Mammalian orthoreovirus Infection is Enhanced in Cells Pre-Treated with Sodium Arsenite


 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Reagents
2.2. Viruses
2.3. Infections
2.4. SG Induction
2.5. Antibodies
2.6. Immunofluorescence
2.7. Immunoblot Assay
2.8. Plaque Assay
3. Results
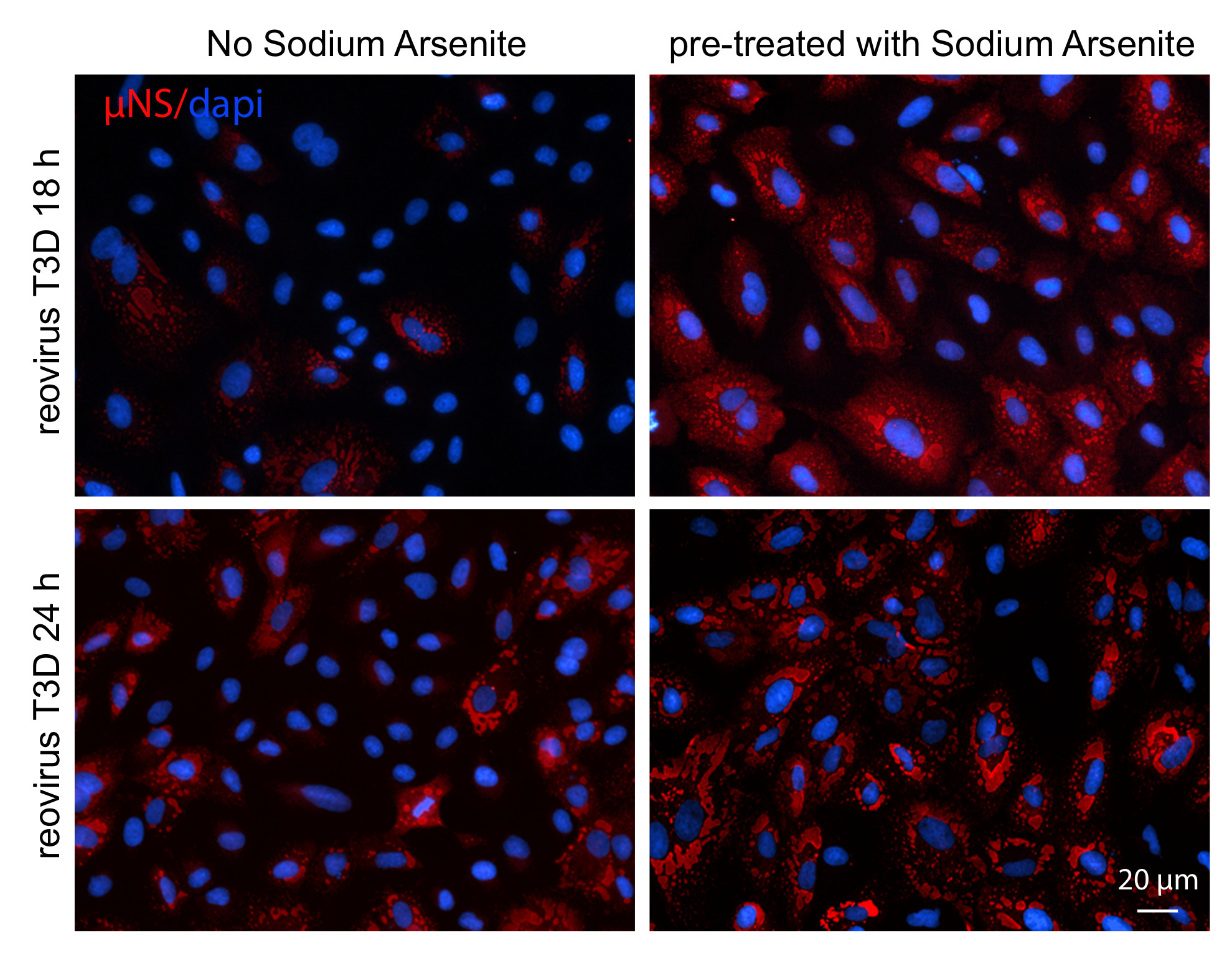
3.1. Infection with Reovirus Induces SG Formation
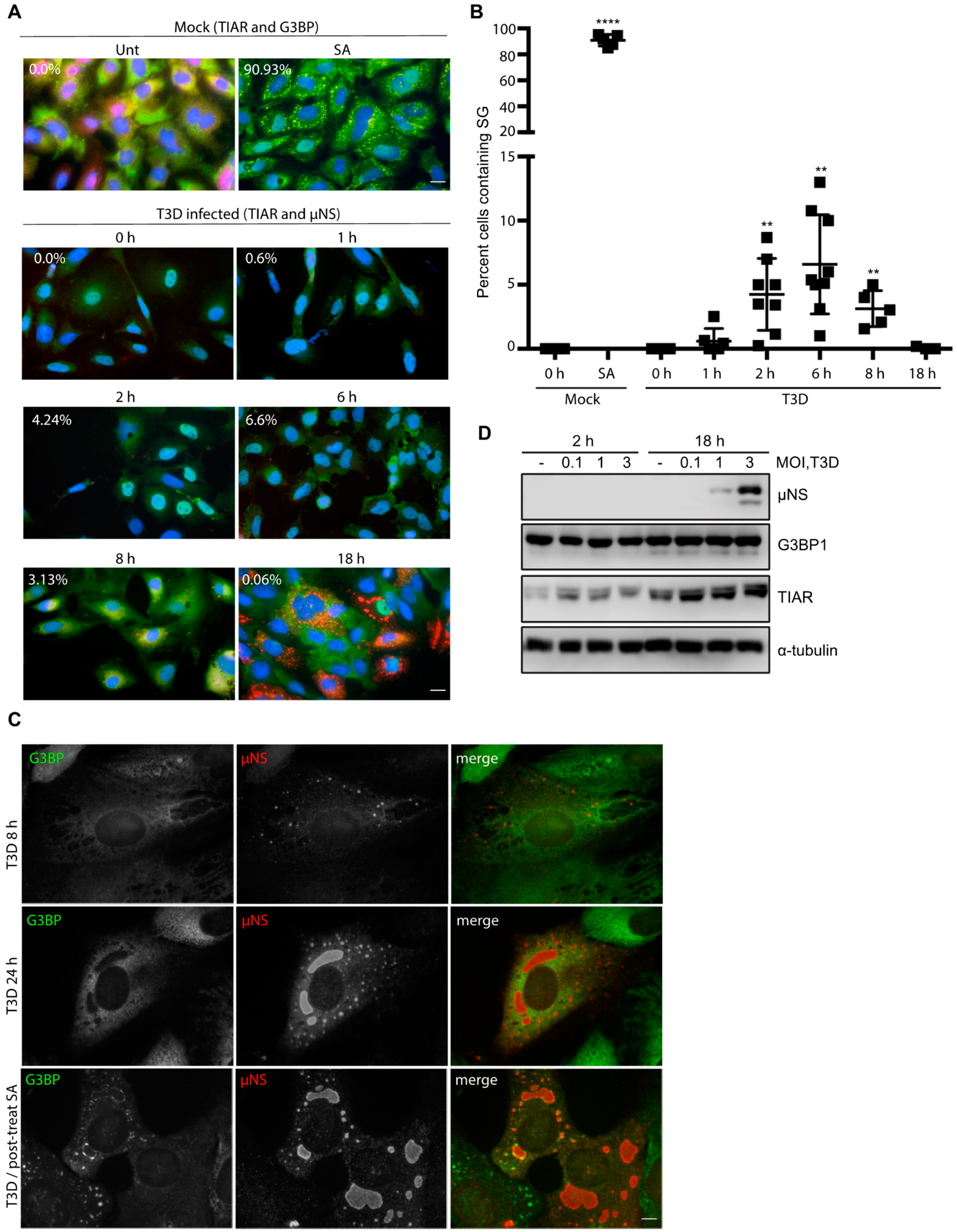
3.2. Pre-Treatment with 0.5 mM SA Enhances Reovirus Infectivity
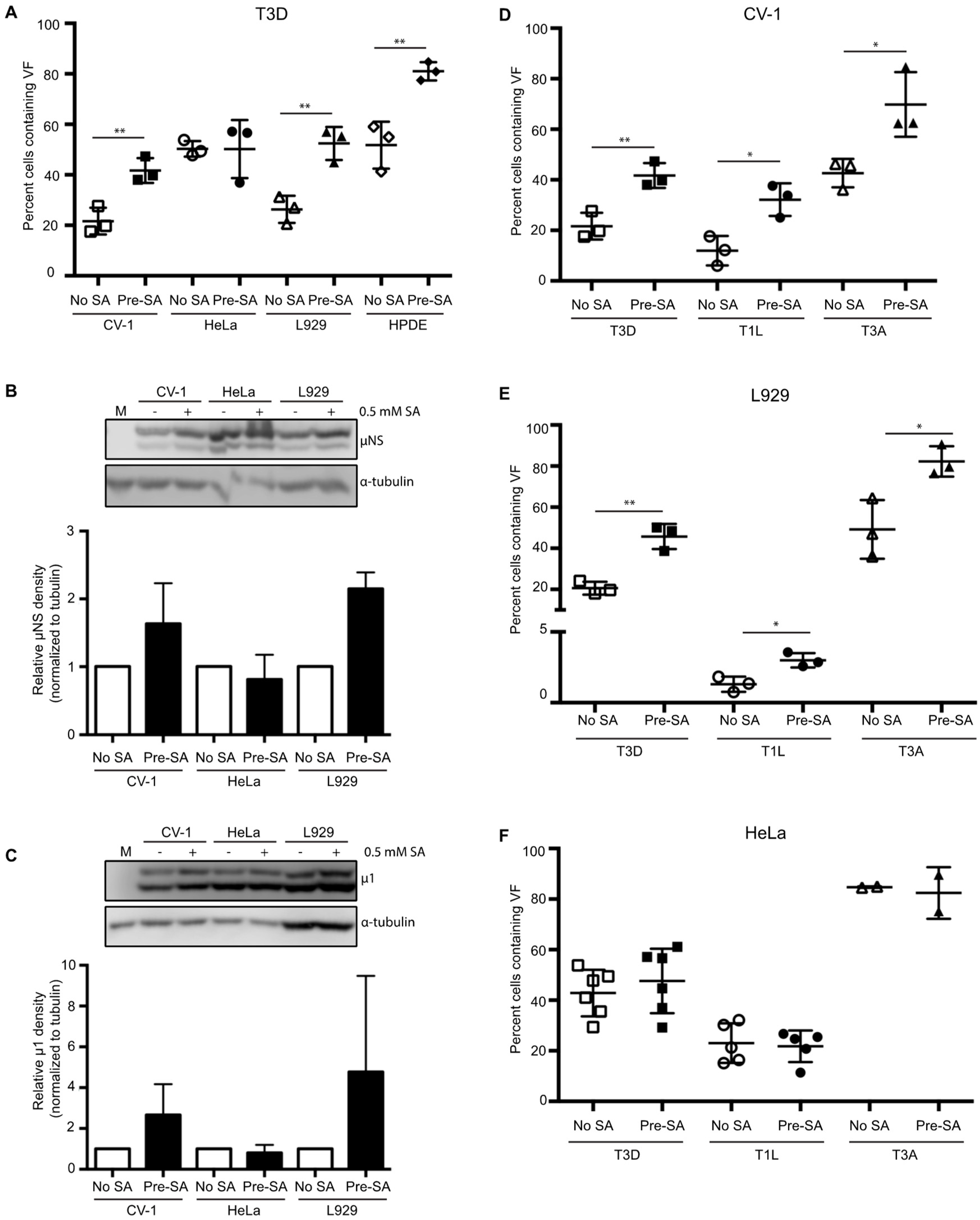
3.3. SA-Induced Enhancement of Reovirus Permissivity is Cell-Type Specific
3.4. SA-Induced Enhancement of Reovirus Permissivity is Strain-Independent
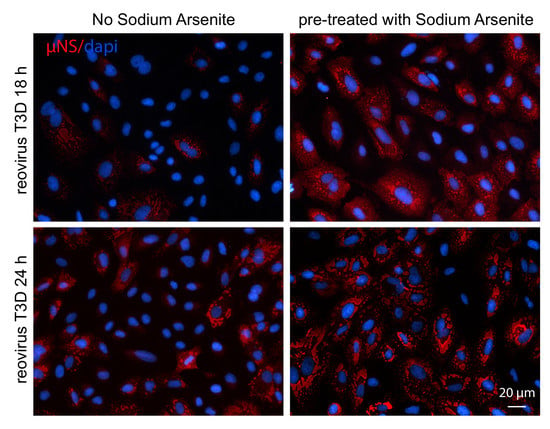
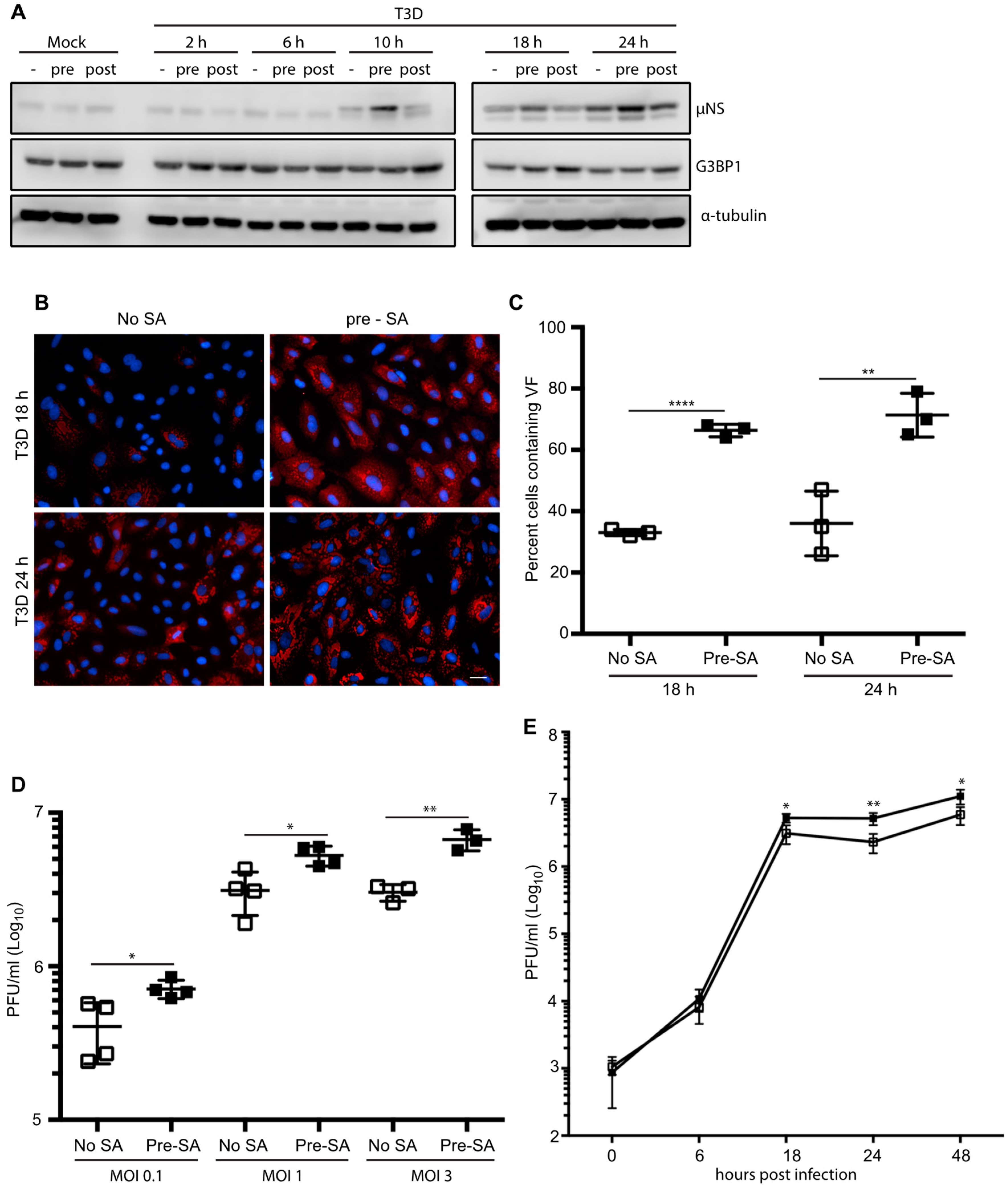
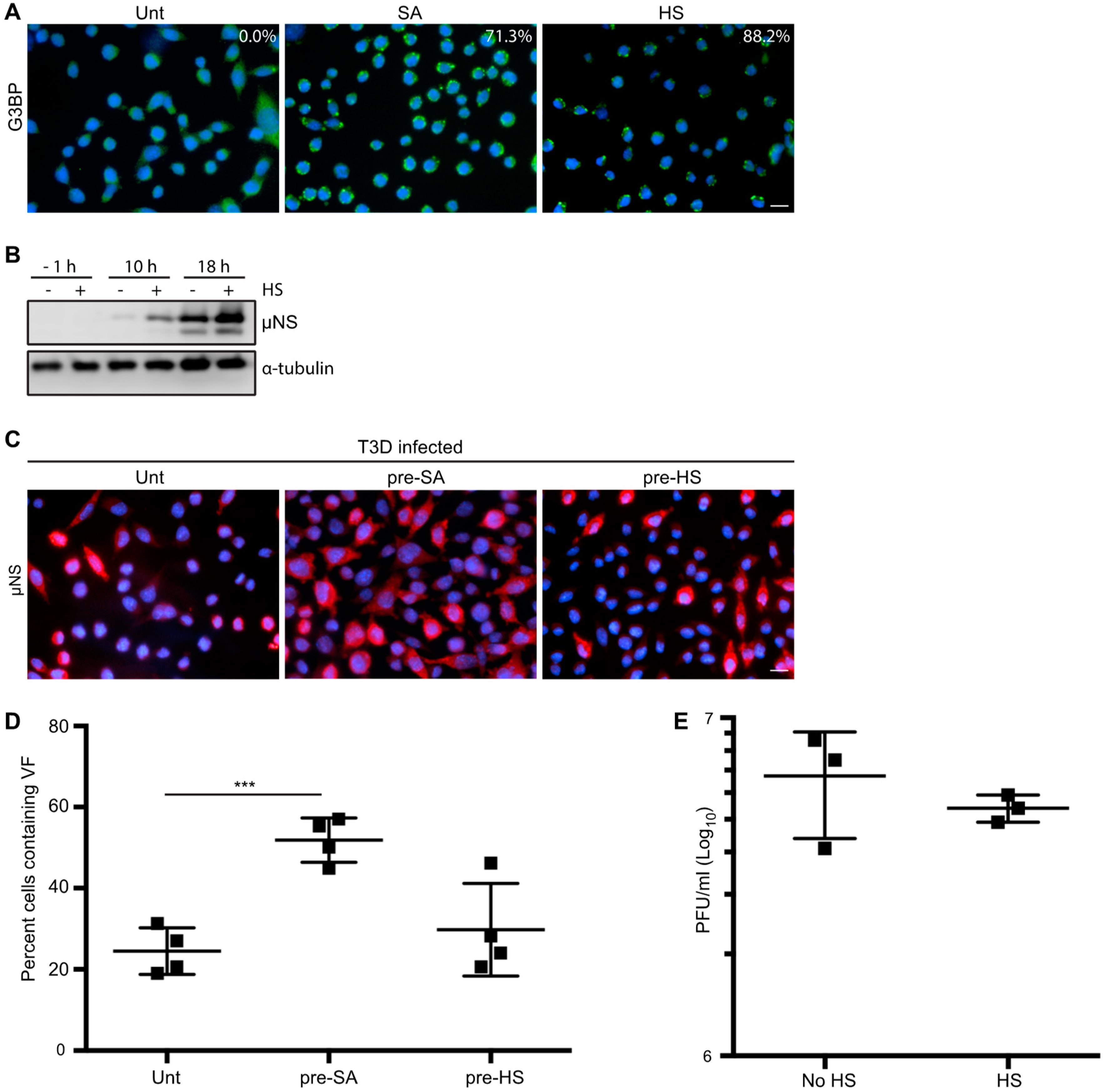
3.5. HS Prior to Infection Enhances Viral Protein Expression in T3D-Infected L929 Cells, but Does not Affect the Percentage of Infected Cells or Viral Yield
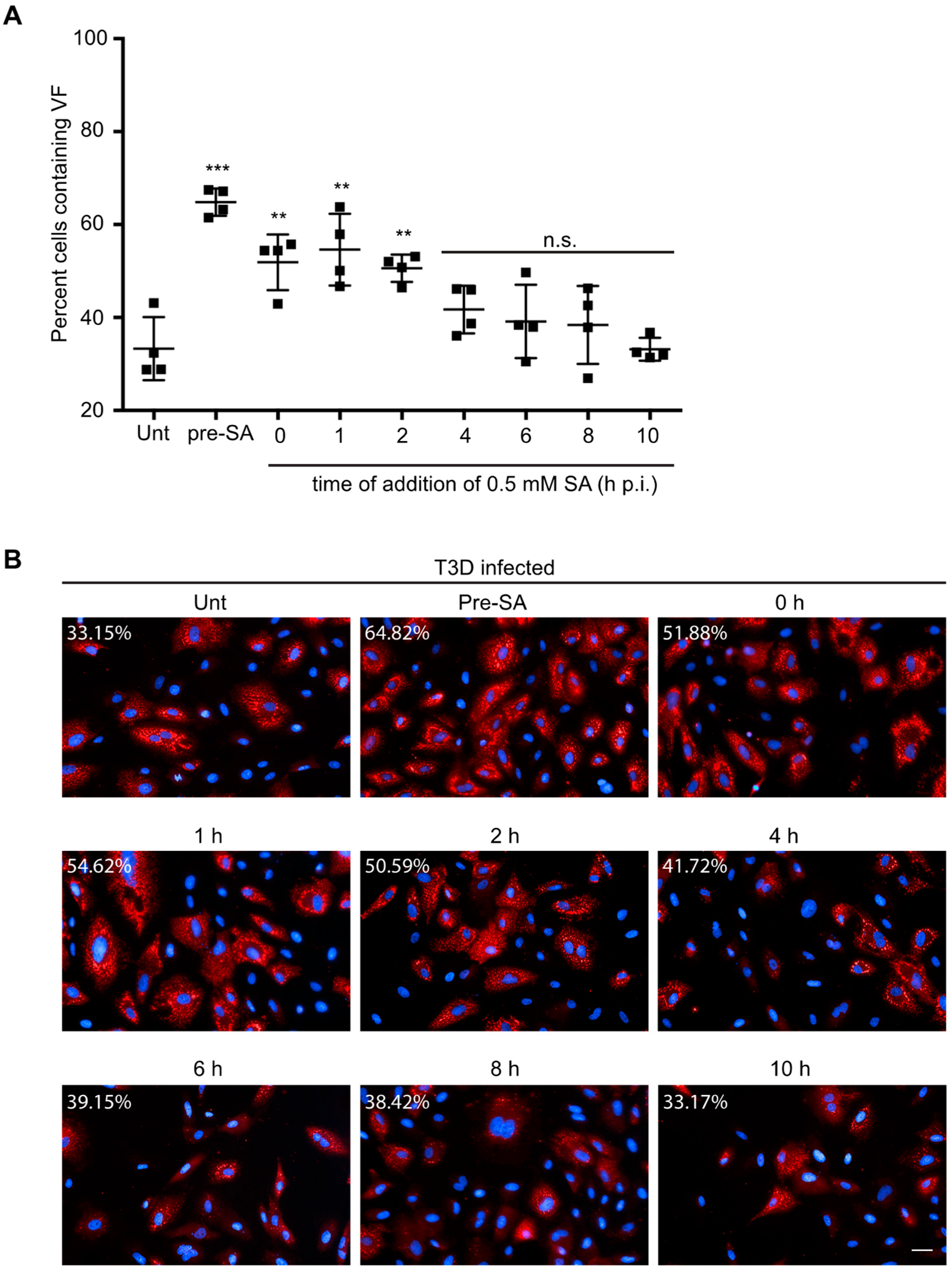
3.6. Addition of SA Prior to 4 h Enhances Reovirus Permissivity
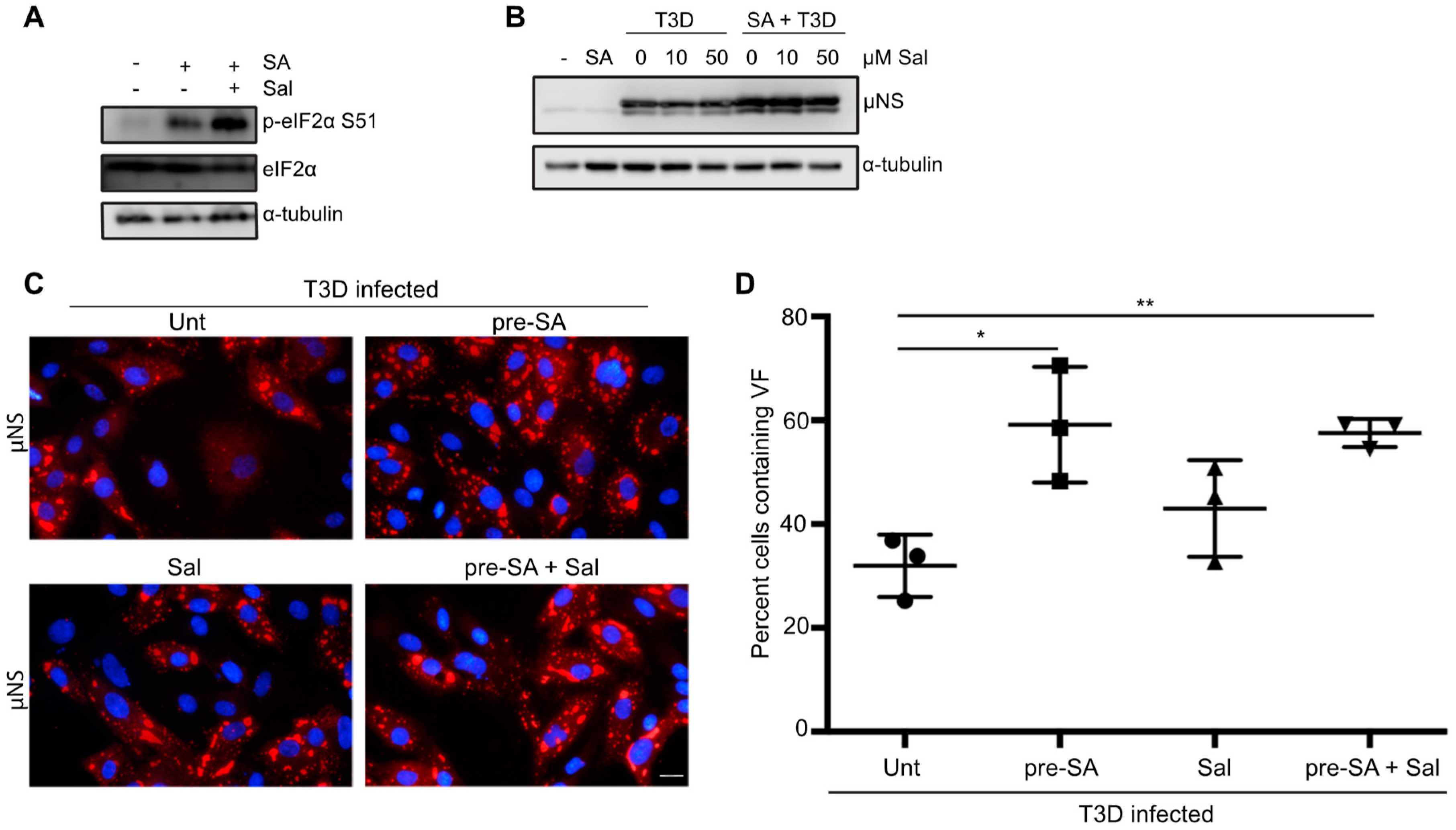
3.7. Preventing eIF2α De-Phosphorylation by Using Salubrinal has no Effect on Reovirus Infectivity
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Garcia, M.A.; Meurs, E.F.; Esteban, M. The dsRNA protein kinase PKR: Virus and cell control. Biochimie 2007, 89, 799–811. [Google Scholar] [CrossRef] [PubMed]
- Berlanga, J.J.; Ventoso, I.; Harding, H.P.; Deng, J.; Ron, D.; Sonenberg, N.; Carrasco, L.; de Haro, C. Antiviral effect of the mammalian translation initiation factor 2α kinase GCN2 against RNA viruses. EMBO J. 2006, 25, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Feng, Z.; He, B. Herpes simplex virus 1 infection activates the endoplasmic reticulum resident kinase PERK and mediates eIF-2α dephosphorylation by the gamma(1)34.5 protein. J. Virol. 2005, 79, 1379–1388. [Google Scholar] [CrossRef] [PubMed]
