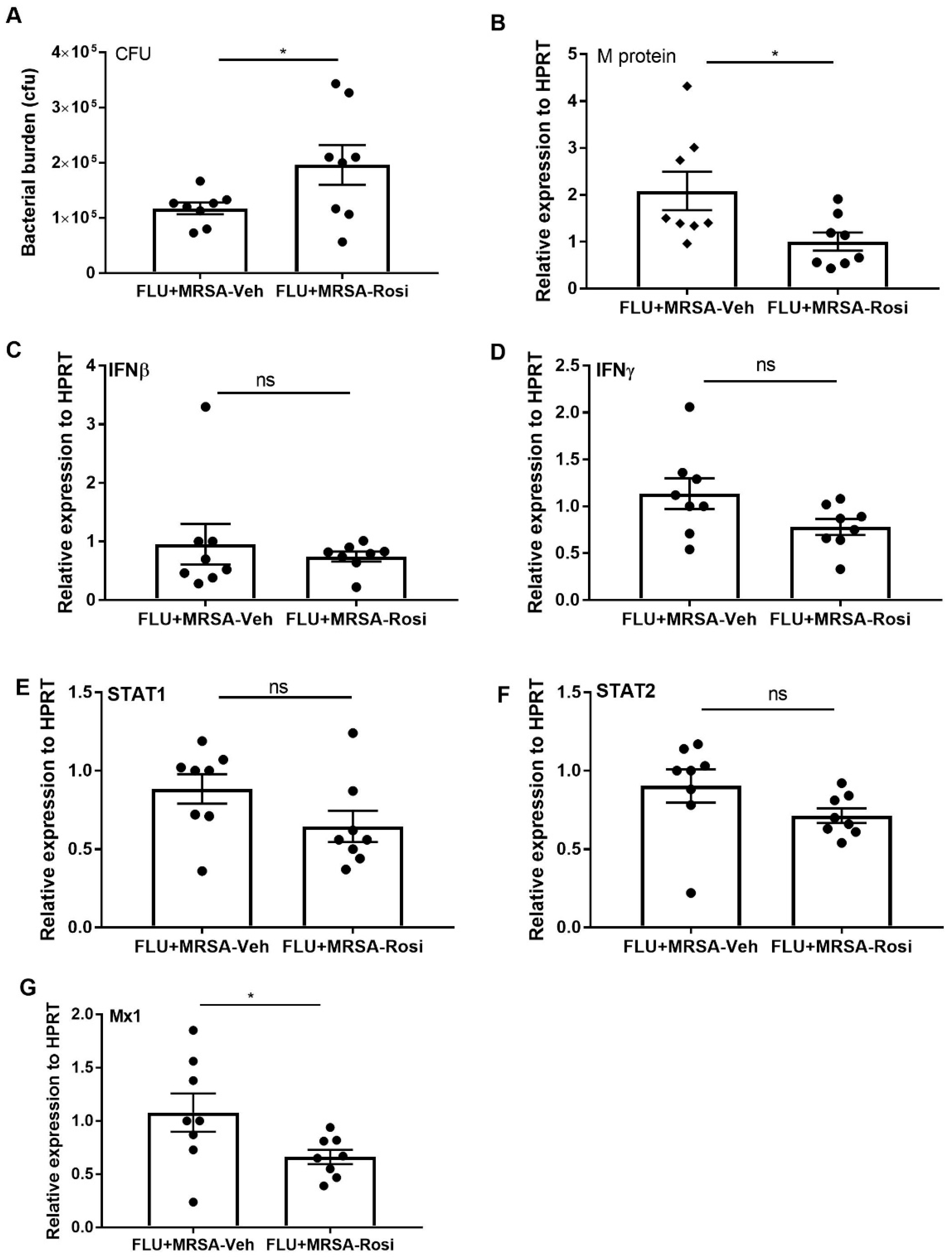
3.4. PPARγ Agonist Treatment Compromises Bacterial Clearance during Influenza-Bacterial Super-Infection
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Iwasaki, A.; Pillai, P.S. Innate immunity to influenza virus infection. Nat. Rev. Immunol. 2014, 14, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Pulendran, B.; Maddur, M.S. Innate immune sensing and response to influenza. Curr. Top. Microbiol. Immunol. 2015, 386, 23–71. [Google Scholar] [PubMed]
- Tripathi, S.; White, M.R.; Hartshorn, K.L. The amazing innate immune response to influenza A virus infection. Innate Immun. 2015, 21, 73–98. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Metcalf, J.P.; Wu, W. Innate immune response to influenza virus. Curr. Opin. Infect. Dis. 2011, 24, 235–240. [Google Scholar] [CrossRef] [PubMed]
- McGill, J.; Heusel, J.W.; Legge, K.L. Innate immune control and regulation of influenza virus infections. J. Leukoc. Biol. 2009, 86, 803–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durbin, J.E.; Fernandez-Sesma, A.; Lee, C.K.; Rao, T.D.; Frey, A.B.; Moran, T.M.; Vukmanovic, S.; Garcia-Sastre, A.; Levy, D.E. Type I IFN modulates innate and specific antiviral immunity. J. Immunol. 2000, 164, 4220–4228. [Google Scholar] [CrossRef] [PubMed]
- Durbin, R.K.; Kotenko, S.V.; Durbin, J.E. Interferon induction and function at the mucosal surface. Immunol. Rev. 2013, 255, 25–39. [Google Scholar] [CrossRef]
- Verhelst, J.; Hulpiau, P.; Saelens, X. Mx proteins: Antiviral gatekeepers that restrain the uninvited. Microbiol. Mol. Biol. Rev. 2013, 77, 551–566. [Google Scholar] [CrossRef]
- Haller, O.; Staeheli, P.; Schwemmle, M.; Kochs, G. Mx GTPases: Dynamin-like antiviral machines of innate immunity. Trends Microbiol. 2015, 23, 154–163. [Google Scholar] [CrossRef]
- Korkmaz, B.; Horwitz, M.S.; Jenne, D.E.; Gauthier, F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol. Rev. 2010, 62, 726–759. [Google Scholar] [CrossRef]
- Korkmaz, B.; Moreau, T.; Gauthier, F. Neutrophil elastase, proteinase 3 and cathepsin G: Physicochemical properties, activity and physiopathological functions. Biochimie 2008, 90, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Damjanovic, D.; Small, C.L.; Jeyanathan, M.; McCormick, S.; Xing, Z. Immunopathology in influenza virus infection: Uncoupling the friend from foe. Clin. Immunol. 2012, 144, 57–69. [Google Scholar] [CrossRef]
- Finelli, L.; Fiore, A.; Dhara, R.; Brammer, L.; Shay, D.K.; Kamimoto, L.; Fry, A.; Hageman, J.; Gorwitz, R.; Bresee, J.; et al. Influenza-associated pediatric mortality in the United States: Increase of Staphylococcus aureus coinfection. Pediatrics 2008, 122, 805–811. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.W.; Geyer, S.M.; Guo, C.Y.; Panoskaltsis-Mortari, A.; Jouvet, P.; Ferdinands, J.; Shay, D.K.; Nateri, J.; Greathouse, K.; Sullivan, R.; et al. Innate immune function and mortality in critically ill children with influenza: A multicenter study. Crit. Care Med. 2013, 41, 224–236. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, E.; Kollef, M.H.; Nathwani, D. Pneumonia caused by methicillin-resistant Staphylococcus aureus. Clin. Infect. Dis. 2008, 46 (Suppl. 5), S378–S385. [Google Scholar] [CrossRef]