- Pakos-Zebrucka, K.; Koryga, I.; Mnich, K.; Ljujic, M.; Samali, A.; Gorman, A.M. The integrated stress response. EMBO Rep. 2016, 17, 1374–1395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamanaka, R.B.; Bennett, B.S.; Cullinan, S.B.; Diehl, J.A. PERK and GCN2 contribute to eIF2α phosphorylation and cell cycle arrest after activation of the unfolded protein response pathway. Mol. Biol. Cell 2005, 16, 5493–5501. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Zeng, H.; Novoa, I.; Lu, P.D.; Calfon, M.; Sadri, N.; Yun, C.; Popko, B.; Paules, R.; et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol. Cell 2003, 11, 619–633. [Google Scholar] [CrossRef]
- Lu, L.; Han, A.P.; Chen, J.J. Translation initiation control by heme-regulated eukaryotic initiation factor 2α kinase in erythroid cells under cytoplasmic stresses. Mol. Cell. Biol. 2001, 21, 7971–7980. [Google Scholar] [CrossRef] [PubMed]
- Anda, S.; Zach, R.; Grallert, B. Activation of Gcn2 in response to different stresses. PLoS ONE 2017, 12, e0182143. [Google Scholar] [CrossRef] [PubMed]
- Pyo, C.W.; Lee, S.H.; Choi, S.Y. Oxidative stress induces PKR-dependent apoptosis via IFN-γactivation signaling in Jurkat T cells. Biochem. Biophys. Res. Commun. 2008, 377, 1001–1006. [Google Scholar] [CrossRef]
- Kedersha, N.L.; Gupta, M.; Li, W.; Miller, I.; Anderson, P. RNA-binding proteins TIA-1 and TIAR link the phosphorylation of eIF-2α to the assembly of mammalian stress granules. J. Cell Biol. 1999, 147, 1431–1442. [Google Scholar] [CrossRef]
- Tourriere, H.; Chebli, K.; Zekri, L.; Courselaud, B.; Blanchard, J.M.; Bertrand, E.; Tazi, J. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 2003, 160, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Dauber, B.; Wolff, T. Activation of the Antiviral Kinase PKR and Viral Countermeasures. Viruses 2009, 1, 523. [Google Scholar] [CrossRef] [PubMed]