- Ghoneim, H.E.; Thomas, P.G.; McCullers, J.A. Depletion of alveolar macrophages during influenza infection facilitates bacterial superinfections. J. Immunol. 2013, 191, 1250–1259. [Google Scholar] [CrossRef] [PubMed]
- Damjanovic, D.; Lai, R.; Jeyanathan, M.; Hogaboam, C.M.; Xing, Z. Marked improvement of severe lung immunopathology by influenza-associated pneumococcal superinfection requires the control of both bacterial replication and host immune responses. Am. J. Pathol. 2013, 183, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Metzger, D.W. Inhibition of pulmonary antibacterial defense by interferon-gamma during recovery from influenza infection. Nat. Med. 2008, 14, 558–564. [Google Scholar] [CrossRef] [PubMed]
- Small, C.L.; Shaler, C.R.; McCormick, S.; Jeyanathan, M.; Damjanovic, D.; Brown, E.G.; Arck, P.; Jordana, M.; Kaushic, C.; Ashkar, A.A.; et al. Influenza infection leads to increased susceptibility to subsequent bacterial superinfection by impairing NK cell responses in the lung. J. Immunol. 2010, 184, 2048–2056. [Google Scholar] [CrossRef] [PubMed]
- Narayana Moorthy, A.; Narasaraju, T.; Rai, P.; Perumalsamy, R.; Tan, K.B.; Wang, S.; Engelward, B.; Chow, V.T. In vivo and in vitro studies on the roles of neutrophil extracellular traps during secondary pneumococcal pneumonia after primary pulmonary influenza infection. Front. Immunol. 2013, 4, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Moltedo, B.; Moran, T.M. Type I interferon induction during influenza virus infection increases susceptibility to secondary Streptococcus pneumoniae infection by negative regulation of gammadelta T cells. J. Virol. 2012, 86, 12304–12312. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Davis, K.M.; Weiser, J.N. Synergistic stimulation of type I interferons during influenza virus coinfection promotes Streptococcus pneumoniae colonization in mice. J. Clin. Investig. 2011, 121, 3657–3665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudva, A.; Scheller, E.V.; Robinson, K.M.; Crowe, C.R.; Choi, S.M.; Slight, S.R.; Khader, S.A.; Dubin, P.J.; Enelow, R.I.; Kolls, J.K.; et al. Influenza A inhibits Th17-mediated host defense against bacterial pneumonia in mice. J. Immunol. 2011, 186, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Gopal, R.; Manni, M.L.; McHugh, K.J.; Mandalapu, S.; Robinson, K.M.; Alcorn, J.F. STAT1 Is Required for Suppression of Type 17 Immunity during Influenza and Bacterial Superinfection. ImmunoHorizons 2017, 1, 81–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, K.M.; McHugh, K.J.; Mandalapu, S.; Clay, M.E.; Lee, B.; Scheller, E.V.; Enelow, R.I.; Chan, Y.R.; Kolls, J.K.; Alcorn, J.F. Influenza A virus exacerbates Staphylococcus aureus pneumonia in mice by attenuating antimicrobial peptide production. J. Infect. Dis. 2014, 209, 865–875. [Google Scholar] [CrossRef]
- Gopal, R.; Lee, B.; McHugh, K.J.; Rich, H.E.; Ramanan, K.; Mandalapu, S.; Clay, M.E.; Seger, P.J.; Enelow, R.I.; Manni, M.L.; et al. STAT2 Signaling Regulates Macrophage Phenotype During Influenza and Bacterial Super-Infection. Front. Immunol. 2018, 9, 2151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinetti, G.; Fruchart, J.C.; Staels, B. Peroxisome proliferator-activated receptors: New targets for the pharmacological modulation of macrophage gene expression and function. Curr. Opin. Lipidol. 2003, 14, 459–468. [Google Scholar] [CrossRef]
- Moraes, L.A.; Piqueras, L.; Bishop-Bailey, D. Peroxisome proliferator-activated receptors and inflammation. Pharmacol. Ther. 2006, 110, 371–385. [Google Scholar] [CrossRef]
- Malur, A.; McCoy, A.J.; Arce, S.; Barna, B.P.; Kavuru, M.S.; Malur, A.G.; Thomassen, M.J. Deletion of PPAR gamma in alveolar macrophages is associated with a Th-1 pulmonary inflammatory response. J. Immunol. 2009, 182, 5816–5822. [Google Scholar] [CrossRef]
- Kliewer, S.A.; Lenhard, J.M.; Willson, T.M.; Patel, I.; Morris, D.C.; Lehmann, J.M. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 1995, 83, 813–819. [Google Scholar] [CrossRef]
- Willson, T.M.; Brown, P.J.; Sternbach, D.D.; Henke, B.R. The PPARs: From orphan receptors to drug discovery. J. Med. Chem. 2000, 43, 527–550. [Google Scholar] [CrossRef] [PubMed]
- Purnell, J.Q.; Hirsch, I.B. New oral therapies for type 2 diabetes. Am. Fam. Physician 1997, 56, 1835–1842. [Google Scholar] [PubMed]
- Mudaliar, S.; Henry, R.R. New oral therapies for type 2 diabetes mellitus: The glitazones or insulin sensitizers. Annu. Rev. Med. 2001, 52, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Yki-Jarvinen, H. Combination therapies with insulin in type 2 diabetes. Diabetes Care 2001, 24, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Birrell, M.A.; Patel, H.J.; McCluskie, K.; Wong, S.; Leonard, T.; Yacoub, M.H.; Belvisi, M.G. PPAR-gamma agonists as therapy for diseases involving airway neutrophilia. Eur. Respir. J. 2004, 24, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Kaundal, R.K.; Sharma, S.S. Amelioration of pulmonary dysfunction and neutrophilic inflammation by PPAR gamma agonist in LPS-exposed guinea pigs. Pulm Pharm. Ther. 2009, 22, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Mendy, A.; Gopal, R.; Alcorn, J.F.; Forno, E. Reduced mortality from lower respiratory tract disease in adult diabetic patients treated with metformin. Respirology 2019. [CrossRef]