- Krishnamoorthy, J.; Mounir, Z.; Raven, J.F.; Koromilas, A.E. The eIF2α kinases inhibit vesicular stomatitis virus replication independently of eIF2α phosphorylation. Cell Cycle 2008, 7, 2346–2351. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Schmechel, S.C.; Williams, B.R.; Silverman, R.H.; Schiff, L.A. Involvement of the interferon-regulated antiviral proteins PKR and RNase L in reovirus-induced shutoff of cellular translation. J. Virol. 2005, 79, 2240–2250. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.A.; Schmechel, S.C.; Raghavan, A.; Abelson, M.; Reilly, C.; Katze, M.G.; Kaufman, R.J.; Bohjanen, P.R.; Schiff, L.A. Reovirus induces and benefits from an integrated cellular stress response. J. Virol. 2006, 80, 2019–2033. [Google Scholar] [CrossRef] [PubMed]
- Qin, Q.; Hastings, C.; Miller, C.L. Mammalian orthoreovirus particles induce and are recruited into stress granules at early times postinfection. J. Virol. 2009, 83, 11090–11101. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.L.; Arnold, M.M.; Broering, T.J.; Hastings, C.E.; Nibert, M.L. Localization of mammalian orthoreovirus proteins to cytoplasmic factory-like structures via nonoverlapping regions of μNS. J. Virol. 2010, 84, 867–882. [Google Scholar] [CrossRef] [PubMed]
- Fields, B.N.; Raine, C.S.; Baum, S.G. Temperature-sensitive mutants of reovirus type 3: Defects in viral maturation as studied by immunofluorescence and electron microscopy. Virology 1971, 43, 569–578. [Google Scholar] [CrossRef]
- Desmet, E.A.; Anguish, L.J.; Parker, J.S. Virus-mediated compartmentalization of the host translational machinery. mBio 2014, 5. [Google Scholar] [CrossRef]
- Choudhury, P.; Bussiere, L.; Miller, C.L. Mammalian Orthoreovirus Factories Modulate Stress Granule Protein Localization by Interaction with G3BP1. J. Virol. 2017, 91, e01298-17. [Google Scholar] [CrossRef] [Green Version]
- Becker, M.M.; Peters, T.R.; Dermody, T.S. Reovirus σNS and μNS proteins form cytoplasmic inclusion structures in the absence of viral infection. J. Virol. 2003, 77, 5948–5963. [Google Scholar] [CrossRef] [PubMed]