- Robinson, K.M.; Choi, S.M.; McHugh, K.J.; Mandalapu, S.; Enelow, R.I.; Kolls, J.K.; Alcorn, J.F. Influenza A exacerbates Staphylococcus aureus pneumonia by attenuating IL-1beta production in mice. J. Immunol. 2013, 191, 5153–5159. [Google Scholar] [CrossRef]
- Welch, J.S.; Ricote, M.; Akiyama, T.E.; Gonzalez, F.J.; Glass, C.K. PPARgamma and PPARdelta negatively regulate specific subsets of lipopolysaccharide and IFN-gamma target genes in macrophages. Proc. Natl. Acad. Sci. USA 2003, 100, 6712–6717. [Google Scholar] [CrossRef]
- Guntur, K.V.; Guilherme, A.; Xue, L.; Chawla, A.; Czech, M.P. Map4k4 negatively regulates peroxisome proliferator-activated receptor (PPAR) gamma protein translation by suppressing the mammalian target of rapamycin (mTOR) signaling pathway in cultured adipocytes. J. Biol. Chem. 2010, 285, 6595–6603. [Google Scholar] [CrossRef]
- Song, Y.; Lin, Q.; Zheng, J.; Zhu, X.; Yang, S. [PPAR-gamma agonist inhibits the expressions of chemokines induced by IFN-gamma and TNF-alpha in renal tubular epithelial cells]. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2014, 30, 673–676. [Google Scholar] [PubMed]
- Genovese, T.; Cuzzocrea, S.; Di Paola, R.; Mazzon, E.; Mastruzzo, C.; Catalano, P.; Sortino, M.; Crimi, N.; Caputi, A.P.; Thiemermann, C.; et al. Effect of rosiglitazone and 15-deoxy-Delta12,14-prostaglandin J2 on bleomycin-induced lung injury. Eur. Respir. J. 2005, 25, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Milam, J.E.; Keshamouni, V.G.; Phan, S.H.; Hu, B.; Gangireddy, S.R.; Hogaboam, C.M.; Standiford, T.J.; Thannickal, V.J.; Reddy, R.C. PPAR-gamma agonists inhibit profibrotic phenotypes in human lung fibroblasts and bleomycin-induced pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2008, 294, L891–L901. [Google Scholar] [CrossRef] [PubMed]
- Ottolini, M.; Blanco, J.; Porter, D.; Peterson, L.; Curtis, S.; Prince, G. Combination anti-inflammatory and antiviral therapy of influenza in a cotton rat model. Pediatr Pulmonol 2003, 36, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Rainsford, K.D. Influenza (“Bird Flu”), inflammation and anti-inflammatory/analgesic drugs. Inflammopharmacology 2006, 14, 2–9. [Google Scholar] [CrossRef]
- Barish, G.D.; Downes, M.; Alaynick, W.A.; Yu, R.T.; Ocampo, C.B.; Bookout, A.L.; Mangelsdorf, D.J.; Evans, R.M. A Nuclear Receptor Atlas: Macrophage activation. Mol. Endocrinol. 2005, 19, 2466–2477. [Google Scholar] [CrossRef]
- Waite, K.J.; Floyd, Z.E.; Arbour-Reily, P.; Stephens, J.M. Interferon-gamma-induced regulation of peroxisome proliferator-activated receptor gamma and STATs in adipocytes. J. Biol. Chem. 2001, 276, 7062–7068. [Google Scholar] [CrossRef]
- Gopal, R.; Alcorn, J.F. IFNγ Suppresses PPARγ Expression during Influenza and Influenza-MRSA Super-Infection in Mice; Department of Pediatrics, UPMC Children’s Hospital of Pittsburgh: Pittsburgh, PA, USA, 2019. [Google Scholar]