- McEwen, E.; Kedersha, N.; Song, B.; Scheuner, D.; Gilks, N.; Han, A.; Chen, J.J.; Anderson, P.; Kaufman, R.J. Heme-regulated inhibitor kinase-mediated phosphorylation of eukaryotic translation initiation factor 2 inhibits translation, induces stress granule formation, and mediates survival upon arsenite exposure. J. Biol. Chem. 2005, 280, 16925–16933. [Google Scholar] [CrossRef] [PubMed]
- Goral, M.I.; Mochow-Grundy, M.; Dermody, T.S. Sequence diversity within the reovirus S3 gene: Reoviruses evolve independently of host species, geographic locale, and date of isolation. Virology 1996, 216, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Parker, J.S.; Broering, T.J.; Kim, J.; Higgins, D.E.; Nibert, M.L. Reovirus core protein μ2 determines the filamentous morphology of viral inclusion bodies by interacting with and stabilizing microtubules. J. Virol. 2002, 76, 4483–4496. [Google Scholar] [CrossRef] [PubMed]
- Virgin, H.W.; Mann, M.A.; Fields, B.N.; Tyler, K.L. Monoclonal antibodies to reovirus reveal structure/function relationships between capsid proteins and genetics of susceptibility to antibody action. J. Virol. 1991, 65, 6772–6781. [Google Scholar] [PubMed]
- Virgin, H.W.; Bassel-Duby, R.; Fields, B.N.; Tyler, K.L. Antibody protects against lethal infection with the neurally spreading reovirus type 3 (Dearing). J. Virol. 1988, 62, 4594–4604. [Google Scholar] [PubMed]
- Reineke, L.C.; Lloyd, R.E. Diversion of stress granules and P-bodies during viral infection. Virology 2013, 436, 255–267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broering, T.J.; Parker, J.S.; Joyce, P.L.; Kim, J.; Nibert, M.L. Mammalian reovirus nonstructural protein microNS forms large inclusions and colocalizes with reovirus microtubule-associated protein μ2 in transfected cells. J. Virol. 2002, 76, 8285–8297. [Google Scholar] [CrossRef] [PubMed]
- Jayasuriya, A.K.; Nibert, M.L.; Fields, B.N. Complete nucleotide sequence of the M2 gene segment of reovirus type 3 dearing and analysis of its protein product μ1. Virology 1988, 163, 591–602. [Google Scholar] [CrossRef]
- Qin, Q.; Carroll, K.; Hastings, C.; Miller, C.L. Mammalian orthoreovirus escape from host translational shutoff correlates with stress granule disruption and is independent of eIF2α phosphorylation and PKR. J. Virol. 2011, 85, 8798–8810. [Google Scholar] [CrossRef]
- Burwick, N.; Aktas, B.H. The eIF2-α kinase HRI: A potential target beyond the red blood cell. Expert Opin. Ther. Targets 2017, 21, 1171–1177. [Google Scholar] [CrossRef] [PubMed]
- Schmechel, S.; Chute, M.; Skinner, P.; Anderson, R.; Schiff, L. Preferential translation of reovirus mRNA by a sigma3-dependent mechanism. Virology 1997, 232, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Munoz, A.; Alonso, M.A.; Carrasco, L. The regulation of translation in reovirus-infected cells. J. Gen. Virol. 1985, 66 Pt 10, 2161–2170. [Google Scholar] [CrossRef]
- Novoa, I.; Zeng, H.; Harding, H.P.; Ron, D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J. Cell Biol. 2001, 153, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Boyce, M.; Bryant, K.F.; Jousse, C.; Long, K.; Harding, H.P.; Scheuner, D.; Kaufman, R.J.; Ma, D.; Coen, D.M.; Ron, D.; et al. A selective inhibitor of eIF2α dephosphorylation protects cells from ER stress. Science 2005, 307, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Kumar, A.; Xu, Z.; Airo, A.M.; Stryapunina, I.; Wong, C.P.; Branton, W.; Tchesnokov, E.; Gotte, M.; Power, C.; et al. Zika Virus Hijacks Stress Granule Proteins and Modulates the Host Stress Response. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catanzaro, N.; Meng, X.J. Porcine reproductive and respiratory syndrome virus (PRRSV)-induced stress granules are associated with viral replication complexes and suppression of host translation. Virus Res. 2019, 265, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Garaigorta, U.; Heim, M.H.; Boyd, B.; Wieland, S.; Chisari, F.V. Hepatitis C virus (HCV) induces formation of stress granules whose proteins regulate HCV RNA replication and virus assembly and egress. J. Virol. 2012, 86, 11043–11056. [Google Scholar] [CrossRef]
- Lafarga, V.; Sung, H.M.; Haneke, K.; Roessig, L.; Pauleau, A.L.; Bruer, M.; Rodriguez-Acebes, S.; Lopez-Contreras, A.J.; Gruss, O.J.; Erhardt, S.; et al. TIAR marks nuclear G2/M transition granules and restricts CDK1 activity under replication stress. EMBO Rep. 2019, 20, e46224. [Google Scholar] [CrossRef]
- Zhang, T.; Delestienne, N.; Huez, G.; Kruys, V.; Gueydan, C. Identification of the sequence determinants mediating the nucleo-cytoplasmic shuttling of TIAR and TIA-1 RNA-binding proteins. J. Cell Sci. 2005, 118, 5453–5463. [Google Scholar] [CrossRef] [Green Version]
- Taupin, J.L.; Tian, Q.; Kedersha, N.; Robertson, M.; Anderson, P. The RNA-binding protein TIAR is translocated from the nucleus to the cytoplasm during Fas-mediated apoptotic cell death. Proc. Natl. Acad. Sci. USA 1995, 92, 1629–1633. [Google Scholar] [CrossRef] [PubMed]
- Esclatine, A.; Taddeo, B.; Roizman, B. Herpes simplex virus 1 induces cytoplasmic accumulation of TIA-1/TIAR and both synthesis and cytoplasmic accumulation of tristetraprolin, two cellular proteins that bind and destabilize AU-rich RNAs. J. Virol. 2004, 78, 8582–8592. [Google Scholar] [CrossRef] [PubMed]
- Mignone, F.; Gissi, C.; Liuni, S.; Pesole, G. Untranslated regions of mRNAs. Genome Biol. 2002, 3. [Google Scholar] [CrossRef] [PubMed]
- Kozak, M.; Shatkin, A.J. Sequences and properties of two ribosome binding sites from the small size class of reovirus messenger RNA. J. Biol. Chem. 1977, 252, 6895–6908. [Google Scholar] [PubMed]