- Cloutier, A.; Marois, I.; Cloutier, D.; Verreault, C.; Cantin, A.M.; Richter, M.V. The prostanoid 15-deoxy-Delta12,14-prostaglandin-j2 reduces lung inflammation and protects mice against lethal influenza infection. J. Infect. Dis. 2012, 205, 621–630. [Google Scholar] [CrossRef]
- Cunard, R.; Eto, Y.; Muljadi, J.T.; Glass, C.K.; Kelly, C.J.; Ricote, M. Repression of IFN-gamma expression by peroxisome proliferator-activated receptor gamma. J. Immunol. 2004, 172, 7530–7536. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, L.; Zhang, M.; Wang, P.; Zhang, L.; Yuan, C.; Qi, J.; Qiao, Y.; Kuo, P.C.; Gao, C. Peroxisome proliferator-activated receptor gamma negatively regulates IFN-beta production in Toll-like receptor (TLR) 3- and TLR4-stimulated macrophages by preventing interferon regulatory factor 3 binding to the IFN-beta promoter. J. Biol. Chem. 2011, 286, 5519–5528. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Appel, S.; Mirakaj, V.; Bringmann, A.; Weck, M.M.; Grunebach, F.; Brossart, P. PPAR-gamma agonists inhibit toll-like receptor-mediated activation of dendritic cells via the MAP kinase and NF-kappaB pathways. Blood 2005, 106, 3888–3894. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.P.; Liu, Y.H.; Chen, J.; Song, T.; You, Y.; Tang, Z.Y.; Li, Y.J.; Zhang, G.G. Rosiglitazone attenuates NF-kappaB-dependent ICAM-1 and TNF-alpha production caused by homocysteine via inhibiting ERK1/2/p38MAPK activation. Biochem. Biophys. Res. Commun. 2007, 360, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Ting, A.T.; Seed, B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998, 391, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Reddy, R.C. Immunomodulatory role of PPAR-gamma in alveolar macrophages. J. Investig. Med. 2008, 56, 522–527. [Google Scholar] [CrossRef]
- Thurlow, L.R.; Joshi, G.S.; Richardson, A.R. Peroxisome Proliferator-Activated Receptor gamma Is Essential for the Resolution of Staphylococcus aureus Skin Infections. Cell Host Microbe 2018, 24, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Gopal, R.; Alcorn, J.F. Rosiglitazone Treatment Decreases the Expression of IFNγ, CXCL9 and CXCL10 in MRSA Infection in Mice; Department of Pediatrics, UPMC Children’s Hospital of Pittsburgh: Pittsburgh, PA, USA, 2019. [Google Scholar]
- Zhang, L.J.; Guerrero-Juarez, C.F.; Hata, T.; Bapat, S.P.; Ramos, R.; Plikus, M.V.; Gallo, R.L. Innate immunity. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 2015, 347, 67–71. [Google Scholar] [CrossRef]
- Bauer, C.M.; Zavitz, C.C.; Botelho, F.M.; Lambert, K.N.; Brown, E.G.; Mossman, K.L.; Taylor, J.D.; Stampfli, M.R. Treating viral exacerbations of chronic obstructive pulmonary disease: Insights from a mouse model of cigarette smoke and H1N1 influenza infection. PLoS ONE 2010, 5, e13251. [Google Scholar] [CrossRef]
- Robinson, K.M.; Ramanan, K.; Clay, M.E.; McHugh, K.J.; Rich, H.E.; Alcorn, J.F. Novel protective mechanism for interleukin-33 at the mucosal barrier during influenza-associated bacterial superinfection. Mucosal Immunol. 2018, 11, 199–208. [Google Scholar] [CrossRef]