- Kozak, M.; Shatkin, A.J. Identification of features in 5′ terminal fragments from reovirus mRNA which are important for ribosome binding. Cell 1978, 13, 201–212. [Google Scholar] [CrossRef]
- Strong, J.E.; Coffey, M.C.; Tang, D.; Sabinin, P.; Lee, P.W. The molecular basis of viral oncolysis: Usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998, 17, 3351–3362. [Google Scholar] [CrossRef]
- Marcato, P.; Shmulevitz, M.; Pan, D.; Stoltz, D.; Lee, P.W. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol. Ther. 2007, 15, 1522–1530. [Google Scholar] [CrossRef]
- Rudd, P.; Lemay, G. Correlation between interferon sensitivity of reovirus isolates and ability to discriminate between normal and Ras-transformed cells. J. Gen. Virol. 2005, 86, 1489–1497. [Google Scholar] [CrossRef]
- Twigger, K.; Roulstone, V.; Kyula, J.; Karapanagiotou, E.M.; Syrigos, K.N.; Morgan, R.; White, C.; Bhide, S.; Nuovo, G.; Coffey, M.; et al. Reovirus exerts potent oncolytic effects in head and neck cancer cell lines that are independent of signalling in the EGFR pathway. BMC Cancer 2012, 12, 368. [Google Scholar] [CrossRef]
- Phillips, M.B.; Stuart, J.D.; Rodriguez Stewart, R.M.; Berry, J.T.; Mainou, B.A.; Boehme, K.W. Current understanding of reovirus oncolysis mechanisms. Oncolytic Virother. 2018, 7, 53–63. [Google Scholar] [CrossRef]
- Liu, B.; Han, Y.; Qian, S.B. Cotranslational response to proteotoxic stress by elongation pausing of ribosomes. Mol. Cell 2013, 49, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Somji, S.; Todd, J.H.; Sens, M.A.; Garrett, S.H.; Sens, D.A. Expression of the constitutive and inducible forms of heat shock protein 70 in human proximal tubule cells exposed to heat, sodium arsenite, and CdCl(2). Environ. Health Perspect. 1999, 107, 887–893. [Google Scholar] [PubMed]