- Morissette, M.C.; Shen, P.; Thayaparan, D.; Stampfli, M.R. Impacts of peroxisome proliferator-activated receptor-gamma activation on cigarette smoke-induced exacerbated response to bacteria. Eur. Respir. J. 2015, 45, 191–200. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Participants | Rosiglitazone | |||
|---|---|---|---|---|
| Characteristics | No | Yes | P | |
| Prevalence, % | 100 | 95.4 | 4.6 | |
| Age groups, % | 0.27 | |||
| 20–39 years | 10.9 | 11.0 | 8.9 | |
| 40–59 years | 38.6 | 38.3 | 44.5 | |
| ≥ 60 years | 50.5 | 50.7 | 46.5 | |
| Gender, % | 0.80 | |||
| Men | 48.3 | 48.3 | 47.4 | |
| Women | 51.7 | 51.7 | 52.6 | |
| Race/ethnicity, % | 0.57 | |||
| Non-Hispanic Whites | 64.0 | 63.8 | 67.0 | |
| Non-Hispanic Blacks | 16.9 | 16.9 | 15.8 | |
| Mexican-Americans | 12.3 | 12.3 | 12.8 | |
| Other | 6.9 | 7.0 | 4.4 | |
| Poverty-income ratio, % a | 0.08 | |||
| ≤1 | 16.8 | 16.9 | 15.4 | |
| 1< & ≤ 3 | 45.7 | 46.0 | 39.1 | |
| >3 | 37.5 | 37.1 | 45.5 | |
| Cigarettes smoking, % b | 53.7 | 54.0 | 48.6 | 0.14 |
| Asthma or COPD, % c | 17.1 | 17.0 | 19.0 | 0.52 |
| Insulin treatment, % | 22.2 | 21.8 | 29.8 | 0.03 |
| Other oral antidiabetic drugs, % | 27.3 | 39.0 | 65.1 | <0.001 |
| Rate of mortality from Influenza and pneumonia (1,000 person-years) | 0.8 | 0.7 | 2.0 | 0.02 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gopal, R.; Mendy, A.; Marinelli, M.A.; Richwalls, L.J.; Seger, P.J.; Patel, S.; McHugh, K.J.; Rich, H.E.; Grousd, J.A.; Forno, E.; et al. Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Suppresses Inflammation and Bacterial Clearance during Influenza-Bacterial Super-Infection. Viruses 2019, 11, 505. https://doi.org/10.3390/v11060505
Gopal R, Mendy A, Marinelli MA, Richwalls LJ, Seger PJ, Patel S, McHugh KJ, Rich HE, Grousd JA, Forno E, et al. Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Suppresses Inflammation and Bacterial Clearance during Influenza-Bacterial Super-Infection. Viruses. 2019; 11(6):505. https://doi.org/10.3390/v11060505
Chicago/Turabian StyleGopal, Radha, Angelico Mendy, Michael A. Marinelli, Lacee J. Richwalls, Philip J. Seger, Shivani Patel, Kevin J. McHugh, Helen E. Rich, Jennifer A. Grousd, Erick Forno, and et al. 2019. "Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Suppresses Inflammation and Bacterial Clearance during Influenza-Bacterial Super-Infection" Viruses 11, no. 6: 505. https://doi.org/10.3390/v11060505
APA StyleGopal, R., Mendy, A., Marinelli, M. A., Richwalls, L. J., Seger, P. J., Patel, S., McHugh, K. J., Rich, H. E., Grousd, J. A., Forno, E., & Alcorn, J. F. (2019). Peroxisome Proliferator-Activated Receptor Gamma (PPARγ) Suppresses Inflammation and Bacterial Clearance during Influenza-Bacterial Super-Infection. Viruses, 11(6), 505. https://doi.org/10.3390/v11060505