- Kaufer, S.; Coffey, C.M.; Parker, J.S. The cellular chaperone hsc70 is specifically recruited to reovirus viral factories independently of its chaperone function. J. Virol. 2012, 86, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Ivanovic, T.; Agosto, M.A.; Chandran, K.; Nibert, M.L. A role for molecular chaperone Hsc70 in reovirus outer capsid disassembly. J. Biol. Chem. 2007, 282, 12210–12219. [Google Scholar] [CrossRef] [PubMed]
- Garaigorta, U.; Chisari, F.V. Hepatitis C virus blocks interferon effector function by inducing protein kinase R phosphorylation. Cell Host Microbe 2009, 6, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Rojas, M.; Arias, C.F.; Lopez, S. Protein kinase R is responsible for the phosphorylation of eIF2α in rotavirus infection. J. Virol. 2010, 84, 10457–10466. [Google Scholar] [CrossRef] [PubMed]
- Raaben, M.; Groot Koerkamp, M.J.; Rottier, P.J.; de Haan, C.A. Mouse hepatitis coronavirus replication induces host translational shutoff and mRNA decay, with concomitant formation of stress granules and processing bodies. Cell. Microbiol. 2007, 9, 2218–2229. [Google Scholar] [CrossRef] [Green Version]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lutz, M.M., IV; Worth, M.P.; Hinchman, M.M.; Parker, J.S.L.; Ledgerwood, E.D. Mammalian orthoreovirus Infection is Enhanced in Cells Pre-Treated with Sodium Arsenite. Viruses 2019, 11, 563. https://doi.org/10.3390/v11060563
Lutz MM IV, Worth MP, Hinchman MM, Parker JSL, Ledgerwood ED. Mammalian orthoreovirus Infection is Enhanced in Cells Pre-Treated with Sodium Arsenite. Viruses. 2019; 11(6):563. https://doi.org/10.3390/v11060563
Chicago/Turabian StyleLutz, Michael M., IV, Megan P. Worth, Meleana M. Hinchman, John S.L. Parker, and Emily D. Ledgerwood. 2019. "Mammalian orthoreovirus Infection is Enhanced in Cells Pre-Treated with Sodium Arsenite" Viruses 11, no. 6: 563. https://doi.org/10.3390/v11060563
APA StyleLutz, M. M., IV, Worth, M. P., Hinchman, M. M., Parker, J. S. L., & Ledgerwood, E. D. (2019). Mammalian orthoreovirus Infection is Enhanced in Cells Pre-Treated with Sodium Arsenite. Viruses, 11(6), 563. https://doi.org/10.3390/v11060563